Rôle de la structure du génome viral sur la réplication du virus de l’hépatite C
319
0
0
Texte intégral
Figure


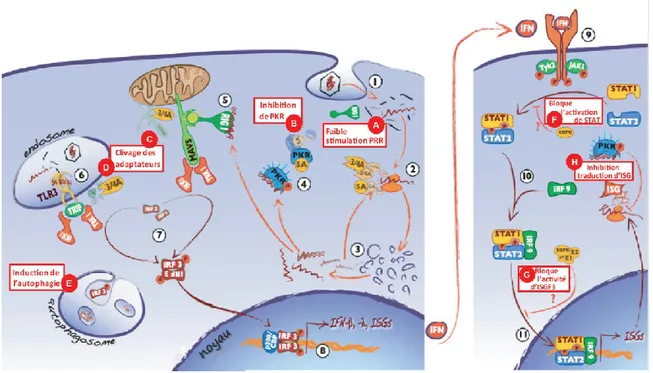

+7
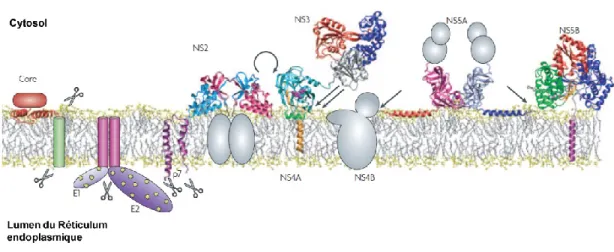

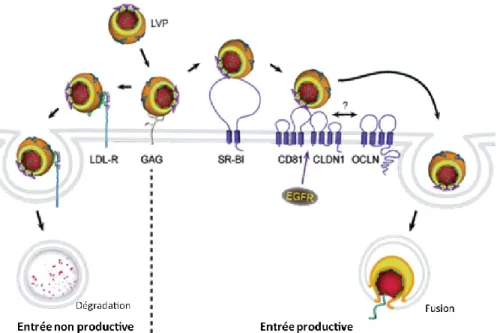
Documents relatifs
La réplication et la ségrégation du génome viral nécessitent 2 facteurs viraux que sont la protéine virale EBNA1 Epstein-Barr Nuclear Antigen 1 et la région oriP sur le génome
Pour appuyer cette hypothèse, j’ai observé que lors d’une infection avec le virus déficient pour l’expression de la protéine C, qui n’induit pas d’autophagie dans les
