Understanding the durability of advanced fibre-reinforced polymer (FRP) composites for structural applications
Texte intégral
Figure
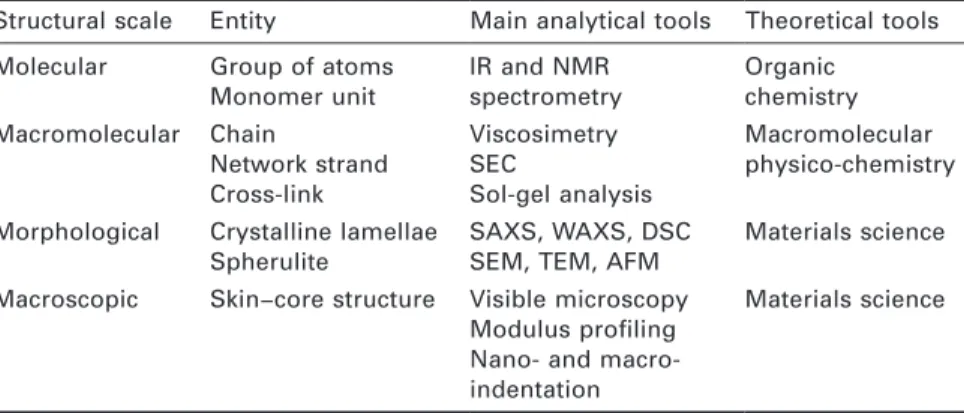
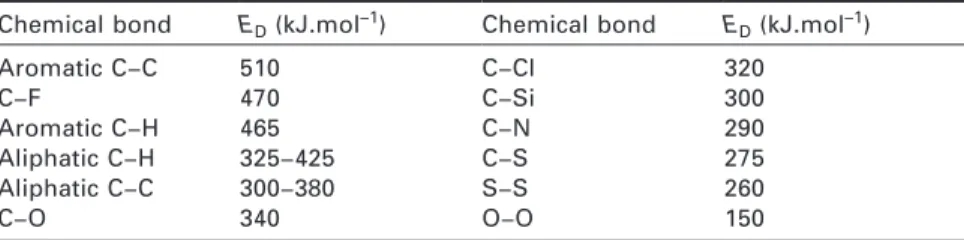
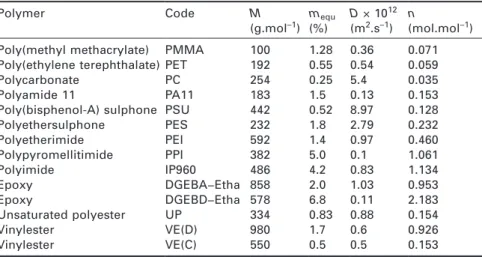

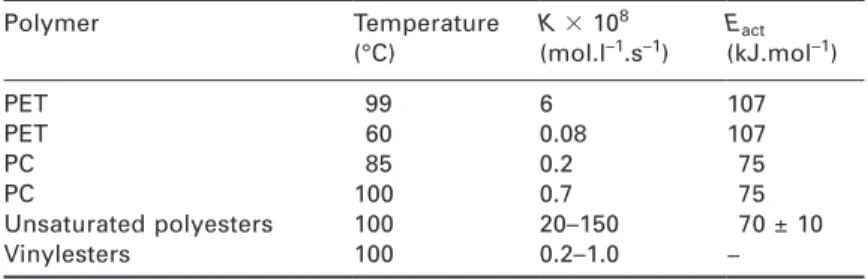

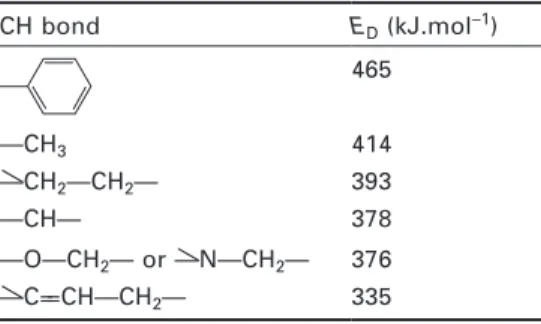

Documents relatifs
This work investigates the effect of repeated loading on moisture absorption in resin transfer molded glass/epoxy composites with 32.4% fiber volume fraction. A set
To study our two measures of growth, employment and revenue, we considered both available resources (financing and support) and dynamic capabilities
3 Number of boundary partners of project Number of boundary partners drawn in project network 4 Number of overall partners drawn Number of all partners drawn in project network
L’ examen des inscriptions révèle donc deux phénomènes en partie contradictoires, mais qui peuvent tout à fait avoir coexisté : d’ une part, les textes mettent en avant les liens
In addition to a strand containing the monofunctional drug phenanthriplatin, DNA strands containing either a site-specific cisplatin lesion or a mismatched base
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Another example is Table 5 which gives an exhaustive list of non reflexive verbs taking tense auxiliary être in standard modern French.
[r]
