Université de Sherbrooke
Rôle des prophages dans la virulence de Clostridioides difficile
et développement d’un logiciel bio-informatique pour améliorer l’analyse des génomes de phages
Par Julian Garneau
Département de microbiologie et d’infectiologie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph. D.)
en microbiologie
Sherbrooke, Québec, Canada Avril, 2020
Membres du jury d’évaluation
Pr Louis-Charles Fortier, Département de microbiologie et infectiologie, directeur de thèse Dr Marc Monot, Département de génomes et génétique, Institut Pasteur, co-directeur Pr Louis Valiquette, Département de microbiologie et infectiologie, co-directeur
Pr Alfredo Menendez, Département de microbiologie et infectiologie, directeur programme Pr Sébastien Rodrigue, Département de biologie, faculté des sciences, Université de
Sherbrooke, évaluateur externe au programme
Pr Steve Charette, Département de biochimie, de microbiologie et de bio-informatique, Faculté des sciences et de génie, Université Laval, évaluateur externe à l’université
RÉSUMÉ
Rôle des prophages dans la virulence de Clostridium difficile
et développement d’un logiciel bio-informatique pour améliorer l’analyse des génomes de phages
Par Julian Garneau
Département de microbiologie et d’infectiologie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph. D.) en microbiologie, Université de Sherbrooke,
Sherbrooke, Québec, Canada, J1H 5N4
Clostridioides difficile, longtemps connue sous le nom de Clostridium difficile, est une bactérie pathogène et résistante à plusieurs antibiotiques capable de coloniser le système digestif des mammifères, dont celui des humains. Des études ont suggéré que certains types de souches (ex. : ribotype 027) étaient reliés à des infections plus graves. Un de nos objectifs était d’éclaircir la situation controversée concernant l’hypervirulence de la souche 027. Nous avons donc réalisé un typage moléculaire discriminant sur une collection de 450 isolats de ribotype 027, afin de vérifier si ces souches pouvaient avoir évolué en sous-groupes génétiques ayant des niveaux de virulence variables. Nos analyses ont permis d’observer que les souches d’un sous-groupe MLVA étaient reliées à des cas cliniques moins graves, suggérant une possible évolution de la souche hypervirulente en souche moins agressive. Les prophages ont la capacité d’influencer l’évolution, l’adaptabilité et la virulence des bactéries, mais leur rôle est relativement peu exploré chez C. difficile. Des travaux non complétés présentés en annexe suggèrent que le prophage phi027, que nous avons retrouvé intégré dans la quasi-totalité des isolats de la souche épidémique et « hypervirulente » (ribotype 027) a la capacité d’augmenter la production de toxines par la bactérie. Dans le cadre d’un volet plus fondamental, nous avons analysé en détail la composition génétique ainsi que la prévalence d’un prophage récemment découvert au laboratoire, nommé le phiCD211. Nous avons constaté que ce phage possédait le plus grand génome parmi les phages infectant C. difficile qui sont connus à ce jour. Nous avons donc analysé en détail le génome de ce phage et nous avons pu identifier plusieurs gènes dont les fonctions sont possiblement associées à la virulence.
Enfin, dans ce projet de doctorat, nous avions également comme objectif d’améliorer et d’accélérer l’étude et la caractérisation des phages de façon globale. Un des mécanismes importants chez les bactériophages est l’étape d’encapsidation de l’ADN viral à l’intérieur de la procapside (capside vide). Plusieurs mécanismes existent selon les différents phages et il est souvent très laborieux de les identifier et de les caractériser en laboratoire. Nous avons donc développé une approche bio-informatique permettant d’identifier ces mécanismes en utilisant les lectures de séquençage de l’ADN génomique de phages. Notre procédure permet aussi d’identifier précisément les extrémités (termini) des génomes.
Mots clés : Clostridium difficile, bactériophage, toxine, virulence, phagemide, phagothérapie, virome
SUMMARY
Investigation of prophage role in virulence of Clostridium difficile and development of a bioinformatics software to improve the analysis of phage genome sequences
By Julian Garneau
Microbiology and Infectiology
Thesis presented at the Faculty of Medicine and Health Sciences to obtain
the philosophiae doctor (Ph. D.) diploma in microbiology, Faculty of medicine and health sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Clostridioides difficile, long known as Clostridium difficile, is a pathogenic bacterium resistant to several antibiotics capable of colonizing the digestive tract of mammals, including humans. Previous studies have suggested that certain types of strains (e.g. ribotype 027) are linked to more serious infections. One of our objectives was to clarify the ambiguous situation concerning the hypervirulence of the 027 strain. We therefore used a highly discriminant molecular typing method to analyze a collection of 450 isolates of ribotype 027, in order to verify if the 027 strain may have evolved into genetic subgroups with varying levels of virulence. Our analyses showed that the isolates belonging to one MLVA subgroup were linked to less serious clinical outcomes, suggesting a possible evolution of the hypervirulent strain into a less aggressive one.
Prophages have the capacity to influence the evolution, the adaptability and the virulence of bacteria, but their role is relatively unexplored in C. difficile. The preliminary work presented in the appendix suggests that the phi027 prophage, which we found integrated into almost all isolates of the epidemic and "hypervirulent" strain (ribotype 027) has the capacity to increase the production of toxins by the bacterium. In a more fundamental part of this project, we aimed to analyze in detail the genetic composition as well as the prevalence of a prophage recently discovered in the laboratory, called phiCD211. We have found that this phage has the largest genome among the phages infecting C. difficile known to date. We therefore analyzed in detail the genome of this phage and we were able to identify several genes with functions that are possibly associated with bacterial virulence.
Lastly, we also had the objective of improving and accelerating the study and characterization of phages in general. One of the important steps in the bacteriophage life cycle is the packaging of its genomic DNA inside the procapsid (empty capsid). Several mechanisms exist according to the different phages and it is often labor-intensive and difficult to characterize those mechanisms in the laboratory. We have therefore developed a rapid bioinformatics approach that identifies the different known mechanisms using the reads obtained after sequencing the genomic DNA of a given phage. Our procedure also makes it possible to precisely identify the ends (termini) of the phage genomes as well as other important details related to the different packaging mechanisms.
Keywords: Clostridium difficile, bacteriophage, toxin, virulence, phagemid, phagotherapy, virome
TABLE DES MATIÈRES
CHAPITRE I ... 1
Introduction ... 1
La bactérie Clostridium difficile ... 1
Les infections à Clostridium difficile, un problème de santé publique important ... 2
Épidémiologie de Clostridium difficile ... 3
La souche épidémique de ribotype 027 (lignée NAP1/BI/027) ... 7
Caractéristiques générales du génome de Clostridium difficile ... 11
Les facteurs de virulence chez Clostridium difficile ... 14
TcdA et TcdB, les facteurs de virulence principaux ... 14
Production de toxines dans la lignée NAP1/BI/027 ... 16
Le PaLoc et la régulation de l’expression des toxines ... 18
Autres facteurs de virulence ... 24
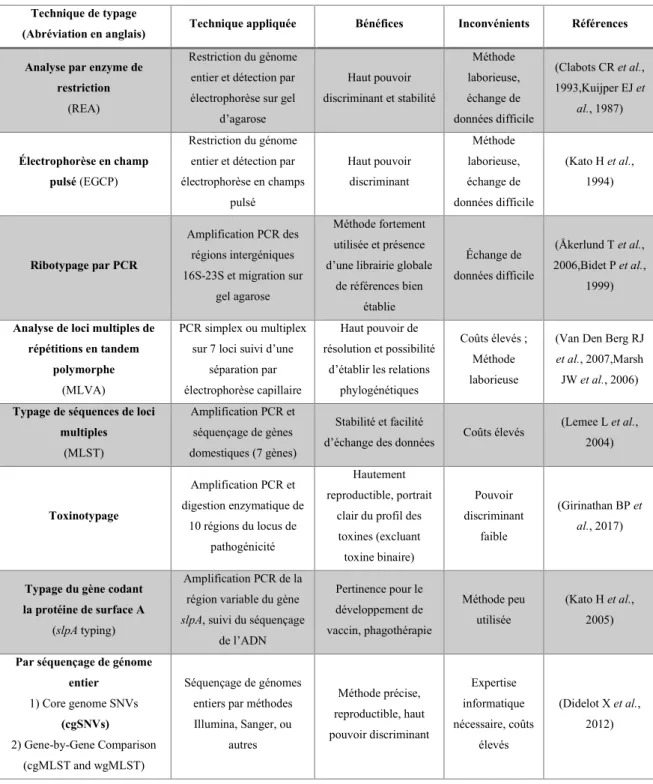
Méthodes de typage moléculaire pour Clostridium difficile ... 27
Méthodes de typage les plus utilisées ... 28
Électrophorèse sur gel en champs pulsé (EGCP) ... 28
Ribotypage par PCR ... 29
Typage par séquençage de loci multiples (MLST) ... 30
Séquençage de génome entier ... 30
Autres méthodes de typage développées et testées pour Clostridium difficile ... 32
Analyse par enzyme de restriction (REA) ... 32
Toxinotypage ... 32
Typage slpA ... 32
Analyse de loci multiples de répétitions en tandem polymorphes ... 33
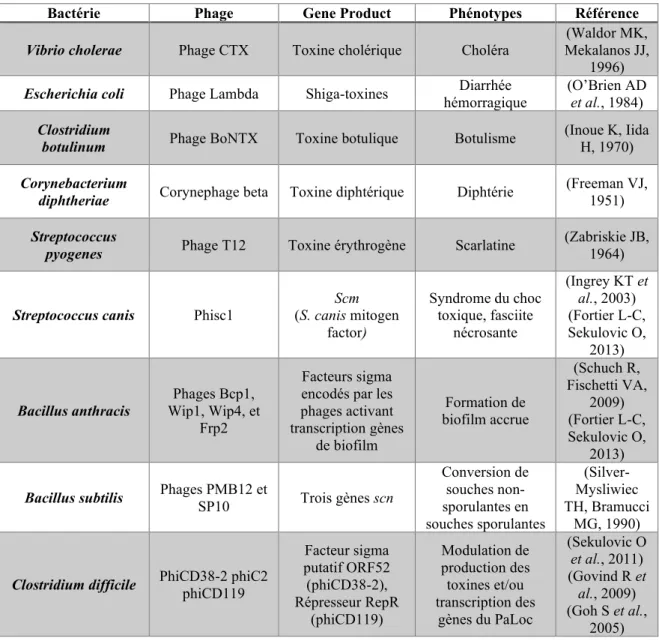
Les bactériophages et leur importance dans la biologie bactérienne ... 34
Système de classification des bactériophages ... 35
Classification selon la morphologie ... 35
Classification selon le cycle de réplication ... 35
Classification selon le mécanisme d’encapsidation de l’ADN génomique ... 37
La conversion lysogénique ... 41
Exemples de conversions lysogéniques avec changements phénotypiques importants ... 42
Impacts connus des prophages sur la bactérie Clostridium difficile ... 43
CHAPITRE II ... 49 Article 1 ... 49 CHAPITRE III ... 71 Article 2 ... 71 CHAPITRE IV ... 99 Article 3 ... 99 CHAPITRE V ... 125 Discussion et perspectives... 125 Remerciements ... 141 Annexe I ... 143 Références ... 159
LISTE DES FIGURES
CHAPITRE I
Figure 1. Distribution des principaux ribotypes de C. difficile à travers le monde selon les
années ... 4
Figure 2. Étapes importantes d’une infection à C. difficile. ... 10
Figure 3. Représentation des différents cycles de réplication chez les bactériophages ... 37
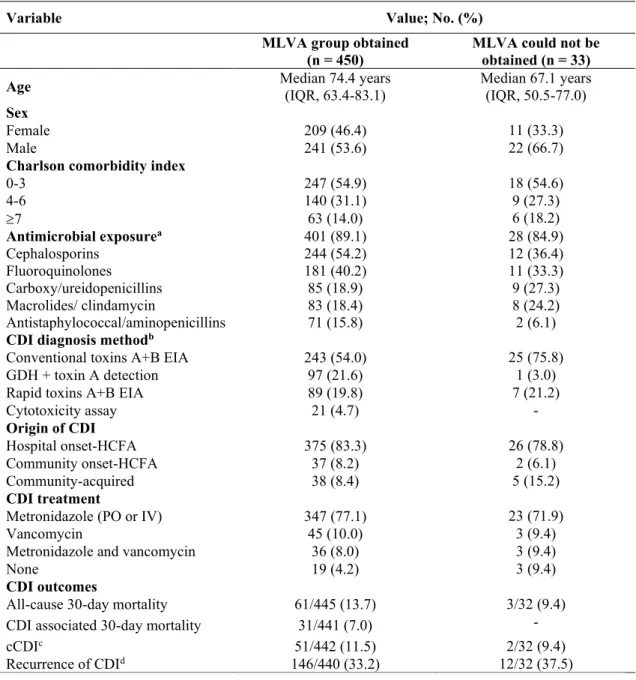
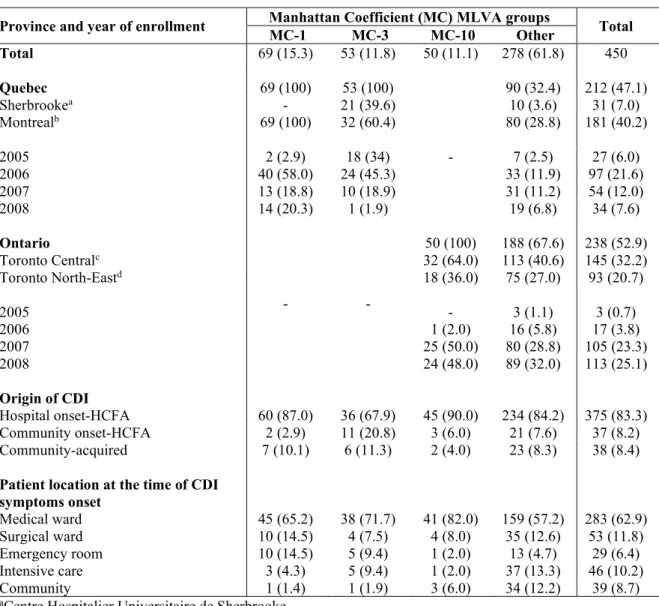
CHAPITRE II Figure 1. Minimum-spanning tree of MLVA data from 450 R027 C. difficile isolates ... 59
CHAPITRE III Figure 1. PhiCD211 morphology as observed by TEM ... 81
Figure 2.Genome organization of phiCD211 and whole genome comparison with other phiCD211-like phages ... 83
Figure 3. Whole genome comparison using Gegenees ... 89
Figure 4. Whole genome alignment of 12 representative phiCD211-like phages using Phamerator ... 90
CHAPITRE IV Figure 1. Biases in the number of reads starting at DNA termini ... 109
Figure 2. Sequence coverage at termini position ... 111
Figure 3. Detection of Mu-like phages ... 117
Figure 4. Bacteriophage P1 presence of secondary termini ... 118
ANNEXE I Figure 1. Whole-genome comparison of C. difficile isolates using Gegenees ... 153
Figure 2. Relative quantification by ELISA of intracellular (A) and extracellular (B) toxin A production ... 154
Figure 3. Growth curves for wild-type and lysogenic strains in TY medium ... 154
LISTE DES TABLEAUX
CHAPITRE I
Tableau 1. Résumé des facteurs de virulence chez C. difficile ... 20 Tableau 2. Principales techniques de typage développées chez C. difficile ... 31 Tableau 3. Exemples de facteurs de virulence encodés par des prophages ... 43 CHAPITRE II
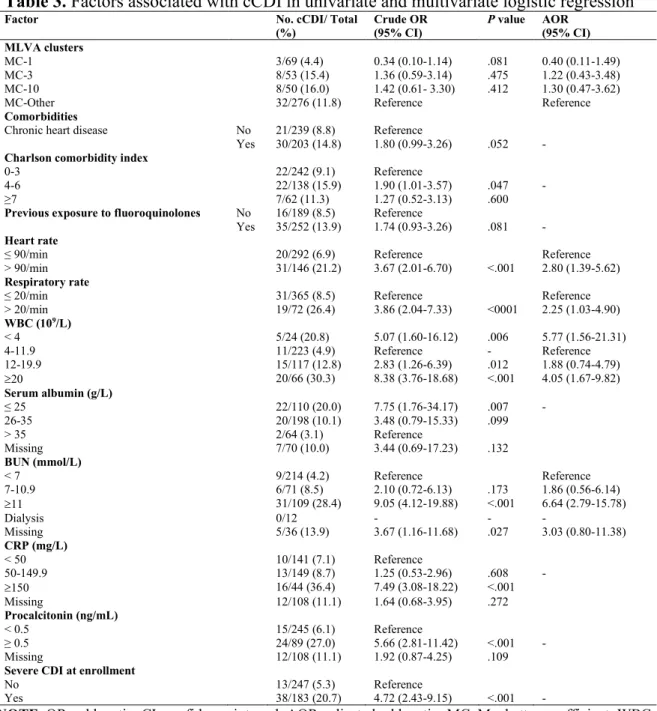
Tableau 1. Characteristics of patients with R027 Clostridioides difficile infection ... 58 Tableau 2. Geographical and temporal distribution of major MLVA groups ... 61 Tableau 3. Factors associated with cCDI in univariate & multivariate logistic regression . 63 CHAPITRE III
Tableau 1. Structural and virion-associated proteins detected by mass spectrometry... 84 Tableau 2. Prevalence of phiCD211 in 2,548 C. difficile isolates ... 88 CHAPITRE IV
Tableau 1. Characteristics of reference phages ... 115 Tableau 2. Summary of PhageTerm results ... 116 ANNEXE I
Tableau 1. Prevalence of phi027 in C. difficile genomes according to SlpA type ... 151 Tableau 2. Ribotype R027 isolates used in this study ... 152 Tableau S1. Oligonucleotides used in this study ... 158
LISTE DES ABRÉVIATIONS
CPM ICSP ICD DACD SNP SNV PaLoc CDT GTD APD CROPS CSPG4 NOX ROS GTP CAMPs PCR REA PLFR EGCP VNTR MLVA MLST WGS cgSNV cgMLST wgMLST HMW LMW SLP FQR sb db ADN ADNg ARN ATP DTR IPP ICDc ACE RBP ICTV SCV Colite pseudomembraneuseInternational Committee on Systematics of Prokaryotes Infections à Clostridium difficile
Diarrhées associées à C. difficile Single nucleotide polymorphisim Single nucleotide variant
Pathogenecity locus
Clostridium difficile transferase Glycosyltransferase domain Autoprotease domain
Combined repetitive oligopeptides Chondroitine sulfate proteoglycan 4 NADPH oxydase
Reactive oxygen species Guanosine triphosphate
Cationic antimicrobial peptides Polymerase chain reaction
Restriction Endonuclease Analysis
Polymorphisme de longueur des fragments de restriction Électrophorèse sur gel en champ pulsé
Variable copy Numbers of Tandem Repeats
Multi-Locus Variable Copy Numbers of Tandem Repeats Analysis Multilocus sequence typing
Whole genome sequencing (Séquençage de génome entier) Core genome single nucleotide variant
Core genome multilocus sequence typing Whole genome multi locus sequence typing High molecular weight
Low molecular weight Surface layer protein Fluoroquinolone resistant Simple brin Double brin Acide désoxyribonucléique ADN génomique Acide ribonucléique Adénosine triphosphate Direct terminal repeats
Inhibiteurs de la pompe à protons
Infections à Clostridium difficile compliquée Allele-Coupled Exchange
Receptor binding protein
International Committee on Taxonomy of Viruses Small Colony Variant
CRISPR
CHAPITRE I
INTRODUCTION
La bactérie Clostridium difficile
La bactérie Clostridium difficile est un des agents pathogènes les plus importants chez l’humain. Elle est responsable d’un grand nombre de cas d’infections à travers le monde (Martin JSH et al., 2016). L’infection de l’humain par cette bactérie peut occasionner une variété de symptômes tels que des diarrhées sévères, des colites pseudomembraneuses (CPM) et peut dans des cas plus graves conduire au décès des sujets infectés. Plusieurs études épidémiologiques récentes permettent cependant de constater que C. difficile ne peut plus être considéré comme un agent pathogène exclusivement nosocomial, car sa propagation et sa présence en milieux communautaires semblent avoir augmentées considérablement au cours des dernières années (Beaugerie L et al., 2003, Guh AY et al., 2017).
C. difficile a pour la première fois été observée et identifiée comme une nouvelle bactérie anaérobie stricte produisant des spores en 1935 par Ivan C. Hall et Elizabeth O’Toole au cours d’une étude portant sur l’analyse des changements bactériens dans le méconium de nouveau-nés nourris au lait maternel (Köpke M et al., 2013). Cet agent pathogène n’a donc pas été isolé en premier lieu en raison d’une manifestation clinique causée par une infection, mais plutôt dans le cadre de recherches fondamentales. En raison de la difficulté à la cultiver et à la manipuler, cette nouvelle bactérie fut, à l’origine, nommée Bacillus difficilis, mais elle a rapidement été renommée Clostridium difficile lors de sa reclassification en 1938. Jusqu’à aujourd’hui, plusieurs tentatives de reclassification ont eu lieu et différentes appellations ont été proposées, telles que Peptoclostridium difficile (Yutin N et Galperin MY, 2013) et plus récemment, Clostridioides difficile (Lawson PA et al., 2016). Cependant, comme aucune décision n’a été prononcée par l’« International Committee on Systematics of Prokaryotes » (ICSP) pour procéder à une invalidation officielle d’appellation, Clostridioides difficile est fortement suggéré et adopté, mais Clostridium difficile demeure aussi une désignation valide et acceptable jusqu’à nouvel ordre (Oren A et Rupnik M, 2018).
Ce n’est qu’en 1978, soit environ 40 ans après son identification, que C. difficile a été clairement suggérée comme agent étiologique causant des diarrhées graves et des CPM (George RH et al., 1978, Larson HE et al., 1978). C’est aussi à cette période que le lien entre les toxines produites par la bactérie et les symptômes caractéristiques des ICD a été établi. Au cours de cette même année, il a aussi été conclu que la vancomycine pourrait s’avérer être un traitement efficace pour éliminer la bactérie et résoudre les CPM (Keighley MRB et al., 1978).
Les infections à Clostridium difficile, un problème de santé publique important
C. difficile est considérée comme la cause la plus fréquente de diarrhées infectieuses associées aux soins de santé dans les pays industrialisés, incluant le Canada (CNISP, 2018). Dans une étude publiée en 2015, le nombre de cas d’ICD au Canada en 2012 a été estimé à 37 800, dont 21% étaient des épisodes de rechutes, et 27% étaient des cas de récurrences (rechutes ou réinfections) (Tang-Feldman Y et al., 2003). Le coût total imposé à la société canadienne par cette maladie est estimé à environ 281 millions CAD (Levy AR et al., 2015).
Aux États-Unis, le nombre total de personnes infectées est évalué à environ 300 000 personnes par année et les coûts associés à la gestion des ICD sont considérables (Hall AJ et al., 2012). Il est difficile de connaître les coûts reliés à la gestion des ICD de façon précise, mais les estimations faites dans ce pays ont évalué ces dépenses à environ 5,4-6,3 milliards USD par année (Zhang S et al., 2016). Dans cette même étude, réalisée sur des données amassées entre 2005 et 2015, il a été estimé que le coût par cas d’ICD était en moyenne de 42 316 USD.
Il est important de mentionner qu’un très grand nombre de cas est potentiellement sous-diagnostiqué à travers le monde. En Europe seulement, les ICD non détectées sont évaluées à près de 40,000 cas chaque année (Davies KA et al., 2014). Il est souvent considéré que les ICD représentent un problème plus fortement présent dans les régions industrialisées ou riches en ressources (Balassiano IT et al., 2012). Cependant, cette notion est fortement mise en doute par le fait que les ICD sont clairement sous-diagnostiquées, particulièrement dans les régions ayant des ressources limitées et ne pouvant pas exécuter les tests cliniques pour
détecter C. difficile comme agent responsable des diarrhées chez les patients hospitalisés. Tel que mentionné par un groupe de chercheurs étudiant la question à Wuhan, en Chine, le fait de ne pas tester pour les ICD ne veut pas dire que les ICD n’existent pas (Galaydick J et al., 2015). Dans cette étude, il a été conclu que la prévalence des ICD dans la ville de Wuhan était beaucoup plus élevée que ce qui était précédemment assumé. Une des conclusions simples de cette étude est que la conscience face au problème des ICD était faible chez le personnel médical, ayant comme conséquence qu’aucune procédure n’était adoptée de façon routinière pour détecter la bactérie. Cette même observation a été rapportée dans une revue récente, dans laquelle les auteurs insistent sur le fait que les ICD sont une importante cause de diarrhées en milieu hospitalier et communautaire dans les pays à ressources limitées (Roldan GA et al., 2018). Leurs analyses suggèrent même que l’incidence générale, la prévalence et la mortalité associées aux ICD sont similaires, voire même plus élevées dans ces pays.
Épidémiologie de Clostridium difficile
Lorsqu’on parle de l’épidémiologie globale de C. difficile, on peut certainement séparer l’histoire de cette bactérie pathogène en trois grandes périodes importantes. Premièrement, il faut mentionner la période qui a précédé les premières grandes épidémies de C. difficile (avant les années 2000). Ensuite est survenue la période épidémique, dont les sommets ou les pics épidémiques ont eu lieu à différents moments dans le monde, mais qui s’est globalement étendue des années 2000 à 2010 (Valiente E et al., 2014). Finalement, bien qu’il n’y ait pas de moment clairement défini, on peut mentionner la période post-épidémie, où les ICD ont persisté, mais tout de même grandement diminué à travers le monde en raison des mesures générales de santé publique adoptées.
Figure 1. Distribution des principaux ribotypes de C. difficile à travers le monde selon les années. Figure adaptée avec l’autorisation de (Valiente E et al., 2014)
Comme mentionné un peu plus tôt, ce n’est qu’autour des années 1980, environ 40 ans après sa première identification, que C. difficile fut clairement reconnue comme la bactérie responsable des diarrhées et des CPM observées chez certains patients malades (George RH et al., 1978,Larson HE et al., 1978). Pour la période précédant les années 2000, qu’on peut caractériser de pré-épidémique, des éclosions d’ICD ont été observées à travers le monde, mais les taux rapportés sont demeurés relativement stables (Pépin J et al., 2004). On peut notamment penser aux éclosions provoquées par la « souche J » (type REA) résistante à la clindamycine dans différents hôpitaux des États-Unis (1989-1992) et du Royaume-Uni (début années 1990) (Bauer MP et al., 2011,He M et al., 2010). Globalement, de 1996 à 2000, l'incidence des ICD dans certains hôpitaux de soins intensifs aux États-Unis a été relativement fixe à environ 30–40 cas pour 100 000 habitants. En 2003, ce nombre a cependant presque doublé passant à 60 cas pour 100 000 habitants (McDonald LC et al., 2006). Au Canada, au Québec plus précisément, sur une période de temps similaire, il a été rapporté que le taux d’ICD avait également plus que doublé passant de 66 cas par 100 000 habitants en 1991 à 156 cas par 100 000 habitants en 2003 (Pépin J et al., 2004). Dans cette même étude, en plus de l’augmentation du nombre d’ICD, il avait été observé que la
proportion de cas compliqués s’était aussi fortement accentuée, passant de 7,1% en 1991 à 18,2% en 2003.
Cette période épidémique ayant commencé au début des années 2000 est caractérisée par l’apparition et la dissémination rapide d’une souche multirésistante et caractérisée comme hypervirulente. Au début, cette souche a été nommée NAP1/B1/027 (North American pulsed-field gel electrophoresis type 1, restriction endonuclease group BI, polymerase chain reaction ribotype 027) en raison des trois méthodes de typage appliquées à l’époque, le EGCP, le REA et le ribotypage par PCR respectivement. Un peu plus tard, en raison de la prédominance du ribotypage par PCR, cette souche s’est souvent vue nommée plus simplement la souche de ribotype 027 (R027).
Dans certaines régions, comme au Québec, cette souche a été isolée dans près de 80% des cas d’ICD (Abou Chakra CN et al., 2015). En plus d’être retrouvée dans la majorité des ICD, il a été observé que la R027 semblait être responsable d’un plus grand nombre de cas sévères et un plus haut taux de mortalité, comparativement aux autres souches plus rarement isolées (c.-à-d. NAP2 et autres types EGCP) (Loo VG et al., 2005,McDonald LC et al., 2005,Pépin J et al., 2004). Le typage instauré de façon plus systématique pour donner suite aux premières épidémies des années 2000 a permis de constater que la souche de R027 a ensuite aussi été la souche prédominante dans plusieurs régions du monde. En effet, cette souche a été un peu plus tard introduite en Europe entre 2002 et 2006, où les premières épidémies documentées ont eu lieu au Royaume-Uni et un peu plus tard dans d’autres régions d’Europe continentale (Valiente E et al., 2014). La souche R027 a été responsable d’épidémies en Asie vers les années 2005-2008. Beaucoup de cas d’ICD causés par la R027 ont aussi été observés plus tard en Amérique du Sud entre 2009-2012. Bien que la prévalence des souches R027 ait diminué au cours des dernières années, cette souche demeure encore très fréquemment isolée dans le monde, surtout en Amérique du Nord (Bauer MP et al., 2011,Davies KA et al., 2016).
Lorsqu’on regarde la période post-épidémie, on comprend maintenant que les souches prédominantes de C. difficile en circulation dans le monde fluctuent au fil du temps (Katz KC et al., 2018). D’autres souches possiblement hypervirulentes de C. difficile semblent faire
leur apparition, comme la souche de ribotype 078 (NAP7/8). Cette souche a vu sa prévalence augmenter et a été associée à des diarrhées sévères et des taux de mortalité élevés aux Pays-Bas, mais aussi dans d’autres pays, incluant le Canada (Goorhuis A et al., 2008). Une étude importante réalisée en 2008, incluant des données de 34 pays d’Europe, a permis de déterminer que les ribotypes les plus prévalents de la période post-épidémique étaient les souches R014, R020, R001, R078, R018, R106, R027 et R002 (Bauer MP et al., 2011). Dans une enquête australienne achevée en 2010, les cinq ribotypes les plus courants étaient les R014, R020, R002, R054, R056 et R070 (Cheng AC et al., 2016). Les données provenant d'autres régions comme l'Amérique du Sud, l'Afrique et le Moyen-Orient sont plutôt restreintes et se limitent à des études sur un faible nombre de centres hospitaliers. En Asie, les souches de ribotype R017, R018, R002 et R001 seraient prédominantes, mais les études à grande échelle pour cette région du monde sont encore rares et attendues (Collins DA et al., 2013). En Amérique du Nord, une étude importante datant de 2011-2013, incluant des échantillons de 32 hôpitaux des États-Unis a révélé que les ribotypes les plus prévalents étaient R027, R014, R020, R106, R001, R053 et R002 (Collins DA et al., 2013).
Bien qu’une quantité importante de données d’épidémiologie moléculaire pour C. difficile se soient accumulées depuis les premières grandes épidémies des années 2000, il y a encore un biais considérable au niveau mondial. La grande majorité des informations sur les ICD nous viennent d’Europe, dont une grande partie du Royaume-Uni, d’Amérique du Nord et d’Australie. Beaucoup d’autres régions et pays sont autant touchées par les ICD (ex : Asie, Amérique du Sud, et Afrique subsaharienne) (Roldan GA et al., 2018), mais n’ont pas les mêmes capacités ou volonté de suivi, de typage moléculaire et d’analyse pour contribuer à fournir un portrait global plus représentatif de la situation (Curcio D et al., 2019). De plus, pour les données présentement rassemblées, il y a aussi un manque de standardisation et une difficulté de comparaison qui sont dus aux différentes méthodes de typage qui ont été choisies dans les différentes régions du monde. Par exemple, le ribotypage PCR a été grandement utilisé en Europe et en Australie, tandis que la méthode de choix en Amérique du Nord a été le EGCP. Bien qu’il existe des correspondances entre les types identifiés par chaque méthode, les recoupements ne sont pas toujours parfaits et la comparaison des données entre les différents lieux est parfois complexe.
La souche épidémique de ribotype 027 (lignée NAP1/BI/027)
La souche R027 a suscité énormément d’intérêt et mené à beaucoup de travaux de recherche dans les deux dernières décennies en raison de sa dissémination très rapide à travers le monde, de son association fréquente aux cas sévères d’infection et au fort taux de mortalité qui lui est attribué. Dans une étude très importante, He et collaborateurs (2013) ont voulu mieux comprendre la structure populationnelle globale des souches R027 (He M et al., 2013). Pour cela, ils ont séquencé le génome de 151 souches de ce ribotype ayant été prélevées entre 1985 et 2010 à travers le monde (Amérique du Nord, Europe, Australie, Japon, Corée, Irlande). Une des conclusions de l’étude a été que la souche pré-épidémique R027 aurait évolué en faisant l’acquisition d’une forte résistance aux antibiotiques de type fluoroquinolone et que cette résistance aurait permis au ribotype 027 de se disséminer et de créer les épidémies mondiales de C. difficile. Une observation surprenante de cette étude est que cette résistance aurait, en fait, été acquise de façon indépendante, à deux reprises (par deux clones distincts) et à des moments différents (entre 1984 et 1999). En effet, exactement la même mutation (Thr82Ile) serait apparue dans la sous-unité A de l’ADN gyrase (gyrA) de la souche R027 pré-épidémique, créant ainsi deux lignées épidémiques distinctes nommées FQR1 et FQR2.
Ces deux lignées épidémiques seraient apparues en Amérique du Nord. La lignée FQR1 serait originaire des États-Unis (99% de probabilité) possiblement de Pittsburgh en Pennsylvanie, tandis que la provenance de la lignée FQR2 est un peu moins certaine, possiblement des États-Unis (59% de probabilité) ou de Montréal au Canada (33% de probabilité). La forte utilisation des fluoroquinolones en Amérique du Nord dans les années 1990-2000 (Linder JA et al., 2005), provoquant alors une énorme pression de sélection sur C. difficile, a été suggérée pour expliquer l’acquisition quasi simultanée de ces deux mutations identiques dans l’ADN gyrase. Bien que les deux lignées FQR1 et FQR2 soient apparues à des moments et endroits rapprochés, elles ont démontré un profil différent de dissémination à travers le monde. Les premières souches FQR1 seraient passées de l’est des États-Unis vers la Corée du Sud et vers la Suisse, tandis que les souches FQR2 auraient plutôt été transférées vers le Royaume-Uni, l’Europe ainsi que l’Australie, où elles ont causé des épidémies importantes (He et al. 2013).
Un élément intéressant à mentionner à propos des lignées épidémiques FQR1 et FQR2 est qu’elles auraient également acquis presque concurremment un nouveau transposon conjugatif très similaire, l’élément CTn5-like Tn6192. La mutation dans l’ADN gyrase et ce transposon sont en somme les deux seuls éléments génétiques majeurs qui distinguent les souches R027 pré-épidémiques des deux lignées épidémiques FQR1 et FQR2, toutes deux R027. Les autres SNP identifié dans les génomes FQR1 et FQR2 ne sont pas partagés par les deux lignées et ont donc été considérés comme des variations génétiques ne pouvant pas expliquer l’hypervirulence de ces souches. Cependant, bien que FQR1 et FQR2 possèdent le transposon conjugatif Tn6192, le rôle qu’aurait pu jouer cet élément génétique mobile dans la dissémination et l’hypervirulence de ces deux lignées reste encore à ce jour inconnu, mais cependant très intrigant.
En analyse bio-informatique, il a été vu que Tn6192 contenait des gènes codant un système de transport ABC, un régulateur transcriptionnel à deux composants et une protéine de liaison à l'ADN (He M et al., 2013). Les systèmes de transduction à deux composants sont fréquemment trouvés chez les bactéries et permettent de détecter, de répondre et donc de s'adapter à différents stimuli environnementaux, à des facteurs de stress ou à différentes conditions de croissance (Stock AM et al., 2000). Le transposon de la lignée FQR2 est quelque peu différent de celui trouvé dans FQR1, car il contient en plus une insertion de 15,7 kb (Tn610531) contenant 14 gènes supplémentaires. Il a été prédit que quatre de ces gènes coderaient des protéines de liaison à l'ADN ou des régulateurs transcriptionnels (Brouwer MSM et al., 2012, 2011). Il a d’ailleurs été souligné que certains de ces régulateurs transcriptionnels possèderaient des motifs HTH_XRE communément trouvés dans les régulateurs transcriptionnels de bactériophages (Brouwer MSM et al., 2011). Cela suggère une possible interaction entre les phages et ce type de transposon retrouvé dans les souches épidémiques. Aucun nouveau phage n’a été mis en évidence dans les souches épidémiques en comparaison avec les souches pré-épidémiques (le prophage phi027 était déjà présent dans les génomes des souches R027 pré-épidémiques). Si on suppose néanmoins que le phage peut être responsable de l’augmentation de la virulence, cela pourrait, entre autres,
s’expliquer par la présence combinée du nouveau transposon et du prophage qui pourraient interagir.
Finalement, il faut mentionner une étude récemment réalisée (2019) au Texas qui est venue apporter plusieurs précisions importantes au niveau de l’émergence des lignées épidémiques résistantes FQR1 et FQR2 du ribotype 027 (Endres BT et al., 2019). Cette étude vient changer quelque peu ce qui a longtemps été assumé, car leurs données supportent que les deux lignées FQR1 et FQR2 résistantes R027 auraient plutôt commencé à émerger et à se disséminer à partir du Texas dans le sud des États-Unis, vers la fin des années 1980 et non au début des années 2000 dans le nord des États-Unis et au Québec. L’émergence correspond néanmoins avec l’augmentation marquée de l’utilisation des antibiotiques de type fluoroquinolones à cette époque.
Cycle de vie de Clostridium difficile
Il est important de savoir que la bactérie C. difficile est capable d’exister sous forme de cellules végétatives actives, mais qu’elle est aussi capable de former et de subsister sous forme de spores. Les spores sont des cellules en dormance qui sont hautement résistantes à différentes conditions environnementales extrêmes et qui sont aussi souvent insensibles à plusieurs traitements couramment utilisés en clinique tels que les agents antimicrobiens et certains désinfectants, même puissants (Paredes-Sabja D et al., 2014). C. difficile se transmet donc par voie féco-orale sous forme de spores. Comme la forme végétative de la bactérie est très peu tolérante à l’oxygène rencontré hors de l’hôte et qu’elle est aussi sensible à l’acidité de l’estomac (Wetzel D et McBride SM, 2020), les spores sont considérées comme le véhicule nécessaire pour la transmission, l’infection et la colonisation par C. difficile (Deakin LJ et al., 2012). Une fois ingérées, les spores sont naturellement acheminées jusqu’au petit intestin par le transit intestinal, là où elles entrent en contact avec des acides biliaires produits par le foie, tel que le taurocholate. La liaison des acides biliaires primaires, tel que le taurocholate, avec le récepteur de germination CspC est nécessaire à l’enclenchement de la germination dans le côlon (Kevorkian Y et al., 2016, Rohlfing AE et al., 2019).
Figure 2. Étapes importantes d’une infection à C. difficile. Reproduit avec l’autorisation de (Janoir C, 2016).
C’est dans le petit intestin qu’il y a une plus forte présence d’acides biliaires primaires, et également une moins grande quantité d’acides biliaires secondaires, ces derniers étant d’ailleurs connus comme inhibiteur de la germination (Bhattacharjee D et al., 2016, Ridlon JM et al., 2006, Theriot CM et Young VB, 2015). Suite à l’initiation de la germination, il y a une cascade protéolytique qui mène à la dégradation du peptidoglycane de la spore. Cela va ultimement mener à la réhydratation de la spore et permettre la croissance sous la forme végétative qui est métaboliquement active. La capacité des cellules végétatives à coloniser efficacement le côlon va être influencée par l’état du microbiote et du métabolome environnant (Buffie CG et al., 2015, Theriot CM et Young VB, 2015). On sait aujourd’hui que les dysbioses (déséquilibres du microbiote), notamment ceux provoqués par la prise d’antibiotiques, favorisent grandement la colonisation et l’infection par C. difficile (Theriot CM et al., 2016). En conditions favorables, les bactéries végétatives peuvent sécréter certaines protéines de surface, telle que l’enzyme mucolytique Cwp84 qui dégrade la muqueuse du côlon (Janoir C et al., 2007). Plusieurs autres protéines de surface produites par la bactérie ont été étudiées et jouent possiblement un rôle déterminant dans l’adhésion
de C. difficile aux cellules épithéliales, suite à la dégradation de la muqueuse (voir tableau 1) (Kovacs-Simon A et al., 2014, Lin YP et al., 2011, Merrigan MM et al., 2013, Tulli L et al., 2013).
C. difficile est une bactérie motile, capable de réguler son passage d’un état de motilité à un état immobile grâce au messager secondaire, di-GMP cyclique (Bordeleau E et Burrus V, 2015, Peltier J et al., 2015). Les cellules végétatives produisent des entérotoxines TcdA et TcdB, qui sont considérées comme indispensables au développement des symptômes, comme les diarrhées associées à C. difficile (DACD) (Rupnik M, 2001,Voth DE et Ballard JD, 2005). C. difficile a également la capacité de coloniser le tractus intestinal sans occasionner de symptômes (colonisation asymptomatique) et cela malgré la production de toxines (Crobach MJT et al., 2018). Bien que très probable, il n’est pas encore tout à fait clair si la formation de biofilm est un élément important dans la colonisation, la persistance et la virulence de C. difficile in vivo, suite à sa germination et à sa croissance au site infectieux.
Il a été démontré in vitro que la bactérie était capable de former des biofilms robustes (Crowther GS et al., 2014, Dapa T et Unnikrishnan M, 2013,Semenyuk EG et al., 2015). Les biofilms pourraient avoir un rôle important dans la récurrence, en raison de leur capacité accrue à résister aux traitements antibiotiques (Mah TFC et O’Toole GA, 2001). Pour terminer le cycle de réplication de la bactérie, on doit mentionner la capacité des cellules végétatives à reformer de nouvelles spores dans le système digestif. La formation des spores s’initie généralement en réponse à des conditions environnementales défavorables aux cellules végétatives et est finement contrôlée par des systèmes de régulations génétiques complexes. Il a été démontré que Spo0A était le « master regulator » de la sporulation. Sans le gène spo0A, C. difficile est incapable de produire ses spores favorisant la transmission et l’infection d’un nouvel hôte (Heap JT et al., 2007,Mackin KE et al., 2013).
Caractéristiques générales du génome de Clostridium difficile
Les génomes bactériens sont souvent décrits selon les termes de « pangénome », de « core genome » (génome de base) et de « génome accessoire ». Le pangénome est le répertoire de
tous les gènes que possède une espèce bactérienne. Le pangénome est donc l’addition du génome de base (les gènes présents chez toutes les souches de l’espèce) et du génome accessoire (les gènes présents chez certaines souches seulement) (Tettelin H et al., 2008). C. difficile possède un seul chromosome d’en moyenne 4 Mpb et sa taille peut varier considérablement selon la présence d’éléments génétiques mobiles intégrés (Knight DR et al., 2015). Il a été estimé que C. difficile possède un génome de base très petit (environ 16% du pangénome) (Janvilisri T et al., 2009, Scaria J et al., 2010). Le génome de base contiendrait entre 947 et 1033 gènes tandis que le pangénome a été estimé à 9640 gènes (Scaria J et al., 2010). Une étude plus récente sur une collection de 207 souches de divers ribotypes (16 ribotypes provenant d’Australie, d’Asie, d’Europe et d’Amérique du Nord) ont permis d’estimer la taille du pangénome à 10 378 gènes et celle du core génome à 2 058 gènes (Knight DR et al., 2019). Ces nouvelles valeurs permettent aussi de mieux comprendre que les estimations de la taille du pangénome et du génome de base peuvent varier considérablement selon la quantité et le type de souches ayant été inclues dans les analyses. De plus, une grande quantité de génomes de C. difficile ont été séquencés, mais plusieurs de ces séquences n’ont pas encore été analysées ou ne sont pas disponible pour le publique.
La grande quantité de gènes accessoires est explicable par le fait que le génome de C. difficile possède un grand nombre d’éléments génétiques mobiles, qui représentent souvent près de 11% du génome bactérien, comme cela a été observé lors du séquençage de la première souche de la bactérie, la souche CD630 (Sebaihia M et al., 2006). Il avait été observé que cette souche possédait, entre autres, un plasmide, 2 prophages, 8 transposons, des IStrons, des « clustered regularly interspersed short palindromic repeat » (CRISPR) et un élément « sigK intervening » (skin) (Sebaihia M et al., 2006). Les IStrons sont des éléments génétiques chimériques composés d'un intron du groupe I associé à une séquence d'insertion (IS) (Braun V et al., 2000). Le séquençage ultérieur de plusieurs génomes de C. difficile a révélé que la présence de ces éléments était plutôt la règle que l’exception (He M et al., 2010). Certaines souches peuvent posséder jusqu’à 6 plasmides, mais certains de ces plasmides peuvent potentiellement être des prophages non intégrés (phage épisomaux) (Mulligan ME et al., 1988). Peu de plasmides de C. difficile ont été séquencés à ce jour, et la majorité est considérée comme cryptique (fonction inconnue).
La plupart des souches de C. difficile possèdent un et souvent plusieurs prophages (bactériophages en dormance) qui peuvent être intégrés au génome bactérien ou non. Tous les prophages identifiés à ce jour sont des phages tempérés et font partie des familles Myoviridae (familles de phages à queues longues contractiles) ou Siphoviridae (famille de phages à queues longues non contractiles) (Hargreaves KR et al., 2013). La plupart des prophages répertoriés à ce jour ont un contenu en G+C similaire à celui de C. difficile (28-30%) et certains possèdent même des gènes reliés aux systèmes CRISPR, ainsi que des protospacers, suggérant une évolution à long-terme avec leur hôte (Knight DR et al., 2015).
C. difficile est considérée comme une espèce bactérienne génétiquement diversifiée et globalement répandue. De multiples projets de séquençage de génomes entiers et des analyses de génomique comparative ont montré que C. difficile avait une structure clonale comportant au moins 6 clades majeurs, soit les clades 1, 2, 3, 4, 5 et un clade cryptique C-I. Un clade représente un groupement de plusieurs embranchements d’un organisme ayant une origine commune. Le clade C-I pourrait en fait être une nouvelle sous-espèce de C. difficile en raison de sa haute divergence phylogénétique en comparaison avec les autres clades connus (Dingle KE et al., 2014). Une réexamination récente des bases de données pour le typage moléculaire de C. difficile montre qu’il y aurait également au moins deux autres clades cryptiques très divergents des clades connus, nommés C-II et C-III (https://pubmlst.org). Le clade 5 est le plus divergent des clades non cryptiques et il est estimé que ce clade aurait divergé des autres il y a entre 1.1 et 85 millions d’années (He M et al., 2010). Il a été observé que des souches provenant de la plupart des différents clades sont capables de causer des ICD chez les humains (Dingle KE et al., 2011, Stabler RA et al., 2012).
Le génome de C. difficile est considéré comme évoluant relativement lentement au niveau de l’acquisition de mutations. Plusieurs études différentes ont estimé un taux évolutif d’environ 1 à 2 SNPs/génome/année (Didelot X et al., 2012,Eyre DW et al., 2013, 2013). Le taux d’événements de recombinaison homologue est aussi considéré comme étant relativement bas, estimé 4 fois plus faible que le taux de SNP (Dingle KE et al., 2011). Il
faut cependant considérer le fait que C. difficile a la capacité de former des spores et que les taux calculés pour des bactéries se répliquant dans l’hôte ne sont donc pas appropriés ou représentatifs de l’évolution sur de longues périodes de temps où les spores restent en dormance et sont inactives (Elliott B et al., 2017).
Les facteurs de virulence chez Clostridium difficile TcdA et TcdB, les facteurs de virulence principaux
La pathogénicité de C. difficile est principalement conférée par deux exotoxines, soit la toxine A (TcdA) et la toxine B (TcdB). Les mécanismes d’action de ces deux toxines sont aujourd’hui mieux compris, mais certaines parties restent encore à éclaircir. Il est bien connu que suite à leur sécrétion par la bactérie, TcdA et TcdB vont se lier et ensuite entrer dans les cellules épithéliales du côlon, ce qui va entraîner la production de cytokines et de chimiokines pro-inflammatoires, provoquer l’infiltration de neutrophiles, la perturbation des jonctions serrées, la sécrétion parfois intense de fluide et la mort éventuelle des cellules épithéliales par apoptose (Shen A, 2012). Les deux toxines sont constituées de quatre domaines fonctionnels, le domaine glycosyltransférase (GTD), le domaine auto-protéase (APD), le domaine « combined repetitive oligopeptides » (CROPS), et un « delivery domain » (Chumbler NM et al., 2016). TcdA et TcdB vont entrer dans les cellules par endocytose utilisant un ou plusieurs récepteurs qui ne sont pas encore clairement identifiés (Papatheodorou P et al., 2010). Plusieurs glycolipides et protéines « glycosylées » ont été suggérés comme récepteurs pour TcdA, mais aucun récepteur spécifique sur les cellules épithéliales n’a encore pu être déterminé avec certitude (Pruitt RN, Lacy DB, 2012). Deux candidats ont été proposés comme récepteurs potentiels pour TcdB, soit le « poliovirus receptor-like protein 3 », aussi appelé nectin 3 (PVRL3) et le chondroïtine-sulfate protéoglycane 4 (CSPG4) (LaFrance ME et al., 2015,Yuan P et al., 2015). Ces deux récepteurs pourraient être utilisés par TcdB à des stades différents de l’infection. PVRL3 pourrait être le récepteur utilisé dans les premiers stades de l’infection, car il semble être fortement exprimé à la surface des cellules épithéliales du côlon. Il a été démontré que PVRL3 et TcdB se retrouvaient colocalisés au niveau des tissus prélevés chez des sujets infectés par C. difficile (Yuan P et al., 2015). L’autre récepteur potentiel CSPG4 est quant à
lui fortement exprimé chez la souris et l’humain, mais au niveau des myofibroblastes sous-épithéliaux de l’intestin, ce qui mène à penser qu’il serait impliqué dans une phase plus tardive de l’infection, subséquemment aux premiers dommages subis par l’épithélium de surface du côlon (Shen A et al., 2011,Yuan P et al., 2015).
Le rôle exact de TcdA et TcdB dans un contexte d’infection a été difficile à établir, mais le développement d’outils de manipulation génétique plus efficaces pour C. difficile a permis d’obtenir des informations supplémentaires sur ces deux facteurs de virulence importants. Plusieurs études, réalisées en utilisant un modèle de hamster ou de souris, sont parvenues à démontrer que TcdB seule (absence de TcdA) était capable de provoquer les phénotypes ou symptômes habituels d’une ICD (Lyras D et al., 2009). Cependant, les résultats sont un peu moins clairs en ce qui concerne la capacité spécifique de TcdA seule (absence de TcdB) à provoquer l’apparition des symptômes et son impact sur la survie des animaux infectés (Kuehne SA et al., 2010, 2014). Les dommages observés après infection avec des souches mutantes TcdA− TcdB+ se sont avérés plus importants que les dommages observés pour des
infections avec des souches mutantes TcdA+TcdB−. Par exemple, dans un modèle de souris,
les infections avec des souches TcdB+ ont montré des dommages sévères aux tissus du côlon
et du caecum, tels que de fortes érosions et disparition des cryptes intestinales, des ulcérations de la muqueuse et même la perte des cellules caliciformes (cellules en gobelet) (Carter GP et al., 2015). Des œdèmes sévères (accumulations anormales de liquide) avec hémorragies, des influx importants de granulocytes dans la « lamina propia » ont aussi été observés dans les tissus de souris infectées par les souches TcdB+. Cependant, cette même
étude a montré que les infections avec des souches TcdA+ TcdB− causaient des dommages
beaucoup moins importants, limités à l’apparition d’œdèmes modérés et l’infiltration de granulocytes. Les dommages tissulaires et les phénotypes précédemment décrits semblent être entièrement en fonction de la présence de TcdB ou TcdA, car les souris infectées par des souches ne produisant aucune des deux toxines (TcdA− TcdB−) ont affiché des coupes
histologiques hautement similaires à celles des souris contrôles non infectés. En considérant ces dernières études et leurs conclusions, il semble compréhensible et logique de penser qu’un très grand nombre de souches TcdA− TcdB+ aient été isolées en clinique suite à des
la prévalence a augmenté au cours du temps et qui sont par moment apparues en proportions considérées comme épidémiques (Drudy D et al., 2007, King AM et al., 2015). Il semble aussi logique que les souches exprimant les deux toxines, telle que la souche épidémique de ribotype 027 (R027), soient tout de même les isolats prédominants qui ont été les plus fréquemment isolés à travers le monde (Rupnik M, 2016). Brièvement, le ribotype d'une souche est obtenu suite à la réalisation du ribotypage par PCR, qui est une méthode basée sur l'hétérogénéité des régions espaceurs "spacers" intergéniques ribosomiques. Ces régions "spacers", situées entre les gènes d'ARNr 16S-23S, vont varier en nombre et en taille selon les différentes souches analysées. Le ribotypage, est une des méthodes préférées pour le génotypage de C. difficile. Plus de détails seront donnés sur cette technique plus loin dans l'introduction (Indra A et al., 2010). Ensuite, une variante de souche produisant seulement TcdA a récemment pu être isolée et identifiée. Cependant il est intéressant de souligner que les cas cliniques d’ICD causés par ce type de souches sont extrêmement rares, probablement parce que moins virulents que les souches exprimant les deux toxines ou seulement TcdB (Monot M et al., 2015).
Production de toxines dans la lignée NAP1/BI/027
La totalité des souches (sauf rares exceptions) de C. difficile capables de causer les symptômes des ICD produisent les toxines A et B, qui sont les facteurs de virulence principaux. Plusieurs études ont proposé que l’émergence et l’hypervirulence des R027 puissent être expliquée par une production accrue de toxines dans ces souches (Loo VG et al., 2005,McDonald LC et al., 2005,Warny M et al., 2005). Cependant, plusieurs études subséquentes sont venues suggérer qu’il n’y avait pas de différence marquée concernant la production de toxine chez les R027 versus d’autres ribotypes (Åkerlund T et al., 2008,Merrigan M et al., 2010). Quelques raisons ont été suggérées pour expliquer ces résultats contradictoires, comme le nombre insuffisant de souches utilisées dans les études, indiquant qu’un nombre plus élevé de souches auraient possiblement permis d’obtenir des résultats avec une significativité convaincante pour soutenir ou invalider la production de toxines accrue des isolats R207 (Sirard S et al., 2011). Une autre raison mentionnée est la variabilité des modèles in vitro et des conditions de laboratoire concernant les évaluations de production de toxines (Freeman J et al., 2006). Par exemple, certains dosages de toxines ont
été faits en essais in vitro sans validation en modèle animal ou autres modèles considérés plus représentatifs des conditions in vivo (ex. : modèle in vitro de chemostat à trois étapes) (Freeman J et al., 2005). Des différences marquées entre certains milieux de culture utilisés pour l’évaluation de la quantité de toxines viennent également ajouter des doutes en ce qui concerne la validité de certains résultats concernant les niveaux de toxines produits dans les différentes études. Cependant, bien qu’il n’y ait pas encore de réponses ou d’explications définitives concernant la surproduction de toxines dans les souches R027, il est encore parfois observé, et souvent suggéré que ces souches ont tendance à produire plus de toxines que la plupart des autres ribotypes (Neely F et al., 2017).
Plusieurs études ont proposé qu’une délétion de 18 pb fréquemment trouvée dans le gène tcdC du PaLoc des souches R027 pourrait être responsable de l’augmentation de la production de toxine dans ce ribotype (Loo VG et al., 2005,McDonald LC et al., 2005,Warny M et al., 2005). Comme TcdC est un régulateur négatif des toxines A et B (encodées par tcdA et tcdB), une délétion dans ce gène est susceptible d’entraîner une régulation à la hausse de la production des toxines. Plus tard, il a aussi été montré qu’une délétion de 1 pb dans ce même gène tcdC était présente dans un grand nombre de souches R027 et que cette mutation était certainement associée à une plus forte production de toxines (Curry SR et al., 2007,Matamouros S et al., 2007). Cependant, il est important de souligner qu’une étude ayant procédé à une analyse d’isolats épidémiques et pré-épidémiques a montré que toutes les souches, incluant les souches pré-épidémiques, contenaient la délétion de 18 pb dans le PaLoc et qu’il serait alors peu probable que l'émergence des souches épidémiques soit explicable par cette délétion (McDonald LC et al., 2005).
Dans une autre étude incluant 151 isolats, He et collaborateurs (2013) ont conclu qu’il n’y avait pas de différence génétique entre les souches R027 pré-épidémiques et épidémiques et qu’il serait donc improbable que les délétions de 18 pb et de 1 pb souvent trouvés dans les R027 soient l’explication de la surproduction de toxines ou de l’hypervirulence de cette lignée (He et al. 2013). Ces observations sont en accord avec une étude antérieure qui avait comparé deux génomes de souches R027 (souche épidémique et hypervirulente R20291 et souche pré-épidémique CD196 non hypervirulente). L’étude suggérait qu’il n’y avait pas de
différence dans les séquences du PaLoc entre les deux souches qui pourrait expliquer la possible hypervirulence ou l’émergence des souches R027 (Stabler R et al., 2009).
Le PaLoc et la régulation de l’expression des toxines
Il est maintenant bien établi que les symptômes de l’ICD sont reliés à la présence du locus de pathogénicité (PaLoc) (Braun V et al., 1996,Hammond GA, Johnson JL, 1995). Sauf dans de rares cas, le locus est situé au même endroit dans le génome bactérien, et on y retrouve les séquences codant pour les deux toxines homologues TcdA et TcdB. Cette région code également pour trois protéines (TcdR, TcdE, TcdC) qui semblent impliquées dans la régulation de la production et la sécrétion des toxines (Awad MM et al., 2015,Smits WK, 2013). TcdR est un facteur sigma alternatif de la famille des « extracytoplasmic function sigma factors » qui est essentiel pour l’initiation de la production de TcdA et TcdB (Mani N, Dupuy B, 2001,Mani N et al., 2002). TcdC est quant à lui un facteur anti-sigma qui régule négativement l’expression des toxines (Dupuy B et al., 2008).
Il est intéressant de souligner que les souches de ribotype 027 affichent une mutation non-sens dans le gène tcdC, causant donc l’inactivation de cette protéine et par conséquent la dérépression de l’expression des gènes de toxines. Il a été suggéré à plusieurs reprises que cette dérépression des toxines pourrait jouer un rôle dans l’hypervirulence associée aux souches de ce ribotype, mais cette proposition est controversée (Warny M et al., 2005). En effet, plusieurs études ont tenté de mieux définir l’impact exact de TcdC dans la production des toxines, mais des conclusions contradictoires ont été obtenues et le rôle fonctionnel de la protéine demeure encore imprécis (Smits WK, 2013).
Le rôle exact de TcdE est également encore incertain, mais il a été observé que la protéine était homologue à plusieurs holines de phages, qui sont impliquées dans le relâchement de virions hors de la cellule bactérienne (Kai Soo Tan et al., 2001). Des études ont donc proposé que TcdE aurait comme fonction de faciliter la sécrétion des toxines TcdA et TcdB hors de la cellule (Govind R, Dupuy B, 2012,Olling A et al., 2012). Ces mêmes études suggéraient que TcdA, tout comme TcdB, ne semblaient pas posséder de séquences « signal » ou « d’exportation », ce qui pousse encore plus à penser qu’elles doivent être exportées par le
biais de la lyse bactérienne, ou par un mécanisme de sécrétion non-classique impliquant TcdE.
Il a été suggéré que l’expression des toxines et donc la pathogenèse de C. difficile était intimement liée au métabolisme et à l’environnement nutritionnel de la bactérie (Bouillaut L et al., 2015). À titre d’exemple, la production de TcdA et TcdB peut être inhibée par la présence de glucose, de butanol, ainsi que par d’autres sources de carbones rapidement métabolisables (Antunes A et al., 2012,Dupuy B, Sonenshein AL, 1998). En effet, certains acides aminés spécifiques, en particulier la cystéine et la proline, ont été reconnus comme étant capables d’inhiber fortement (100 fois) la production des toxines A et B dans la souche VPI10463. À l’inverse, la présence d’acide butyrique, un acide gras à chaîne courte, est capable de stimuler la synthèse des toxines A et B (Bouillaut L et al., 2015). Il a également été observé que certains antimicrobiens (ex : ciprofloxacine, co-amoxiclav) en concentrations sous-inhibitrices pouvaient aussi stimuler la production de toxines (Chilton CH et al., 2012,Mani N et al., 2002).
Il a été démontré que le facteur sigma SigD, qui est habituellement relié au contrôle des gènes de motilité, peut aussi influencer l’expression des toxines. Sa liaison à un promoteur en amont de tcdR résulte en une augmentation de la transcription de tcdR, qui régule positivement l’expression des toxines (El Meouche I et al., 2013). Il a été vu que Spo0A, considéré comme régulateur maître de la sporulation dans les espèces Clostridium et Bacillus, pouvait aussi réguler l’expression des toxines chez C. difficile, mais seulement dans les souches de ribotype 027. Dans les souches de ribotype 027 (R027), Spo0A a été décrit comme un régulateur négatif de la production de TcdA et TcdB (Mackin KE et al., 2013). Cette régulation, qui s’avère différente chez les souches R027, démontre encore une fois la nature hétérogène des différents isolats de C. difficile et qu’il est important d’étudier en détails les souches provenant de différentes lignées évolutives (Knetsch CW et al., 2012, Mackin KE et al., 2013).
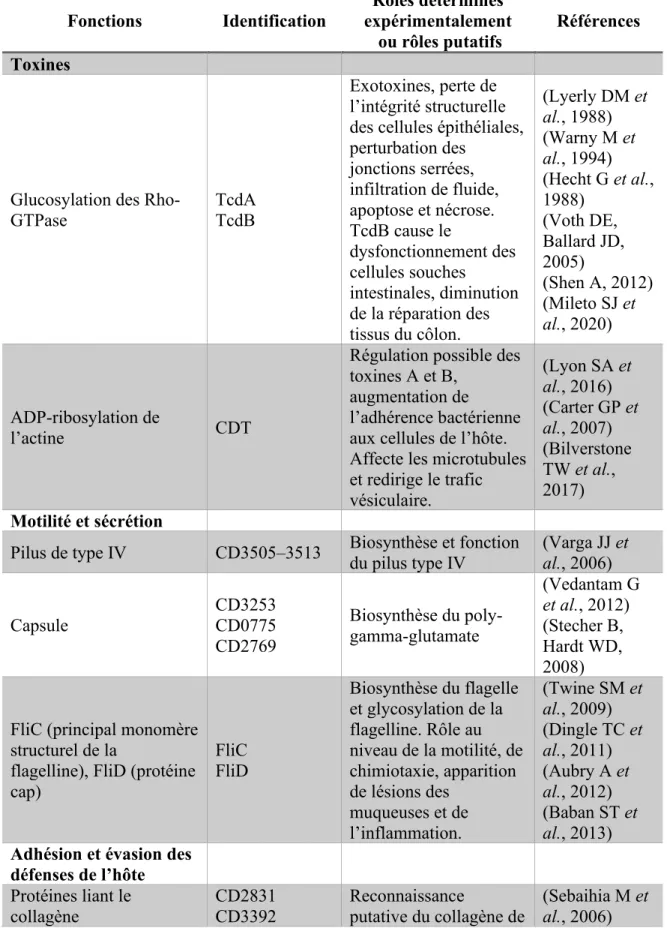
Tableau 1. Résumé des facteurs de virulence chez C. difficile.
Fonctions Identification expérimentalement Rôles déterminés ou rôles putatifs
Références Toxines
Glucosylation des Rho-GTPase
TcdA TcdB
Exotoxines, perte de l’intégrité structurelle des cellules épithéliales, perturbation des jonctions serrées, infiltration de fluide, apoptose et nécrose. TcdB cause le dysfonctionnement des cellules souches intestinales, diminution de la réparation des tissus du côlon. (Lyerly DM et al., 1988) (Warny M et al., 1994) (Hecht G et al., 1988) (Voth DE, Ballard JD, 2005) (Shen A, 2012) (Mileto SJ et al., 2020) ADP-ribosylation de l’actine CDT
Régulation possible des toxines A et B,
augmentation de
l’adhérence bactérienne aux cellules de l’hôte. Affecte les microtubules et redirige le trafic vésiculaire. (Lyon SA et al., 2016) (Carter GP et al., 2007) (Bilverstone TW et al., 2017) Motilité et sécrétion
Pilus de type IV CD3505–3513 Biosynthèse et fonction du pilus type IV (Varga JJ et al., 2006)
Capsule CD3253 CD0775 CD2769 Biosynthèse du poly-gamma-glutamate (Vedantam G et al., 2012) (Stecher B, Hardt WD, 2008)
FliC (principal monomère structurel de la
flagelline), FliD (protéine cap) FliC FliD Biosynthèse du flagelle et glycosylation de la flagelline. Rôle au niveau de la motilité, de chimiotaxie, apparition de lésions des muqueuses et de l’inflammation. (Twine SM et al., 2009) (Dingle TC et al., 2011) (Aubry A et al., 2012) (Baban ST et al., 2013) Adhésion et évasion des
défenses de l’hôte Protéines liant le collagène CD2831 CD3392 Reconnaissance putative du collagène de (Sebaihia M et al., 2006)
CD0386 CbpA la matrice extracellulaire (Tulli L et al., 2013) Protéines liant la fibronectine FbpA CD2797 Fbp68 Reconnaissance putative de fibronectine (Barketi-Klai A et al., 2011) (Lin YP et al., 2011) (Hennequin C et al., 2003) Protéine avec domaine
Thrombospondine CD3145 Reconnaissance putative du fibrinogène (matrice extracellulaire) (Sebaihia M et al., 2006) von-Willebrand Factor binding proteins CD3038 CD2248 CD0323
Protéines liant le facteur von-Willebrand
(Sebaihia M et al., 2006) Sortase CD2718 Sortase de classe B (Sebaihia M et al., 2006)
Protéine majeure de surface SlpA Protéine majeure de surface, Liaison à la muqueuse intestinale, interaction avec TLR4, interaction avec protéines de la matrice extracellulaire. Capacité de liaison avec cellules CaCo-2. (Calabi E et al., 2001) Cystéine protéase Cwp84 Cystéine protéase, clivage de SlpA, Enzyme mucolytique, dégradation de matrices extracellulaires, dégradation de la muqueuse du côlon (de la Riva L et al., 2011) (Janoir C et al., 2007)
Adhésine Cwp66 CD630_28310 Adhésine, adhérence aux cellules de l’hôte (Waligora AJ et al., 2001) Hémagglutinine/Adhésine CD0514 Hémagglutinine
putative
(Sebaihia M et al., 2006) Protéine de la paroi reliée
à la variation de phase CwpV Protéine de surface « phase-variable », auto-agrégation bactérienne, évasion immunitaire, résistance aux phages (Reynolds CB et al., 2011) (Sekulovic O et al., 2015) Lipoprotéine CD630_08730 Lipoprotéine, pour liaison aux cellules Caco-2
(Kovacs-Simon A et al., 2014)
Ligands ou récepteurs spécifiques à la surface des entérocytes. Deux protéines de 40–45 kDa (pas encore identifiées précisément)
Adhésion aux cellules du côlon, persistance de la bactérie après traitement (Paredes-Sabja D, Sarker MR, 2012) Heat-shock protein GroEL GroEL Protéine GroEL, adhésion cellulaire, colonisation (Hennequin C et al., 2001) Protéase Zmp1 Contrôlerait l’activité de l’adhésine CD630_28310. Protéase pouvant cliver les protéines liant le collagène (CD28310). Résultant en diminution de l’adhésion bactérienne à l’hôte. (Janoir C, 2016) Estérification Opéron Dlt
Estérification des acides téichoïques menant à la réduction de l’efficacité des CAMPs dirigés contre la bactérie (Augmentation de la charge positive de la surface bactérienne). Évasion de la réponse immunitaire précoce. Résistance aux CAMPs.
(Revilla-Guarinos A et al., 2014) (McBride SM, Sonenshein AL, 2011)
Désacétylation Déacetylase PdaV (CD630_15560) Désacétylation des résidus N-Acétylglucosamine médiée par la désacétylase PdaV (CD630_15560). Résistance accrue au lysozyme (hydrolase du peptidoglycane) produit par les cellules
épithéliales.
(Ho TD et al., 2014)
Transporteur ABC CprABC
Résistance aux CAMPs médiée par les
transporteurs ABC. Le transporteur CprABC de C. difficile a évolué pour reconnaître (Revilla-Guarinos A et al., 2014) (McBride SM, Sonenshein AL, 2011)
plusieurs substrats permettant de se défendre contre plusieurs toxines produites par le microbiote intestinal. Régulateurs transcriptionnels AgrA Agr quorum-signaling system Régulateurs transcriptionnels, régulation de la production de TcdA et TcdB, synthèse de di-GMP cyclique, assemblage du flagelle, impact sur la colonisation et rechute (Darkoh C et al., 2015,Martin MJ et al., 2013) Autres protéines
Hémolysine CD1546 Protéine hémolysine-like, lyse cellulaire (Sebaihia M et al., 2006)
Protéase CD1228 Protéase spécifique au collagène, dégradation du collagène (Sebaihia M et al., 2006) Composition du génome Contenu en dinucléotides CpG, (Génome entier)
Souches ayant une fréquence réduite de CpG seraient moins immunogéniques. Plus faible activation des toll-like receptor 9 (TLR-9), évasion du système immunitaire inné, meilleure efficacité de traduction. (Kamuju V et al., 2018) Autres Spores
Adhérence aux cellules épithéliales et capacité à tuer les macrophages
(Paredes-Sabja D et al., 2012)
Autres facteurs de virulence
On connait maintenant un peu mieux le rôle et l’impact des toxines A et B sur l’hôte et dans le processus d’infection, mais l’implication et l’importance des autres facteurs de virulence présents chez C. difficile sont moins bien comprises. Certains facteurs de virulence peuvent avoir des impacts plus subtils que celui des toxines, alors leur étude représente parfois un défi considérable. Bien que l’impact de certains de ces facteurs soit plus évasif à notre compréhension, il est possible que le succès épidémique et virulent de certaines souches en dépende grandement. On peut mentionner à titre d’exemples les bactéries pathogènes Yersinia et Salmonella qui sont capables d’atténuer le système immunitaire de l’hôte en produisant des protéines effectrices capables d’imiter les fonctions accomplies par les protéines de l’hôte (tyrosine phosphatases) (Van Avondt K et al., 2015). La liaison des protéines effectrices bactériennes à des adaptateurs signalétiques chez les neutrophiles (SLP-76) va avoir pour effet de diminuer la dégranulation de ces derniers, ce qui va réduire l’élimination des bactéries dans l’hôte. Ce type de mécanisme adaptatif chez plusieurs bactéries montre qu’il est important d’approfondir les recherches au-delà de l’expression des toxines, qui n’arrive pas à expliquer les variations de virulence observées chez les différentes souches de C. difficile en circulation. Les facteurs de virulence sont souvent séparés en deux grandes catégories : une première catégorie qui considère les dommages créés à l’hôte et la deuxième qui considère plutôt la capacité de colonisation et la résistance aux différents systèmes de défense de l’hôte. Plus haut, nous avons couvert la première catégorie en discutant des deux toxines majeures produites par C. difficile. Maintenant, nous allons considérer les facteurs de virulence qui contribuent à la colonisation et à la persistance de la bactérie dans l’hôte. Sommairement, on peut considérer que la colonisation du système digestif est en grande partie gouvernée par la capacité de la bactérie à adhérer aux muqueuses de l’hôte ainsi qu’à sa capacité à résister aux défenses dirigées contre elle (Janoir C, 2016).
La toxine binaire
Certaines souches de C. difficile encodent une troisième toxine appelée la « C. difficile transferase » (CDT ou toxine binaire). Un grand intérêt est porté à cette toxine, car on la retrouve dans un nombre croissant de souches d’origine humaine et animale (Gerding DN et al., 2013), et sa prévalence est considérée comme élevée dans les souches communément