Time-dependent wave-packet treatment of the Si4+ + He collision
Texte intégral
Figure
Documents relatifs
The estimated cost to a searcher is the number of sec- onds needed to peruse the suggested jump-in points. There are three cases. 1) If the suggested jump-in point does not
Structures of vibrational origin were observed at energies below 1 eV, well below the 2 shape resonance, in the cross sections for the excitation of vibrational overtones and
• Lets reflect on years of experience and take everything we know and combine it into a new modern language... Prominent differences
In order to find similar content in the image, a comparative analysis of the methods that work with the key points, namely: ORB, BRISK, AKAZE, FAST, respectively, based on
As the TBRS model predicted, the longer attentional capture induced by the close condition had a detrimental effect on maintenance and resulted in poorer recall performance than
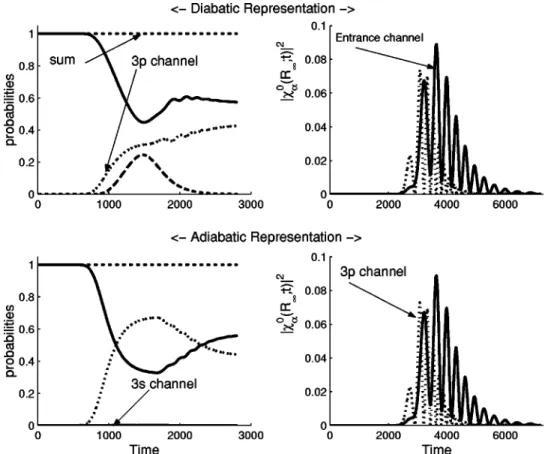
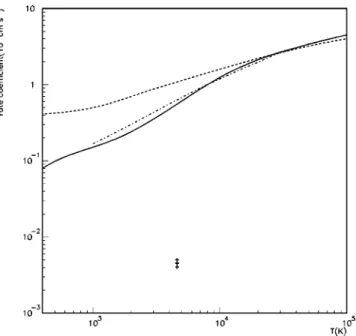
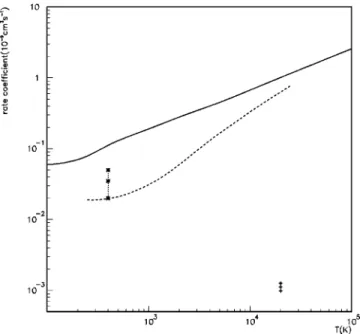
1 and 2, we present the total cross sections for excitation of the He + ground state into the 2s and 2p states, respectively, obtained using the two approaches described above
in Figure 2a that corresponds to the total number of events for one-electron capture in the same data set.. First we note that the value obtained for the double electron capture
Comparing these results with mass spectrometer measurements of Carbone /I/, we conclude that the slow changes of the electric field, the wave number and the frequency of
