AN ABINITIO STUDY OF THE ISOMERIZATION AND FRAGMENTATION OF CHO2+ IONS - AN EXAMPLE OF SPIN-CONTROLLED REACTIONS
Texte intégral
Figure
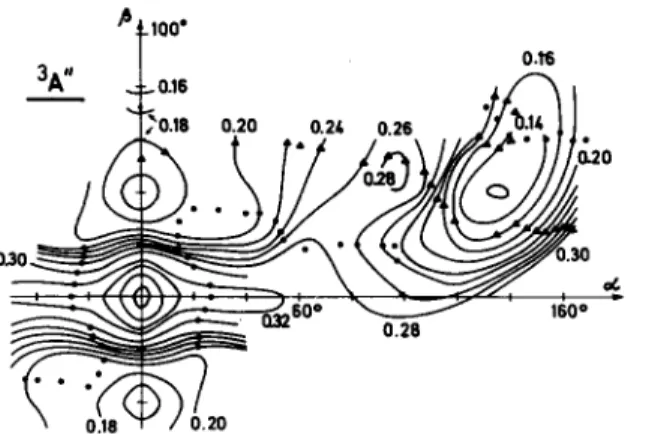
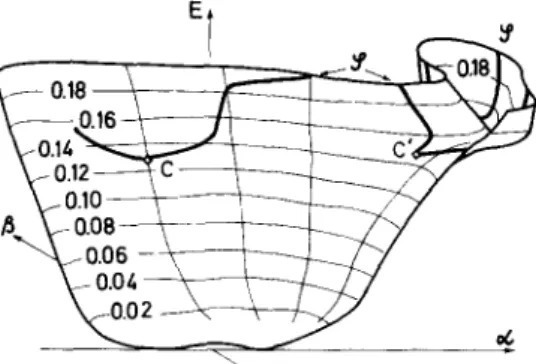
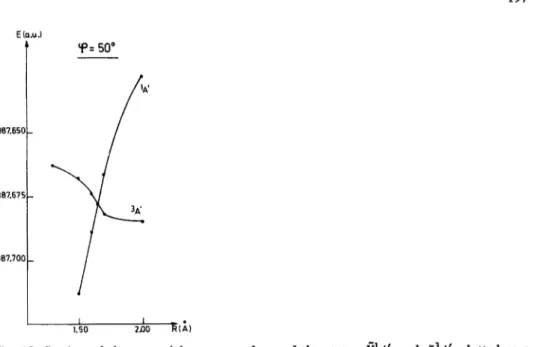
![Fig. 1. Geometry of the [CHOT ] ion in the system of coordinates ( OL, /?)](https://thumb-eu.123doks.com/thumbv2/123doknet/5461950.129222/4.757.107.304.715.903/fig-geometry-chot-ion-coordinates-ol.webp)


Documents relatifs
But the results of [11] and [13] show that small deformations of LeBrun metrics on 4CP 2 with (C ∗ ) 2 -action which have 6-dimensional moduli always drop algebraic dimension of
- Magnetic properties are studied of a system having two types of electronic singlet-ground-state ions, d-ion (singlet-doublet) and t-ion (singlet-triplet), by taking account
However, the MRSDCI/DZP oscillator strengths and radiative lifetimes differ significantly from both the MRCI and EOM-CCSDT data (see Table 2), probably due to the
Direct atomic recombination and three particle recombination are also negligible at the electron densities in the expanding plasma.. We first give an overview of the
An AES study of damage induced by inert gas ions at SiO2 surfaces : influence of ion mass and
2014 The dipole polarizability of rare gas atoms, positive alkali ions and negative halogen ions has been calculated in the local density approximation (LDA) to the
De plus, des énergies relativistes moyennes sont obte- nues pour toutes les configurations envisagées.. Ces écarts sont en très bon accord avec les valeurs
In this case, mechanisms involving electron tunneling can be excluded If the photon energy, which leads to desorption is comparable to the ionization energy of the adsorbate,
