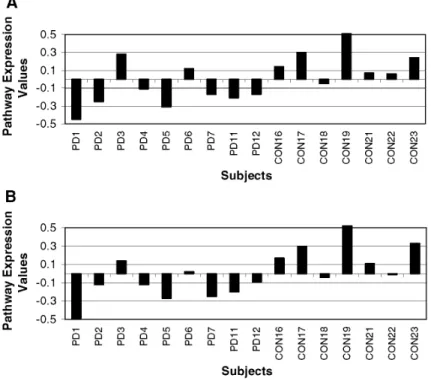
Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson's disease.
Texte intégral
Figure

Documents relatifs
As for changes in spindle activity associated with mild AD, AD patients do not show any significant decrease in the number of total sleep spindles, nor of spindle intensity if slow
In this study, we were interested in the effect of gender on the results of the voice localizer and we asked an explorative research question of whether brain activation
At the discrete level, this property is obtained by using in the discretization of the body forces a local, divergence-preserving velocity reconstruction operator whose normal
Pre- and mature RNase P from the above strains expressing the 33HA-tagged protein subunits were iso- lated using Sephadex G-200 as an affinity resin + The eluates were then split
The particle habit is also sensitive to radiance uncertainties, which is not surprising as the single scattering properties of some habits, like two- and three-dimensional rosettes,
Retinoic acid-treated (adipogenic conditions) or not (skeletal myogenic conditions) EBs from wild type CGR8 ES cells were induced to differentiate and treated or not by 100 mM
2)) L L''iin nd diiccaatteeu urr m meen nssu ueell d dee ll''eem mp pllo oii iin nttéérriim maaiirree een n ffiin n d dee m mo oiiss est élaboré par la Dares à partir
Après avoir significativement reculé en 2008 (-1,2 point) et progressé modestement en 2009 (+0,3 point), le taux de rotation (défini comme la moyenne des taux d’entrée et de
