Détermination de la stabilité cinétique des
complexes uranium-substances humiques dans le
bassin versant du fleuve St-Laurent
Mémoire
Kenny Nadeau
Maîtrise en chimie
Maître ès sciences (M. Sc.)
Québec, Canada
© Kenny Nadeau, 2014
iii
RÉSUMÉ
Le bassin versant du St-Laurent (BVSL), constitué des Grands Lacs et du fleuve Saint-Laurent, est un écosystème d’eau douce unique puisqu’il est le seul dans le monde à avoir accueilli tous les stades du cycle du combustible nucléaire. La présence d’activités nucléaires sur les rives de l’écosystème nécessite une connaissance approfondie du comportement environnemental des radionucléides. Comme la plupart des actinides ont une origine anthropique, ils vont se comporter selon des paramètres thermodynamiques qui peuvent être estimés selon divers modèles. Le cas des TENORM (matériaux présentant une radioactivité naturelle renforcée), comme l’uranium (U) irradié, est différent, puisque ces matériaux auront à compétitionner avec l’uranium naturel déjà à l’équilibre dans l’environnement. Dans une telle situation, non seulement les paramètres thermodynamiques mais également certains paramètres cinétiques, influeront sur la distribution et la spéciation des isotopes.
Ce mémoire présente les travaux effectués sur la grande partie du projet de maîtrise, soit la détermination de la stabilité cinétique des complexes uranium-substances humiques. Les travaux accomplis au cours de cette maîtrise incluent l’optimisation des paramètres cinétiques dans un milieu test artificiel (pH, température et concentration), ainsi que l’approfondissement de nos connaissances sur l’utilisation de techniques de séparation (dialyse, ultrafiltration et chromatographie d’exclusion stérique). De plus, les concentrations d’uranium ont été analysées par spectrométrie de masse couplée à un plasma inductif (ICP-MS).
v
ABSTRACT
The watersheds composed of the Great Lake basin (GLB) and the St-Lawrence River basin (SLRB) is unique in that it is one of the only freshwater ecosystems in the world that is home of all stages of the nuclear fuel cycle. The presence of nuclear activities on the shore of these watersheds requires a thorough knowledge of the environmental behaviour of radionuclides. Considering that most actinides are anthropogenic, they behave following thermodynamic parameters that can be estimated by diverse models. The case of technologically enhanced naturally occurring radioactive material (TENORM), such as irradiated uranium, is slightly different since it competes, upon release, with naturally occurring uranium which is already at equilibrium in the environment. In this situation, the thermodynamic stability of the system is involved and the kinetic stability could influence its environmental distribution.
This master’s thesis presents the main results of a master’s degree study on : the determination of the kinetic stability of the complex humic substances-uranium.
The accomplished work during this master’s degree include the optimization of kinetic parameters on an artificial test environment (pH, temperature and concentration) and deepening our knowledge on the use of separation techniques (dialysis, ultrafiltration and size exclusion chromatography). In addition, uranium concentrations were analyzed by inductively coupled plasma-mass spectrometry (ICP-MS).
vii
TABLES DES MATIÈRES
RÉSUMÉ ... iii
ABSTRACT ... v
LISTE DES TABLEAUX ... ix
LISTE DES FIGURES ... xi
ABRÉVIATIONS ... xv REMERCIEMENTS ... xix 1. Introduction ... 1 1.1. Mise en contexte ... 1 2. Théorie ... 7 2.1. L’uranium ... 7
2.2. Introduction d’uranium dans l’environnement ... 13
2.3. Les substances humiques ... 14
2.3.1. Caractérisation élémentaire ... 19
2.3.2. Groupements fonctionnels ... 21
2.3.3. Masses ... 21
2.4. Interaction avec les SH ... 22
2.4.1. Types d’interactions ... 22
2.4.2. Évaluation des interactions ... 23
2.4.3. Impact de divers facteurs environnementaux ... 26
2.5. Impact de la complexation sur la toxicité ... 28
2.6. Complexation des isotopes de l’uranium avec les AH ... 31
3. Techniques ... 33
3.1. Techniques de séparation ... 33
3.1.1. La dialyse ... 33
3.1.2. L’ultrafiltration ... 35
3.1.3. La chromatographie d’exclusion stérique ... 37
3.1.4. Le fractionnement par couplage flux-force asymétrique ... 39
3.1.5. Comparaison entre les diverses techniques de séparation ... 41
3.2. Mesure du ratio isotopique d’uranium ... 42
3.2.1. La spectrométrie-alpha ... 42
3.2.2. La spectrométrie de masse couplée à un plasma induit ... 43
3.2.3. Comparaison des performances analytiques des deux approches pour la mesure des ratios isotopiques de l’uranium. ... 45
3.3. Sommaire ... 47
4. Méthodologie et équipement ... 49
4.1. Réactifs et instrumentation utilisés pour la séparation des fractions d’uranium libre et lié. ... 49
viii
4.1.1. Substances humiques et autres réactifs pour la préparation des standards et des
échantillons ... 49
4.1.2. Préparation d’un échantillon synthétique d’eau du FSL ... 49
4.1.2. Étude de l’impact de la température ... 50
4.1.3. Dialyse ... 50
4.1.4. Ultrafiltration ... 53
4.1.5. Chromatographie d’exclusion stérique ... 55
4.2. Analyse des ratios isotopiques de l’uranium ... 58
4.2.1. Choix des isotopes pour la fortification des échantillons et pour l’analyse par spectrométrie de masse. ... 58
4.2.2. Instrumentation ... 59
5. Résultats et discussion ... 63
5.1. Préservation du paramètre de pH en fonction du temps ... 63
5.2. Étude de la stabilité cinétique de complexe U-AH séparé par dialyse ... 64
5.2.1. Équilibre de l’238U-AH ... 64
5.2.2. Comportement de l’uranium en absence d’acide humiques ... 66
5.2.3. Équilibre de l’uranium en présence de carbonate ... 67
5.2.4. Équilibre de U-AH en variant le ratio 233U/238U ... 69
5.2.5. Équilibre de l’U-AH en fonction de la température ... 70
5.3. Étude de la stabilité cinétique de complexe U-AH séparé par ultrafiltration ... 72
5.4. Étude de la stabilité cinétique de complexe U-AH séparé par SEC ... 74
5.4.1. Éluants et étalons de chromatographie ... 74
5.4.2. Temps d’élution des analytes en SEC ... 77
6. Conclusion et perspectives ... 103 RÉFÉRENCES ... 107 ANNEXE 1 ... 115 Protocole de la dialyse ... 115 ANNEXE 2 ... 117 Protocole de l’ultrafiltration ... 117 ANNEXE 3 ... 119 Procédure de l’ultrafiltration ... 119 ANNEXE 4 ... 121
Protocole pour la SEC ... 121
ANNEXE 5 ... 123
Paramètres de la SEC ... 123
ANNEXE 6 ... 125
ix
LISTE DES TABLEAUX
Tableau 1. Répartition de l’uranium dans l’environnement.12 ... 7
Tableau 2. Caractéristiques des différents isotopes de l’uranium naturel.20,21 ... 12
Tableau 3. Valeurs de constantes de complexation (log K) entre l’uranyle et divers anions à pH 8. ... 13
Tableau 4. Composition élémentaire (en %) de différentes substances humiques en lien avec leur provenance (adapté de Tipping).33 ... 17
Tableau 5. Composition (en %) des groupements fonctionnels de différentes substances humiques.33 ... 21
Tableau 6. Constantes de complexation de métaux usuels trouvés dans les cours d’eau en fonction du pH. ... 24
Tableau 7. Constantes de complexation de SH avec l’uranyle variant en fonction du pH et de leur provenance. ... 25
Tableau 8. Comparaison générale des techniques de séparation. ... 41
Tableau 9. Paramètres pris en compte dans la composition de la solution fluviale. ... 50
Tableau 10. Paramètres d’élution optimisés de la méthode SEC. ... 57
Tableau 11. Limites de détection et de quantification de l’233U et l’238U obtenues avec l’ICP-MS 8800 ICP Triple Quad. ... 59
Tableau 12. Paramètres de l’ICP-MS 800MS de Bruker® optimisés en haute sensibilité pour les analyses de 233U, 238U et 205Tl. ... 60
Tableau 13. Paramètres de l’ICP-MS 8800 ICP Triple Quad (ICP-QQQ) d’Agilent® optimisés pour les analyses de 233U, 238U et 205Tl... 61
Tableau 14. Masse moléculaire des AH selon la courbe d’étalonnage avec un éluant de NH4Cl 10 mM à pH 6. ... 76
Tableau 15. Récupération de l’uranyle utilisant du (NH4)2C2O4 10 mM comme éluant. .... 78
xi
LISTE DES FIGURES
Figure 1. Schématisation de l’incident de Tricastin.3 ... 1
Figure 2. Exemple de carte isométrique des densités de pollution radioactive pour l’incident de Tchernobyl en 1986.5 ... 2
Figure 3. Introduction d’uranium d’origine anthropique dans un milieu naturel déjà à l’équilibre. ... 3
Figure 4. Activités nucléaires dans la région des Grands Lacs en 1991 (adapté de S.R. Joski).9 ... 4
Figure 5. Schéma d’une réaction de fission (235U) ainsi que le fonctionnement d’une centrale nucléaire de type CANDU.14 ... 8
Figure 6. Diagramme de Pourbaix dans un système avec la présence des atomes U-H-O-C.15 ... 9
Figure 7. Prédiction de la spéciation de l’uranium en fonction du pH pour un milieu aqueux avec et sans la présence d’acides fulviques (AF) et de carbone organique dissout (COD).16 ... 10
Figure 8. Fractionnement entre l’234U et l’238U résultants de l’effet de recul présent lors de la désintégration de l’238U en 234Th dans l’eau (A) et le sol (B). ... 14
Figure 9. Représentation des catégories de la matière organique dissoute (adapté de S. Findlay, R. L. Sinsabaugh).31 ... 16
Figure 10. Représentation des couches du sol et localisation de l’humus, source majoritaire de SH (tiré de Belin SVT 6°).32 ... 16
Figure 11. Provenance et séparation des différentes fractions des SH. ... 18
Figure 12. Principales caractéristiques des familles de SH (adapté de Belin SVT 6°).32 .... 19
Figure 13. Structures de squelettes chimiques proposées pour les trois catégories de SH. . 20
Figure 14. Trois différentes approches pour expliquer la complexation/chélation entre un métal et les groupements carboxyliques et phénoliques (adapté de Manahan).47 ... 23
Figure 15. Graphique démontrant la relation entre la concentration de métal libre et la concentration de complexe métal-site actif d’une cellule cible.67 ... 29
Figure 16. Modèle du ligand biotique tel que démontré par l’auteur D.M. Di Toro (traduit de Environ. Toxicol. and Chem.).7 ... 30
Figure 17. Scénarios envisagés pour expliquer la cinétique de complexation entre deux isotopes d’uranium et des SH. ... 32
Figure 18. Principe de la dialyse. ... 34
Figure 19. Principe de la technique d’ultrafiltration. ... 36
xii
Figure 21. Concept général du FFFA.76 ... 39 Figure 22. Exemple d’un spectre d’uranium obtenu grâce à la spectrométrie alpha. ... 43 Figure 23. Principales composantes et schématisation simplifié d’un ICP-MS... 44 Figure 24. Graphique représentant la limite de détection de la spectrométrie alpha et de
l’ICP-MS. ... 46
Figure 25. Schéma du fonctionnement du modèle Float-A-Lyzer® (A) ainsi que du modèle
Spectra/Por (B) de la compagnie Spectrum Labs®. ... 51
Figure 26. Schématisation du protocole ayant servi à réaliser les tests de la dialyse,
l’ultrafiltration et la chromatographie d’exclusion stérique. ... 52
Figure 27. Composantes d’une cellule d’ultrafiltration de la compagnie Amicon®. ... 54 Figure 28. Schéma représentant le système de SEC, présentant les principales
composantes. ... 56
Figure 29. Structure du sulfonate de polystyrène. ... 56 Figure 30. Stabilité du pH de la solution fluviale synthétique en fonction du temps et de la
quantité de NaOH 0,1 M ajoutée. ... 63
Figure 31. Temps nécessaire pour atteindre l’équilibre en dialyse avec le complexe U-AH.
... 65
Figure 32. Comportement des isotopes d’U avec la membrane à dialyse. ... 67 Figure 33. Comportement des isotopes d’U avec les AH en absence de CO32- séparés par
dialyse. ... 68
Figure 34. Comportement des isotopes d’U avec les AH en variant le ratio isotopique de 233U/238U. ... 69 Figure 35. Équilibre entre les isotopes d’U et les AH en fonction des températures à 4 °C,
10 °C, 22 °C séparés par dialyse. ... 71
Figure 36. Équilibre entre les isotopes d’U et les AH séparés par ultrafiltration. ... 73 Figure 37. Courbe d’étalonnage de la masse moléculaire équivalent au maximum du pic
(Mp) en fonction du temps de rétention calculer en SEC avec étalons PSS et AH dans le NH4Cl 10 mM à pH 6. ... 75 Figure 38. Élution de l’238U dans le (NH
4)2C2O4 10 mM à pH 6. Le spectre d’absorption
avec résolution temporelle de la SEC (A) est illustré en parallèle avec les
mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 79
Figure 39. Spectre d’absorption avec résolution temporelle de l’élution des AH dans le
(NH4)2C2O4 10 mM à pH 6. ... 80 Figure 40. Élution du complexe d’UO2-AH dans le (NH4)2C2O4 10 mM à pH 6. Le spectre
d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). .. 81
Figure 41. Élution du complexe d’UO2-AH dans le CO32- avec un gradient d’eau et de
xiii de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 83
Figure 42. Élution du mélange d’UO22+ et d’AH avec un gradient d’eau et de (NH4)2C2O4
10 mM à pH 6. Le spectre d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 84
Figure 43. Élution du mélange d’U et d’AH (Pahokee) avec un gradient d’eau et de
(NH4)2C2O4 10 mM à pH 6. Le spectre d’absorption avec résolution temporelle
de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 85
Figure 44. Élution du mélange d’U et d’AF (Pahokee) dans l’eau et élué avec un gradient
d’eau et de (NH4)2C2O4 10 mM à pH 6. Le spectre d’absorption avec résolution
temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 86
Figure 45. Élution du mélange d’U et de MON (Suwannee) avec un gradient d’eau et de
(NH4)2C2O4 10 mM à pH 6. Le spectre d’absorption avec résolution temporelle
de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 87
Figure 46. Élution du complexe La-AHA dans du NaOH 0,05 M à pH 8 avec un gradient
de NH4Cl 10 mM à pH 8 et de (NH4)2C2O4 10 mM à pH 8. Le spectre
d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). .. 89
Figure 47. Élution du mélange de La et d’AH dans l’eau à pH 8 avec un gradient de NH4Cl
10 mM à pH 8 et de (NH4)2C2O4 10 mM à pH 8. Le spectre d’absorption avec
résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 90
Figure 48. Étude de l’impact de la présence de l’ion ammonium sur le profil d’élution du
La et des AH. Le spectre d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 91
Figure 49. Étude de l’impact de la préparation de l’échantillon pour solubiliser les analytes
sur le profil d’élution du La et des AH. Le spectre d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 92
Figure 50. Étude de l’impact de la concentration de chacun des analytes sur le profil
d’élution du La et des AH. Le spectre d’absorption avec résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 93
Figure 51. Élution du complexe U-AH dans l’eau à pH 8 avec un gradient de NH4Cl 10
mM à pH 8 et de (NH4)2C2O4 10 mM à pH 8. Le spectre d’absorption avec
résolution temporelle de la SEC (A) est illustré en parallèle avec les mesures d’ICP-MS de chaque fraction (B) et le gradient utilisé (C). ... 95
xiv
Figure 52. Profil d’élution du 233U- 238U-AH dans l’eau à pH 8 avec du NH
4Cl 10 mM à
pH 8 et du (NH4)2C2O4 10 mM à pH 8 à T = 0 h. Le spectre d’absorption avec
résolution temporelle de la SEC (A) est illustré en parallèle avec le gradient utilisé (B). ... 96
Figure 53. Profil d’élution du 233U- 238U-AH dans l’eau à pH 8 avec du NH
4Cl 10 mM à
pH 8 et du (NH4)2C2O4 10 mM à pH 8 à T = 502 h. Le spectre d’absorption
avec résolution temporelle de la SEC (A) est illustré en parallèle avec le
gradient utilisé (B). ... 96
Figure 54. Graphique de la tendance de complexation des isotopes d’238U/233U avec les AH
en fonction du temps. ... 98
Figure 55. Graphique de la tendance des isotopes d’238U/233U libre en fonction du temps. 99 Figure 56. Graphique de la tendance de complexation des isotopes d’235U/233U avec les AH
en fonction du temps. ... 100
Figure 57. Graphique de la tendance des isotopes d’235U/233U libre en fonction du temps.
xv
ABRÉVIATIONS
A Activité
AF Acides fulviques
AH Acides humiques
AHA Acides humiques acétylées ASN Autorité de sureté nucléaire BLM « Biotic ligand model »
Bq Becquerel
BVSL Bassin versant du Saint-Laurent CANDU Canada Deutérium Uranium
cm Centimètre
COD Carbone organique dissous cps Coups par secondes
Da Dalton
FFFA Fractionnement par couplage flux-force asymétrique
fg Femtogramme
FIAM « Free-ion activity model » FSL Fleuve Saint-Laurent
g Gramme
h Heure
HNO3 Acide nitrique
ICP-MS Spectrométrie de masse couplée à un plasma inductif
IHSS « International Humic Substances Society » (Société International des substances humiques) IR Infrarouge KeV Kiloélectron-volt Kg Kilogramme KW Kilowatt L Ligand L Litre
xvi
LDM Limite de détection de méthode Log K Constante de complexation
m Masse MeV Milliélectron-volt mg Milligramme min Minutes mL Millilitre mm Millimètre MM Masse atomique mM Millimolaire mmol Millimole
MOD Matière organique dissoute
mol Mole
MON Matière organique naturelle
mV Millivolt
NA Nombre d’Avogadro
NaClO4 Perchlorate de sodium
NaCO3 Carbonate de sodium
ng Nanogramme
(NH4)2C2O4 Oxalate d’ammonium
NH4OH Ammoniaque
nm Nanomètre
(NO3)2UO2 Nitrate d’uranyle
OMS Organisation mondiale de la santé PDI Indice de polydispersité
PEEK « Polyether ether ketone » pKa Constante de protonation ppb Partie par milliard ppm Partie par million ppt Partie par trillion
xvii PSS Sulfonates de polystyrène
RMN Résonnance magnétique nucléaire rpm Tour par minute
s Seconde
SEC « Size exclusion chromatography » (Chromatographie d’exclusion stérique)
SH Substances humiques
T1/2 Temps de demi-vie
TENORM Matériaux présentant une radioactivité naturelle renforcée Torr Millimètre de mercure
TRU Colonne d’extraction d’actinide
μg Microgramme
μL Microlitre
μm Micromètre
μmol Micromole
u.a. Unité d’absorbance
UO2 Uranyle
UV-Vis Ultraviolet-Visible
V Volt
xix
REMERCIEMENTS
J’aimerais tout d’abord remercier mon directeur de recherche, M. Dominic Larivière. Ses généreux conseils, sa patience sans limite et ses idées innovantes m’ont permis d’avancer grandement dans un projet en développement. De même, je tiens à remercier M. Serge Groleau qui m’a également beaucoup aidé tout au long de ma maîtrise avec les divers appareils, notamment l’ICP-MS qui nous en a fait voir de toutes les couleurs.
J’aimerais également remercier mes collègues de travail qui m’ont permis d’avancer dans mon projet avec leurs nombreuses suggestions et surtout, leur bonne humeur et leurs sujets de conversations variés. Un merci particulier à M. Nicolas Guérin qui a été un mentor lors de mes stages au baccalauréat ainsi qu’à Mme Sabrina Potvin, Mme Laurence Whitty-Léveillé et M. Julien Légaré Lavergne qui ont effectué moult tâches et m’ont permis d’obtenir de nombreux résultats lors de leur passage comme étudiant stagiaire au laboratoire.
J’en profite aussi pour remercier les membres de ma famille, qui m’ont toujours soutenu, encouragé et permis de me détendre dans les moments plus difficiles.
Finalement, j’aimerais remercier mes amis Jimmy, Benjamin et Maxime qui m’ont permis de me changer les idées et d’obtenir des idées variées. J’aimerais terminer mes remerciements avec la personne qui m’a le plus soutenu, aidé et même enduré tout au long de ma maîtrise, ma copine Marie-Pier. Merci d’être qui tu es et merci pour tout ce que tu as fait pour moi.
1
1. Introduction
1.1. Mise en contexte
Le 8 juillet 2008, dans un complexe français aux abords du Rhône, survient un déversement d’une solution aqueuse à la suite du lavage d’une cuve de rétention. Une trentaine de mètres cubes de la solution ont été déversés dans le ruisseau de la Gaffière (Figure 1) qui traverse le site industriel, et une partie a pu atteindre la nappe phréatique. Même si en apparence anodin et d’ampleur limitée, cet incident fut hautement médiatisé. L’origine de cette frénésie médiatique réside dans le type d’activité effectué sur ce site, soit l’enrichissement d’uranium naturel en isotope 235 fissible pour alimenter, entre autres, le parc des réacteurs nucléaires de la France en combustible nucléaire. En effet, ladite solution provenait d’une cuve de rétention de la centrale nucléaire de Tricastin et était largement contaminée à l’uranium, atteignant des concentrations approximatives de 12 g/L.1
Cet incident, bien que classifié comme minimal sur l’échelle des événements nucléaires (1 sur une échelle de 8) par l’autorité de sureté nucléaire (ASN), a suscité de nombreuses craintes et inquiétudes de la part du public. Le verdict final confirmera que l’événement a causé plus de peur que de mal, puisque les intervenants auront finalement découvert, en référence à un long processus de prélèvements et de mesures périodiques, que seuls deux endroits situés au sud du site nucléaire présentaient des concentrations supérieures à la limite de 15 μg/L imposée par l’organisation mondiale de la santé2 (OMS).
2
Généralement, les motifs de distribution des radionucléides dans l’environnement sont basés sur des modèles de dispersions atmosphériques pour l’évaluation des doses et teneurs, comme ce fut le cas à Tchernobyl (Figure 2) et Fukushima, en 1986 et 2011, respectivement.4 Or, très peu d’études et de modèles biogéochimiques et géologiques sont
disponibles pour prédire les impacts d’une dispersion liquide contenant divers contaminants potentiels tels que l’uranium. De plus, rares sont les stratégies de modélisation qui impliquent la présence simultanée de contaminants d’origine naturelle et anthropique, comme c’est le cas avec l’uranium.
En effet, l’uranium se retrouve naturellement dans l’environnement à des teneurs variables selon le compartiment de l’environnement étudié. Cet uranium élémentaire possède une isotopie caractéristique qui est de 99,274 %, 0,7200 % et 0,0056 %, pour l’238U, 235U et 234U, qui sont les trois isotopes d’origine naturelle de cet élément. À l’opposé, l’uranium
utilisé dans l’industrie nucléaire doit, pour certains types de réacteurs, comme les réacteurs à eau pressurisée, être enrichi afin d’augmenter la teneur en isotopes fissibles. Ce combustible possède donc, après son traitement, une teneur en isotope 235U qui peut être
enrichi jusqu’à 5 %.
Figure 2. Exemple de carte isométrique des densités de pollution radioactive pour l’incident de
3 Étant donné la variation isotopique de matériaux présentant une radioactivité naturelle renforcée (TENORM), l’impact sur la faune et la flore se révèle être beaucoup plus complexe à étudier. Effectivement, l’uranium possède, comme tous les autres radioisotopes, une ambivalence par rapport à sa toxicité, puisqu’il possède à la fois une toxicité chimique, qui provient de sa composition élémentaire, et une toxicité radiologique, attribuable au caractère radioactif de l’ensemble de ses radioisotopes. Bien que sa toxicité chimique demeure stable en fonction du ratio isotopique, sa toxicité radiologique pourrait grandement varier, selon le degré d’enrichissement et d’activation neutronique dans le cas de combustibles nucléaires neufs et usés. Puisque les divers radioisotopes de l’uranium possèdent une activité spécifique qui leur est propre, la dose reçue par un organisme sera influencée par l’isotopie de l’uranium. Par conséquent, l’effet sur l’organisme résultant d’une contamination dépendra grandement de la composition isotopique du matériel déversé et des voies d’expositions.
Il existe des modèles utilisés afin de déterminer la spéciation d’actinides,6,7 aidant ainsi à
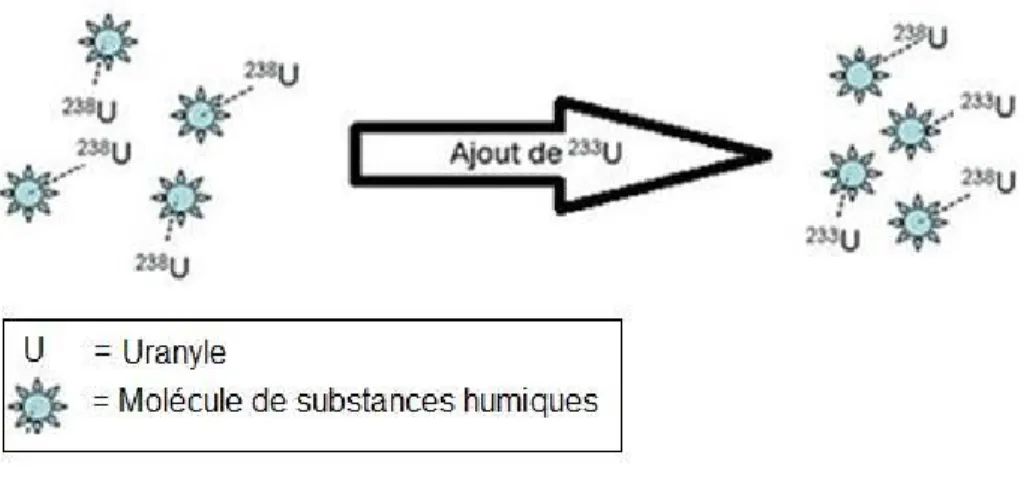
prédire le comportement chimique d’un élément dans un environnement dont les paramètres chimiques et physiques sont connus. Ces modèles se basent sur des données thermodynamiques pour estimer, à des conditions de pH et de températures données, les diverses espèces présentes. Par contre, le cas des TENORM diffère de celui des autres métaux, puisque l’introduction d’espèces incluses dans ces matériaux engendrera un processus de compétition avec celles déjà présentes à l’équilibre dans l’environnement (Figure 3).
4
Dans une telle situation, en plus des paramètres thermodynamiques qui influenceront la distribution et la spéciation des isotopes, certains paramètres cinétiques pourraient également contribuer à modifier l'isotopie du milieu. À l’exception de l’étude de Marx et coll.8 portant sur la différence de stabilité des complexes formés avec deux isotopes
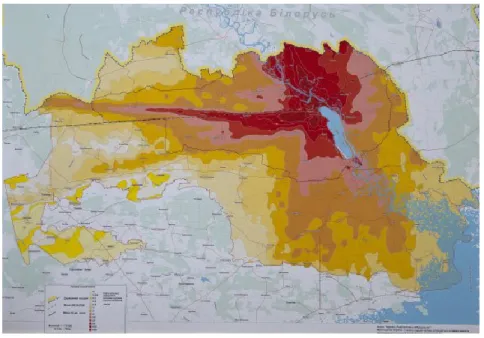
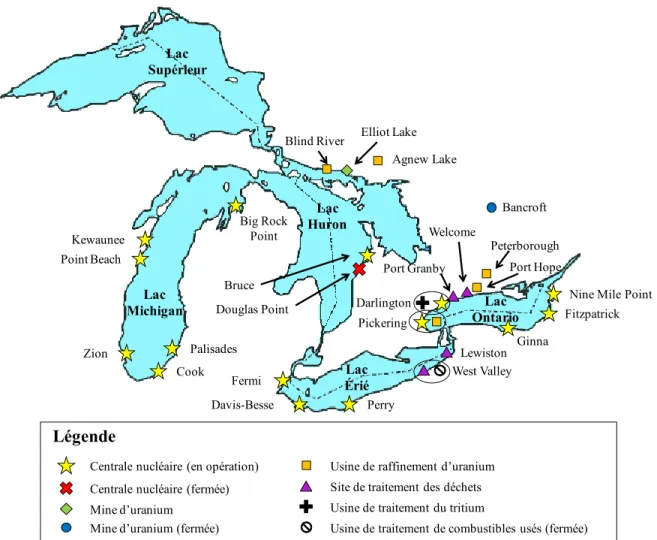
différents, bien peu de chercheurs se sont intéressés à l’impact des paramètres cinétiques sur la spéciation d’ions inorganiques. Nous croyons que l’étude de ces paramètres est d’autant plus pertinente pour l’uranium dont la composition isotopique est fréquemment altérée pour assouvir les besoins énergétiques de la population. De plus, puisque le bassin versant du fleuve Saint-Laurent (BVSF) a accueilli, au cours des soixante dernières années, tous les stades du cycle du combustible nucléaire (Figure 4), il représente un écosystème d’intérêt pour ce type d’étude.
Figure 4. Activités nucléaires dans la région des Grands Lacs en 1991 (adapté de S.R. Joski).9 Centrale nucléaire (en opération)
Centrale nucléaire (fermée)
Mine d’uranium (fermée) Mine d’uranium
Usine de raffinement d’uranium Site de traitement des déchets Usine de traitement du tritium
Usine de traitement de combustibles usés (fermée)
Légende Lac Supérieur Lac Michigan Lac Huron Lac Érié Lac Ontario Kewaunee Point Beach Cook Zion Palisades Big Rock Point Fermi Davis-Besse Perry Ginna Lewiston Fitzpatrick Nine Mile Point Peterborough Port Hope Welcome Port Granby Darlington Bancroft Pickering West Valley Elliot Lake Agnew Lake Blind River Bruce Douglas Point
5 Ce mémoire a donc pour objectif l’étude de la stabilité des complexes d’uranium avec les substances humiques (SH), qui sont des complexants naturels importants. L’étude tentera de reproduire un milieu mimant les conditions chimiques et physiques du fleuve Saint-Laurent (FSL). Des techniques de séparation telles que la chromatographie d’exclusion stérique (SEC), la dialyse et l’ultrafiltration seront comparées et les ratios isotopiques seront analysés à l’aide de la spectrométrie de masse couplée à un plasma inductif (ICP-MS).
7
2. Théorie
2.1. L’uranium
L’uranium est un élément de la famille des actinides qui comprend l’ensemble des éléments inclus entre l’actinium et le lawrencium. C’est le 48e élément naturel, en termes
d’abondance, présent dans la croûte terrestre avec des teneurs, généralement de l’ordre du mg/kg, très variables selon le type de sol.10 Sa grande solubilité, en comparaison avec
plusieurs des autres actinides tel que le thorium, lui confère une omniprésence, au niveau de trace, dans l’ensemble de l’environnement, y compris dans les cours d’eau, comme les fleuves et les océans.11 Sa répartition dans l’environnement est présentée au Tableau 1.
Comme mentionné précédemment, l’abondance isotopique naturelle de l’uranium est de :
238U à 99,274 %, 235U à 0,7200 % et 234U à 0,0056 %. Le Canada étant le 2e plus grand pays
producteur d’uranium dans le monde, la compréhension des processus biogéochimiques de cet élément revêt donc un intérêt scientifique particulier.
Tableau 1. Répartition de l’uranium dans l’environnement.12
Compartiment environnemental mg/kg d'U % de la répartition planétaire de l’U
Gisement riche à 2% U 20000 95,20
Gisement pauvre à 0,1% U 1000 4,76
Roche acide type granite 4 0,02
Croûte continentale 2,8 0,01
Roche sédimentaire 2 0,01
Eau de mer 0,003 <0,01
Historiquement, l’uranium était utilisé dans diverses applications telles que la pigmentation dans la verrerie, la catalyse de certaines réactions, l’analyse qualitative et quantitative du sodium et la fabrication de munitions militaires. Cependant, de nos jours, son utilisation s’oriente essentiellement vers l’industrie nucléaire, où il sert de combustible.13 En effet,
8
fission nucléaire, ce dernier étant le seul isotope naturel fissible de l’uranium. De ce fait, sa teneur dans le combustible est dictée par le type de réacteur où il sera utilisé.
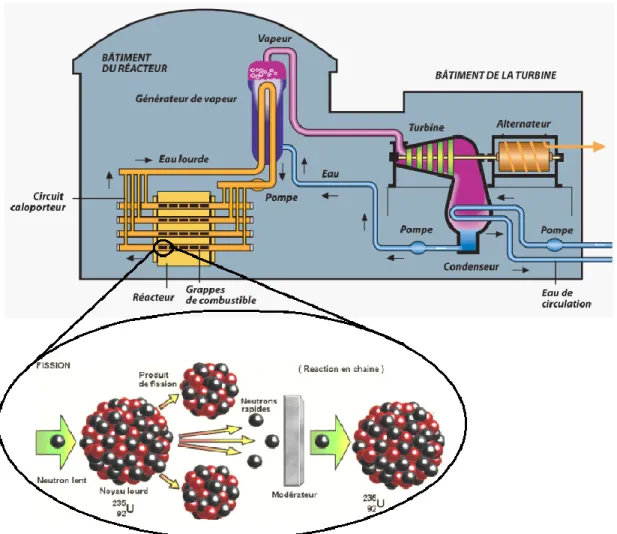
Le caractère fissible de l’235U signifie que lorsqu’il est bombardé par un flux de neutrons,
son noyau captera ces particules élémentaires et se scindera en deux noyaux atomiques de masses plus faibles tout en libérant de l’énergie et des neutrons (Figure 5). Dans un réacteur nucléaire, cette énergie sera ensuite transférée à un liquide appelé le caloporteur, dont le rôle est de convertir cette chaleur absorbée en vapeur. Cette dernière sera utilisée pour créer une pression suffisante permettant d’actionner une turbine à vapeur qui générera de l’électricité à l’aide d’un alternateur.
Figure 5. Schéma d’une réaction de fission (235U) ainsi que le fonctionnement d’une centrale
9 Dans un réacteur nucléaire, l’uranium se retrouve sous forme chimique UO2, soit sous
forme tétravalente (U4+). Bien que quatre valences existent pour l’uranium (+III, +IV, +V
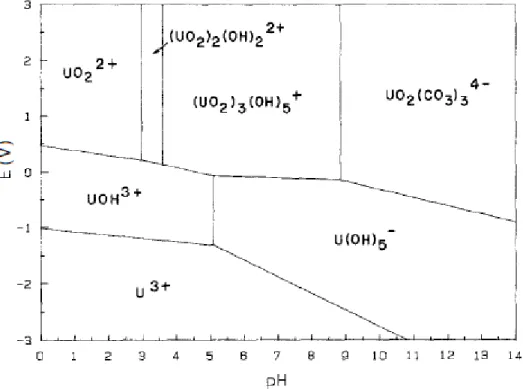
et +VI), on le retrouve presque exclusivement sous la forme +IV et +VI dans l’environnement. Il est à noter que la valence de cet élément dépend grandement du potentiel d’oxydoréduction et du pH du milieu dans lequel on le retrouve (Figure 6).
Figure 6. Diagramme de Pourbaix dans un système avec la présence des atomes U-H-O-C.15
Basé sur les teneurs de chacune des espèces présentes dans un écosystème (ex. CO32-,
acides humiques (AH), OH-), tel un cours d’eau, il est possible d’estimer la spéciation de
10
Figure 7. Prédiction de la spéciation de l’uranium en fonction du pH pour un milieu aqueux avec et
11 Comme mentionné précédemment, l’uranium possède un double statut en matière de toxicité, soit sa chimio-toxicité et sa radio-toxicité. En effet, le caractère chimiotoxique des espèces solubles de l’uranium se traduit, à teneurs élevées dans l’organisme, par des complications rénales. Le seuil de toxicité chimique de l’uranium est estimé à 70 µg par kg de poids corporel. L’ingestion d’espèces d’uranium insolubles se traduit, quant à elle, par la déposition de ce dernier dans la structure squelettique.17 Santé Canada recommande une
concentration maximale acceptable de 20 µg/L dans l’eau de consommation.18 Du point de
vue de la toxicité radiologique, celle-ci se manifeste à la suite de l’émission de particules alpha lors du réarrangement nucléaire. Ces particules alpha émises possèdent une énergie de l’ordre du million d’électronvolts. Si ces dernières venaient à être émises depuis l’intérieur de l’organisme, elles auraient le pouvoir d’exciter et d’ioniser un grand nombre de molécules, ce qui pourrait entraîner des altérations biologiques potentiellement néfastes. Ces altérations pourraient se traduire, sur une échelle de temps de l’ordre de quelques dizaines d’années, en cancer.
Puisque les isotopes naturels de l’uranium ont des temps de demi-vie (T½) longs, leur
transformation en d’autres radioéléments sera lente. Le temps de demi-vie représente le temps nécessaire pour observer la diminution de l’activité initiale par un facteur deux. En appliquant la formule de l’activité massique :
𝐴
𝑚= 𝑁𝐴∗ 𝑙𝑛2 ∗ 𝑀𝑀 −1∗ 𝑇
1/2−1,
où NA est le nombre d’Avogadro et MM est la masse atomique de l’isotope, on remarque que
l’activité (A) d’un isotope, pour une masse donnée, est inversement proportionnel à son T1/2.Bien que l’isotope 238U ait un temps de demi-vie plutôt long (4,5x109 années), donc
une activité massique d’environ 12 445 Bq/g, c’est l’isotope 234U qui a un temps de
demi-vie plus court (2,4 x105 années), donc une activité massique plus importante (~231 300
000 Bq/g). Ainsi, pour 1 g d’uranium naturel par exemple, l’234U a pratiquement la même
12
Tableau 2. Caractéristiques des différents isotopes de l’uranium naturel.20,21
Isotope 234U 235U 238U
Temps de demi-vie (années) 2,5 x 105 7,1 x 108 4,5 x 109
Masse molaire (g/mol) 234,04 235,05 238,05
Pourcentage massique 0,0056 0,720 99,274
Pourcentage en radioactivité 48,9 2,2 48,9
Type de rayonnement Alpha, gamma Alpha, gamma Alpha
Énergie de la particule alpha (MeV) 4,8 4,4 4,2
Activité massique (Bq/g) 2,31 x 108 8,0 x 104 1,24 x 104
Dans l’environnement, l’uranium se retrouve généralement, dans les minerais et la croûte terrestre, avec un degré d’oxydation +IV. Étant donné la solubilité limitée de l’oxyde d’uranium tétravalent (log K de 60,2 pour la solubilité d’UO2),22 c’est sous sa forme
ionique hexavalente que l’uranium est majoritairement retrouvé dans l’eau. Dépendant des conditions chimiques et physiques du milieu, l’ion uranyle (UO22+) pourra être lié à
d’autres molécules telles que les carbonates, les sulfates, les phosphates, les hydroxydes et la matière organique. Puisque la spéciation de l’ion uranyle dans l’eau est influencée par de nombreux facteurs, il est préférable de définir certains paramètres chimiques afin d’effectuer des comparaisons valables. Par exemple, pour une eau à pH 8, une valeur de pH comparable à celle du fleuve Saint-Laurent,23 les composés carbonatés dominent largement
les processus de complexation avec l’UO22+. Le Tableau 3 permet de comparer les
constantes de complexation des hydroxyles, des AH et des carbonates (Log K, où K représente la constante de formation d’un complexe :
{𝑀 + 𝐿 ↔ 𝑀𝐿; 𝐾 =[𝑀][𝐿][𝑀𝐿]}),
où M = métal et L = ligand. Par contre, d’autres molécules de type organique (les substances humiques) présentent également des propriétés complexantes non négligeables en milieu aqueux, principalement en milieu acide (Figure 7).
13
Tableau 3. Valeurs de constantes de complexation (log K) entre l’uranyle et divers anions à pH 8.
UO22+- Lx Log K pH Références
[UO2(AH)x]Y 5,75 8 Riggle et coll.24
UO2(OH)2 12,00 8 Grenthe et coll.25
[UO2(CO3)3]4- 24,00 8 Ciavatta et coll.26
2.2. Introduction d’uranium dans l’environnement
Malgré que l’uranium soit omniprésent dans l’environnement avec des proportions isotopiques naturelles, certains processus anthropiques peuvent altérer les teneurs et l’abondance des isotopes d’uranium. Une des voies possibles d’introduction d’uranium dans l’environnement peut provenir de son utilisation dans le domaine nucléaire. Bien que cette industrie soit régie par des règles et des normes strictes, elle n’est pas à l’abri de certains incidents (Tchernobyl et Fukushima). Par exemple, dans le cas de l’accident de Tchernobyl, sept tonnes d’UO2 en fusion et de particules contenant de nombreux produits
de fission ont été larguées dans l’environnement.27
Par ailleurs, le démantèlement des installations nucléaires, représente également un risque potentiel de fuite d’eau ou de déchets contaminés dans l’environnement et constitue donc une autre voie possible d’introduction d’uranium d’origine anthropique dans l’environnement. Cette voie d’introduction est d’autant plus à considérer pour le bassin versant du Fleuve St-Laurent, puisque le sort de la centrale nucléaire en arrêt de Gentilly-2 n’est pas encore connu.
Finalement, l’exploitation minière peut également servir de vecteur d’introduction de l’uranium dans l’environnement. En effet, durant le processus d’exploitation, l’extraction du minerai du sol peut entraîner la dispersion de l’uranium dans les nappes phréatiques adjacentes aux sites d’exploitation, si aucune mesure de précautions n’est prise. C’est notamment cette dispersion potentielle qui inquiète les habitants de la région du lac Kachiwiss, près de Sept-Îles, puisqu’un site potentiel d’exploitation minier appartenant à la compagnie Terra Ventures est présentement au stade d’exploration.28 Cependant, dans ce
14
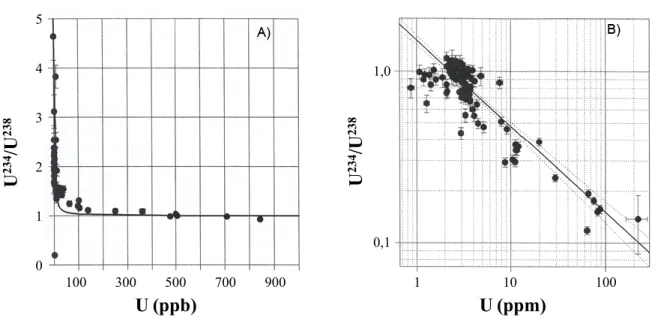
études29 ont montré un faible fractionnement, de l’ordre d’une partie par million, entre
l’234U et l’238U résultant de l’effet de recul présent lors de la désintégration de l’238U en 234Th. L’impact de cet effet se traduit par l’enrichissement de l’isotope 234U dans l’eau
(Figure 8A) et l’appauvrissement de ce dernier dans le sol (Figure 8B). Cet effet est influencé par la concentration d’uranium, qui dicte le degré de solubilité des ions dans l’eau.
Figure 8.Fractionnement entre l’234U et l’238U résultants de l’effet de recul présent lors de la
désintégration de l’238U en 234Th dans l’eau (A) et le sol (B).
D’un point de vue scientifique et toxicologique, l’enjeu est de connaître l’impact que pourrait avoir l’ajout d’uranium présentant un ratio isotopique différent des ratios naturels dans un environnement donné. C’est cet aspect qui sera abordé dans ce mémoire.
2.3. Les substances humiques
Pour bien comprendre en détail le comportement de l’uranium dans un environnement aqueux, un cours d’eau par exemple, il est essentiel de tenir compte des espèces présentes dans ce milieu. Parmi celles-ci, on compte la matière organique, qui est fabriquée par des
100 300 500 700 900 0 1 2 3 4 5 U 234 /U 238 U (ppb) U 234 /U 238 U (ppm) 100 10 1 0,1 1,0
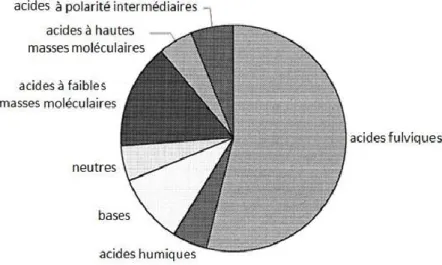
15 procédés émanant des êtres vivants (animaux, végétaux et micro-organismes) et se compose principalement d’une combinaison d’eau, de carbone et d’autres éléments, dont l’hydrogène, l’oxygène et l’azote. La sous-catégorie de la matière organique qui est dissoute dans l’eau (MOD) représente une famille qui englobe un bon nombre de molécules organiques, principalement constituées de carbone. Ces molécules proviennent principalement de la dégradation de la faune et la flore qui sont composées en grande majorité de matières organiques (Figure 9).
Ces molécules interviennent dans un grand nombre de procédés des écosystèmes aquatiques, principalement grâce à leur propriété à complexer les métaux, ce qui assure leur transport et leur distribution dans un écosystème. La plus importante sous-catégorie des MOD, qui représente une proportion de 40 à 60 % (Figure 9), est la famille des SH. Celles-ci sont des macromolécules organiques complexes provenant de la décomposition et de la dégradation de la matière organique. Elles partagent plusieurs ressemblances par rapport à leur structure avec les tannins et les lignines,30 qui sont des molécules retrouvées à l’état
naturel dans le sol, dans l’écorce de certains arbres et dans les plantes ligneuses. Le terme SH ne fait pas référence à une seule molécule mais bien à une famille chimique de molécules qui partagent plusieurs similitudes. En effet, les SH sont divisées en trois grandes catégories, soit les humines, les acides humiques (AH) et les acides fulviques (AF). Ces trois groupes de SH partagent des caractéristiques analogues quant à leur structure chimique, leur masse moléculaire et leur provenance, mais présentent des divergences en ce qui à trait à leur degré de solubilité en solution. C’est notamment cette caractéristique qui permet de les scinder en différentes catégories.
16
Figure 9. Représentation des catégories de la matière organique dissoute (adapté de S. Findlay, R.
L. Sinsabaugh).31
La provenance des SH est hautement reliée au cycle du carbone organique. Puisque les SH sont obtenues à la suite de dégradation des plantes, elles se retrouvent en abondance dans les sols, plus précisément dans la couche appelée humus. Cette couche se retrouve dans la partie supérieure du sol, comme le démontre la Figure 10 et elle est entretenue par la décomposition de la matière organique, grâce à l’efficacité combinée des animaux, des bactéries et des champignons du sol.
Figure 10. Représentation des couches du sol et localisation de l’humus, source majoritaire de SH
17 Par contre, il est aussi possible d’en retrouver dans les eaux de surfaces, les eaux usées, le compost, les sédiments de la mer et des lacs, les tourbières, les schistes charbonneux et les lignites30 en raison du cycle du carbone. Étant obtenues de sources distinctes, au cours de
processus de dégradation plus ou moins complets, les SH ont une composition élémentaire unique et hautement caractéristique du milieu duquel elles proviennent (Tableau 4).
Tableau 4. Composition élémentaire (en %) de différentes substances humiques en lien avec leur
provenance (adapté de Tipping).33
Substances humiques n C H O N S AH du sol plusieurs 52,8 - 58,7 2,2 - 6,2 32,8 - 38,3 0,8 - 4,3 0,1 - 1,5 AF du sol plusieurs 40,7 - 50,7 2,8 - 7,0 39,7 - 49,8 0,9 - 2,3 0,1 - 2,6 AH d'eau sous-terraine 5 65,5 5,2 24,8 2,4 1,0 AF d'eau sous-terraine 5 60,4 6,0 32,0 0,9 0,7 AF d'eau marine 1 51,8 7,0 37,7 6,6 0,5 AH d'eau de rivière 15 52,2 4,9 41,7 2,1 - AF d'eau de rivière 15 52,7 5,1 40,9 1,1 0,6 AF d'eau de lac 3 54,8 5,5 41,1 1,4 1,1 Humine du sol 2 55,9 5,8 32,8 4,9 - Humine de tourbière 2 56,3 5,1 36,5 2,1 - Humine marine 2 56,2 7,0 31,7 5,2 -
n = le nombre d’échantillons considérés
Bien que ces molécules possèdent des structures similaires, il existe une propriété chimique qui permet de les isoler les unes des autres. En effet, c’est par une méthode classique d’extraction utilisant des solutions aqueuses que furent historiquement classifiées ces molécules en trois catégories. La Figure 11 démontre, qu’en traitant de l’humus à l’aide d’une solution basique, il est possible de solubiliser une quantité de matières organiques. On obtient ainsi une fraction insoluble, nommée humine et une fraction soluble. En traitant cette dernière avec une solution acide à un pH inférieur à 2, il est possible de faire coaguler une matière brunâtre, correspondant aux AH. Finalement, la matière soluble dans cette fraction acide correspond à l’AF (Figure 11).34
18
Le degré de solubilité des SH en fonction du pH s’explique grâce à la structure complexe de ces molécules. En effet, les AF, ont un nombre plus élevé de groupements carboxyliques comparativement à celui des AH, ainsi qu’une constante de protonation (pKa) aux alentours
de 2 comparativement à 4 pour les AH, ce qui rend les AF plus stables en solution acide. La Figure 12 résume les principales caractéristiques physiques et chimiques des trois grandes catégories qui seront abordées dans les sections subséquentes.
19
Figure 12. Principales caractéristiques des familles de SH (adapté de F.J. Stevenson).35 2.3.1. Caractérisation élémentaire
La composition chimique de ces groupes de molécules comprend principalement des atomes de carbone, d’oxygène, d’hydrogène, d’azote; mais elles peuvent aussi contenir du soufre et du phosphore.36 En faisant différentes combinaisons de ces éléments structuraux
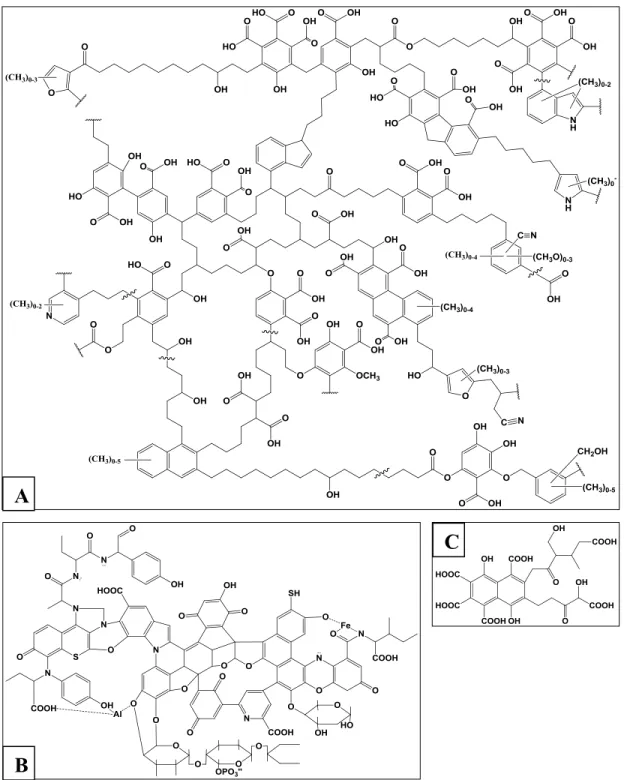
et en faisant varier leurs proportions, il est possible d’obtenir une multitude de molécules ayant un squelette différent, comme le démontre la Figure 13. Ces arrangements moléculaires sont possibles en raison de la capacité du carbone à former des liaisons avec d’autres atomes de carbone ainsi qu’avec bons nombres d’éléments tels que l’oxygène, l’hydrogène et l’azote. Étant donné que ces molécules sont très complexes, la caractérisation élémentaire de celles-ci est difficile, mais est tout de même réalisable à l’aide d’une analyse élémentaire (analyseur C-H-N).
20
Figure 13. Structures de squelettes chimiques proposées pour les trois catégories de SH. A) Humines, B) Acides humiques et C) Acides fulviques.
A
B
21
2.3.2. Groupements fonctionnels
Étant produits lors de la dégradation de la matière organique, il est fréquent de retrouver des groupements aromatiques complexes tels que des acides aminés, des sucres, des peptides et des groupements aliphatiques servant de liaison entre les groupements aromatiques des SH. Chez les groupements fonctionnels présents, on retrouve principalement des groupements phénoliques et carboxyliques, ce qui confère aux SH leur caractère diacide ou triacide. On peut également retrouver des groupements tels que les cétones, les amides ou les hydroxyles (Tableau 5). En raison de leurs différents stades de dégradation, les SH présentent des compositions atomiques différentes. Par exemple, on observe une différence notable par rapport à la teneur en oxygène des AF, qui sont les molécules les plus dégradées des trois familles. En utilisant diverses techniques comme la spectroscopie UV-Visible (UV-Vis), la spectrométrie infrarouge (IR) et la spectrométrie de résonnance magnétique nucléaire (RMN), il est possible de déterminer et de quantifier les différents groupements fonctionnels de ces molécules.
Tableau 5.Composition (en %) des groupements fonctionnels de différentes substances humiques.33 Substances humiques n Aliphatiques non saturés N-Alkyl
méthoxyles Sucres Aromatiques Carboxyles Cétones
AH du sol 8 17 - 30 4 - 9 12 - 18 24 - 42 12 - 18 4 - 7
AF du sol 1 22 5 20 26 24 4
AH aquatique 4 23 - 30 5 - 6 9 - 21 29 - 36 14 - 17 6 - 8 AF aquatique 4 30 - 40 5 - 7 10 - 18 14 - 18 16 - 19 5 - 11 n = le nombre d’échantillons considérés
2.3.3. Masses
Les SH sont des molécules ayant une large gamme de masses allant d’une centaine à quelques centaines de milliers de Daltons. Par exemple, Perminova et coll.37 ont démontré à
l’aide de la chromatographie d’exclusion stérique que les AF aquatiques ont généralement des masses moléculaires variant entre 300 Da (Moscow River, Russie) et 16 730 Da (Lac Mekkojaervi, Finlande) tandis que les AH aquatiques ont des masses supérieures de
22
l’ordre de 3150 Da (Goeta River, Suède) à 28 910 Da (Lac Mekkojaerv, Finlande). Cependant, Ishiwatari38 rapporte des valeurs de masses d’AH mesurées à l’aide de la
filtration sur gel beaucoup plus étendues, allant de moins de 700 Da à 200 000 Da (Lac Haruna et lac Kizaki, Japon). Ces deux publications illustrent bien le fait que les plages de masses sont relatives et sont grandement dépendantes de la provenance des SH ainsi que de la technique utilisée pour faire la mesure. C’est donc dire que l’instrument et la provenance des SH sont des données importantes à prendre en compte afin d’approfondir leurs impacts sur la géochimie d’un élément.
2.4. Interaction avec les SH 2.4.1. Types d’interactions
Les SH jouent depuis longtemps un rôle clé dans de nombreux processus se produisant dans le sol et les cours d’eau. Par exemple, elles interviennent dans les processus d’altération des sols, de nutrition des plantes, de tampon pour le pH, de biodisponibilité, de dégradation et de transport de produits chimiques organiques hydrophobes, de la production hétérotrophe dans les écosystèmes d'eaux noires et du transport/toxicité des métaux en traces.39 En effet, c’est ce dernier paramètre, via des processus de complexation
avec différents métaux en traces, qui est le plus fréquemment étudié. Ce processus a des répercussions importantes sur la biodisponibilité des métaux et, ainsi, altère leur toxicité lorsque ceux-ci sont dissous dans l’eau. Précisons qu’un métal est dit biodisponible lorsqu’il peut être assimilé et induire ainsi un effet sur les organismes. Ce mécanisme se produit le plus souvent lorsque le métal se retrouve sous une forme ionique libre.40
Inversement, si ce dernier n’est pas libre mais bien complexé avec un chélatant, comme les SH, l’arrangement moléculaire ainsi formé devient beaucoup plus difficilement assimilable pour les êtres vivants. De ce fait, la présence de SH dans un écosystème aqueux permet de réduire la concentration d’ions métalliques libres et conséquemment, leur biodisponibilité.41,42,43
23 Les SH contiennent un grand nombre de groupements carboxyliques et phénoliques (Figure 13 et Tableau 5), qui offrent une majorité de sites de complexations aux métaux.44,45,46 Même si leur efficacité de complexation diffère grandement des groupements
mentionnés précédemment, la présence des groupements azotés et sulfurés40 au sein des SH
jouent également un rôle dans la séquestration des ions métalliques. Les SH étant des molécules complexes et de structures variables, il est difficile de proposer un mécanisme de complexation universel, mais trois approches plausibles pour expliquer les interactions avec les groupements carboxyliques et les groupements phénoliques ont été proposées pour des ions divalents (Figure 14).47
Figure 14. Trois différentes approches pour expliquer la complexation/chélation entre un métal et
les groupements carboxyliques et phénoliques (adapté de Manahan).47 2.4.2. Évaluation des interactions
Puisque les SH complexent efficacement les métaux, elles sont parfois qualifiées de dépolluants naturels. On estime que leur capacité totale de liaisons avec les ions métalliques dans les eaux est de l’ordre de 200 à 600 µmol/g.40 Bien que certaines espèces
métalliques ont une préférence de sites de liaisons, comme le mercure pour les groupements sulfurés ou le cuivre pour les groupements carboxyliques, une certaine compétition entre les divers métaux subsiste aux nombreux sites de liaisons. La constante de complexation permet de déterminer lequel des métaux sera favorisé et deux facteurs influencent cette constante, soit la température et la pression. De plus, plusieurs autres facteurs dictent la valeur de la constante de complexation conditionnelle, comme le pH, la nature des SH, la présence d’ions tiers ou encore, la force ionique du milieu. Afin d’alléger le texte, le terme constante de complexation est également utilisé lorsqu’il est question de constante de complexation conditionnelle.
1. Chélation entre un
groupement carboxylique et un groupement phénol
2. Chélation entre deux
groupements carboxyliques
3. Complexation avec
un groupement carboxylique
24
Du fait que de nombreux facteurs entre en ligne de compte, il est extrêmement laborieux de comparer les constantes de complexation entre les ions présents dans les cours d’eau et les SH retrouvées dans un écosystème donné. Quelques unes de ces constantes sont présentées au Tableau 6. Il est important de bien prendre en compte les conditions chimiques et physiques d’un écosystème avant de faire des comparaisons avec les différentes valeurs de constante de complexation entre les métaux et les AH.
Tableau 6. Constantes de complexation de métaux usuels trouvés dans les cours d’eau en fonction
du pH.
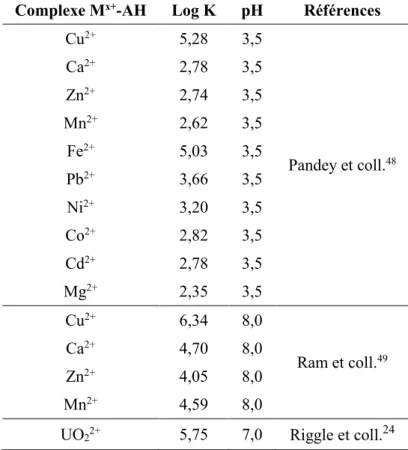
Complexe Mx+-AH Log K pH Références
Cu2+ 5,28 3,5 Pandey et coll.48 Ca2+ 2,78 3,5 Zn2+ 2,74 3,5 Mn2+ 2,62 3,5 Fe2+ 5,03 3,5 Pb2+ 3,66 3,5 Ni2+ 3,20 3,5 Co2+ 2,82 3,5 Cd2+ 2,78 3,5 Mg2+ 2,35 3,5 Cu2+ 6,34 8,0 Ram et coll.49 Ca2+ 4,70 8,0 Zn2+ 4,05 8,0 Mn2+ 4,59 8,0 UO22+ 5,75 7,0 Riggle et coll.24
Puisque les SH jouent un rôle prépondérant dans le processus de dispersion de plusieurs métaux, il est intéressant de comparer leurs affinités avec d’autres métaux en traces pouvant se retrouver dans l’environnement, soit les éléments radioactifs. Il est également intéressant de se demander si la provenance des SH pourrait avoir un effet sur le comportement de complexation de certaines espèces, comme l’uranium entres autres. Plusieurs auteurs ont analysé les interactions SH-UO2, qui sont observables à pH légèrement acide et une
25
Tableau 7. Constantes de complexation de SH avec l’uranyle variant en fonction du pH et de leur
provenance.
Type d’AH complexé avec UO22+ Log K pH Références
193 MAR 4,06 3,0 Lubal et coll.50 193 MAR 5,15 3,5 193 MAR 5,20 4,0 193 MAR 6,13 5,0 317 MAR 5,29 3,5 346 MAR 4,90 3,5 193 MAR 5,15 3,5 AH FLUKA 4,39 3,5 316 MAR 4,33 3,5 Gohy-573 6,20 4,0 Choppin et coll.51 LBHA 5,11 4,0 LBHA 6,80 4,9 Reed et coll.52 LBHA 7,40 6,1 Gohy-573 6,80 4,8 Gohy-573 7,70 6,1 SRFA 5,10 4,8 SRFA 6,60 6,2
AH Aldrich 6,00 - 12,00 5 - 10 Glaus et coll.53
N/D 4,70 - 5,07 4 Saito et coll.54
AH synthétiques 6,10 - 6,56 4 Pompe et coll.55
LHA 5,84 7 Riggle et coll.24
Gohy-573 6,16 4 Czerwinski et coll.56
193 MAR = Schistes charbonneux, Bílina, République Tchèque; 317 MAR = Schistes charbonneux, Vršany, République Tchèque; 346 MAR = Tourbière, Chotěšov, République Tchèque;
FLUKA = Fluka Chemika company, Buchs, Switzerland; 316 MAR = Tourbière, Vršany, République Tchèque; Gohy-573 = Eau sous-terraine Gorleben, Allemagne; LBHA = Lac Bradford, Tallahassee, FL, États-Unis;
SRFA = Rivière Suwannee, Parc National Okefenokee, GA, États-Unis; Aldrich = St. Louis, MO, États-unis;
26
On remarque l’importance de la provenance des substances humiques sur la constante de complexation, puisque cette constante tend à varier en fonction de ce paramètre. Bien que les valeurs de constante de complexation soient du même ordre de grandeur (Tableau 7), il est à noter que bien d’autres facteurs environnementaux jouent également un rôle important sur la valeur de Log K de ces espèces.
2.4.3. Impact de divers facteurs environnementaux
Les facteurs environnementaux ont une influence marquée sur la complexation entre les SH et les métaux. Dans cette section, l’effet de cinq facteurs sera présenté, soit le pH, la concentration de métal, la concentration d’AH, la température et la force ionique.
L’influence du pH sur la complexation de l’uranyle avec les substances humiques joue un rôle important. Effectivement, il est répertorié qu’à bas pH, l’uranyle est sous sa forme libre (non complexé aux AH) et au fur et à mesure que le pH augmente, la complexation entre les AH et l’UO2 sera favorisée. Ceci peut être expliqué par le fait qu’à un pH élevé, une
plus grande déprotonation des AH entraîne une charge négative plus grande et une configuration plus étendue. Le changement de configuration moléculaire des AH, qui engendre généralement des molécules plus ouvertes en raison de la répulsion des charges négatives, favorisera la diffusion des cations et accélèrera ainsi la complexation des cations, comme le rapporte Baker et coll.57 Par contre, la présence de contres anions, tel le
carbonate, et l’augmentation constante d’hydroxyde en lien avec l’augmentation du pH altéreront vraisemblablement cette tendance. Ces faits ont notamment été observés par Zeh et coll.58 pour des valeurs de pH entre 1 à 4. Ils observent par la suite une diminution de la
complexation pour les pH supérieurs à 4,5, principalement en raison de l’apparition d’espèces tertiaires causées par la présence croissante de carbonate à pH plus élevé.
La concentration de métal influence également la complexation et une relation très simple peut être établie. En effet, à une concentration d’AH et un pH constant, plus la concentration d’uranyle sera élevée, plus il sera complexé aux AH. Et ce, jusqu’à atteindre la capacité de liaison maximale des AH, variant généralement de 0,15 à 1,50 mmol/g avec des éléments comme les actinides.59 Cette expérience a notamment été réalisée par Li et
27 coll.60 avec l’uranyle sous sa forme U(VI) et sa forme U(IV) et a permis de démontrer par
le fait même que les SH avaient plus d’un site de liaisons possible.
La concentration de SH joue un rôle différent que celui joué par la concentration de métal dans la relation établie entre ces deux espèces. En réalité, pour établir une relation entre la complexation de métal avec les AH, il faut d’abord considérer les mécanismes de liaison de ces AH avec les métaux. Comme mentionné précédemment, les groupements carboxyliques offrent un site de liaison de prédilection pour les métaux (section 2.4.1.) donc la proportion de groupement -COOH dictera la relation avec l’uranyle. C’est donc dire que plus la composition des AH contiendra un nombre d’équivalent élevé en groupements carboxyliques, plus le nombre de molécules d’uranyle liées sera élevé. C’est notamment ce que démontre Nefedov et coll.,61 Munier-Lamy et coll.,62 ainsi que Pashalidis et coll.63
La température est aussi un facteur qui influence la complexation d’AH et UO22+. Par
contre, peu d’études s’intéressent et rapportent directement ce type d’information. Il y a tout de même Liao et coll.64 qui rapportent que la stabilité de la complexation entre l’UO
22+
et AH a tendance à décroître de 0°C à 20°C. Cet effet est principalement causé par la coagulation des substances humiques lorsque que la température augmente.65 Par contre, ils
ont également découvert qu’à des températures de 30°C à 40°C la constante de complexation augmente. L’hypothèse apportée est que pour ces hautes températures, les AH polymérisent entres elles avant d’effectuer le processus de complexation. Cette polymérisation d’AH aiderait à former des complexes plus stables. Par ailleurs, d’après le climat québécois, les rivières et les fleuves ne risquent pas d’atteindre cette fourchette de température, ce qui n’est donc pas à prendre en considération pour notre étude.
La force ionique est également un paramètre à tenir en compte lors de la détermination de la constante de complexation du complexe UO2-AH. En effet, lorsque la charge ionique est
augmentée de manière croissante en y ajoutant du sel, comme le NaClO4, une diminution
de la constante de complexation (Log K) est observée.66 Ceci est un phénomène observé
dans les réactions de complexation impliquant des molécules de charges opposées, où l’ajout de sel de charge neutre atténue l’attraction électrostatique.33 Par contre, pour
certaines techniques de séparation impliquant une membrane, l’application d’une force ionique est nécessaire, entre autres pour contrer l’effet Donnan.33 Cet effet survient
28
lorsqu’une trop faible force ionique est appliquée ou qu’une trop grande concentration de SH est ajoutée. Il en résulte alors d’une migration des ions libres faussée en raison de l’attraction exercée par les SH, portant une charge négative. Si cette charge n’est pas atténuée avec une force ionique adéquate, les cations seront plus retenus du même côté de la membrane que les AH, en dialyse et en ultrafiltration par exemple, et l’équilibre de complexation sera faussement modifié.
Une fois les principaux facteurs susceptibles de modifier la complexation identifiés, il est important d’aborder l’impact de cette complexation entre un métal toxique et les SH.
2.5. Impact de la complexation sur la toxicité
Lorsqu’une étude de spéciation d’un métal lourd dans l’environnement est réalisée, certains outils sont à notre disposition, comme des modèles qui peuvent être utilisés afin de déterminer lesquelles des espèces présentes dans un milieu complexeront avec un métal. Certains de ces modèles sont même conçus pour pousser l’étude encore plus loin et aideront à prédire l’effet qu’auront certains actinides d’origine anthropique sur le biote en fonction de leur spéciation.
Par exemple, le « Free-ion activity model » (FIAM) permet de prédire les flux de bio-absorption à l’extérieur de la cellule. Ce modèle se base sur différentes hypothèses et trois équations pour aider à prédire le comportement des métaux divalents en fonction de la concentration de métaux libres et de ligands à un pH donné.67 La Figure 15 montre la
tendance observée qui est en fonction de la concentration de métaux libres. C’est notamment avec ce modèle que Morel et coll.6 ont démontré que les organismes
unicellulaires évoluant dans des milieux externes sont influencés par la chimie qui est gouvernée principalement par des processus géochimiques. C’est donc dire que ces micro-organismes seront plus influencés par la présence d’une concentration x de métaux sous forme libre et que la toxicité d’un ion métallique libre changera en fonction de sa concentration. Cette concentration fluctuera en fonction de la présence de ligands, comme les AH.
29
Figure 15. Graphique démontrant la relation entre la concentration de métal libre et la
concentration de complexe métal-site actif d’une cellule cible.67
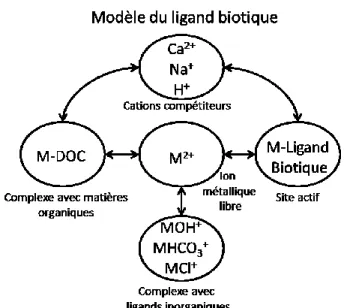
Un autre modèle grandement inspiré du FIAM est le « Biotic ligand model » (BLM). C’est avec ce modèle que Di Toro et coll.7 ont travaillés, ce qui leur a permis d’obtenir certaines
informations sur la toxicité que peut amener la complexation de métaux toxiques avec un ligand biotique, endroit cible où le métal peut se lier et y provoquer l’intoxication d’un organisme. Dans le cas des poissons par exemple, le ligand biotique est un type de protéines qui se retrouve à la surface des branchies et qui sert à régulariser la composition ionique du sang. Ces protéines servent généralement à faire passer le calcium et le sodium, élément vitaux pour le poisson. Par contre, si ces protéines sont complexées avec un métal toxique à une concentration critique, elles ne pourront plus remplir leur fonction et l’organisme mourra.
La Figure 16 démontre que la complexation du métal toxique avec le ligand biotique est en relation directe avec la présence de plusieurs autres facteurs, dont la présence de MOD (matières organiques dissoutes). Bien que ces modèles offrent des informations utiles et pertinentes, ils ne prennent pas en compte l’isotopie particulière que peut présenter certains matériaux présentant une radioactivité naturelle renforcée, notamment dans le cas de l’uranium appauvri et enrichi. De plus, ces modèles se basent exclusivement sur des