HAL Id: dumas-01863941
https://dumas.ccsd.cnrs.fr/dumas-01863941
Submitted on 29 Aug 2018
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
marché des spécialités pharmaceutiques à usage humain
Caroline Roudet
To cite this version:
Caroline Roudet. Les différentes étapes de la mise en place de la procédure multi-étatique d’autorisation de mise sur le marché des spécialités pharmaceutiques à usage humain. Sciences phar-maceutiques. 1988. �dumas-01863941�
AVERTISSEMENT
Ce document est le fruit d'un long travail approuvé par le
jury de soutenance et mis à disposition de l'ensemble de la
communauté universitaire élargie.
Il n’a pas été réévalué depuis la date de soutenance.
Il est soumis à la propriété intellectuelle de l'auteur. Ceci
implique une obligation de citation et de référencement
lors de l’utilisation de ce document.
D’autre part, toute contrefaçon, plagiat, reproduction illicite
encourt une poursuite pénale.
Contact au SID de Grenoble :
bump-theses@univ-grenoble-alpes.fr
LIENS
LIENS
Code de la Propriété Intellectuelle. articles L 122. 4
Code de la Propriété Intellectuelle. articles L 335.2- L 335.10
http://www.cfcopies.com/juridique/droit-auteur
U.F.R. DE PHARMACIE
Domaine de la Merci - LA 1RONCHE
ANNEE 1988
N°d'ORDR~O
LES DIFFERENTES ETAPES DE LA MISE EN PLACE
DE LA PROCEDURE MULTI-ETATIQUE D'AUTORISATION
DE MISE SUR LE MARCHE DES SPECIALITES PHARMACEUTIQUES
A USAGE HUMAIN
THE SE
Présentée à l'UNIVERSITE Joseph FOURIER - GRENOBLE I
pour obtenir le grade de : DOCTEUR EN PHARMACIE par
Mademoiselle Caroline
ROUD ETCette Thèse sera soutenue publiquement le 27 juin 1988 devant:
Madame le Professeur
A.
VERAIN, Président du Jury Madame M. DELETRAZ, PharmacienMadame N. BERAUD, Industriel _ PhOv<I'Y\o..~~
U.F.R. DE PHARMACIE Domaine de la Merci -LA TRONCHE
ANNEE 1988 N°d'ORDRE
LES DIFFERENTES ETAPES DE LA MISE EN PLACE
DE LA PROCEDURE MULTI-ETATIQUE D'AUTORISATION
DE MISE SUR LE MARCHE DES SPECIALITES PHARMACEUTIQUES
A USAGE HUMAIN
THE SE
Présentée à l'UNIVERSITE Joseph FOURIER - GRENOBLE I
pour obtenir le grade de : DOCTEUR EN PHARMACIE par
Mademoiselle Caroline ROUD ET
Cette Thèse sera soutenue publiquement le 27 juin 1988 devant:
Madame le Professeur A. VERAIN, Président du Jury Madame M. DELETRAZ, Pharmacien
Madame N. BERAUD, Industriel
En témoignage de mon amour et de ma gratitude. Qu'ils trouvent à travers ce travail mes pJus vifs remerciements
pour le soutien qu'ils m'ont apporté tout au long de mes études.
A :.a famille,
dont j'ai pu apprécier tout au long de mes études la compétence et les qualités humaines. Qu'elle soit remerciée de m'avoir fait 1 'honneur de présider cette thèse.
Qu'elle trouve ici l'expression de ma profonde reconnaissance.
Qu'elle soit assuree de profonde reconnaissan.=e peur sa disponibilité et l'aide qu'elle m'a apportée de parsa haute competence, et trouve ici, l'expression demes plus
Qui m'a fait l'honneur de me confier ce travail et d'en suivre la réalisation pas à
pas.
Qu'elle trouve ici l'expression de mes sentiments dévoués et très respectueux.
9
Il y a 30 ans que le traité de Rome signé le 25 mars 1957 instituait la Communauté Economique Européenne ou CEE qui devait au fil des années intégrer économiquement et politiquement 12 états membres et 320 millions d'habitants.
Grâce à des mécanismes originaux de transfert de compétence établis par les traités constitutifs, les institut ions communautaires <Conseil des Ministres, Commission, Parlement européen et Cour de Justice) créent, appliquent et interprètent le droit communautaire, qui a la primauté sur les législations nationales.
Dans le but de construire l'Union Economique, l'objectif essentiel de cette communauté est d'établir un marché commun, c'est
à dire de supprimer tout ce qui empêche la libre circulation des hommes, des marchandises, des services et des capitaux, de garantir la libre concurrence ainsi que la coordination des politiques économiques nationales, dans les pays signataires du traité de Rome. En particulier, cette libre circulation ne doit pas être entravée par des obstacles tarifaires ou non tarifaires <75).
Dans la mesure où des disparités entre les législations nationales créent des obstacles aux échanges, celles-ci doivent être modifiées en vue de 1' harmonisation des législations s'appliquant dans toute la Communauté.
Les divers Etats européens ont pris conscience de l'étroitesse de leurs marchés respectifs et du frein qu'elle constitue à l'expansion de leurs entreprises.
Pour permettre aux entreprises européennes de concurrencer à armes égales les géants américains et l'industrie japonaise, et leur assurer la place à laquelle elles pouvaient prétendre dans les affaires du monde, il fallait s'unir pour établir un marché unique, étendu, adapté aux exigences de l'économie moderne, facteur de prospérité et d'élévation du niveau de vie des nations intéressées.
Dans le secteur pharmaceutique, l'industrie européenne a besoin de ce grand marché. La recherce pharmaceutique étant de plus en plus coûteuse et aléatoire, il convient donc de créer un environnement réglementaire favorable à une amélioration constante des thérapeutiques, et à une ouverture des marchés qui passent par le développement d'un esprit communautaire de la pharmacie et du médicament (76).
Dans ce but, un programme de 12 mesures spécifiques au secteur pharmaceutique figure parmi les 300 actions annoncées par la Commission en 1985 dans son "livre blanc" sur l'achèvement du marché
intérieur (48).
Nous nous proposons dans ce travail, d'étudier les efforts d'harmonisation effectués dans le cadre du Traité de Rome, et les difficultés rencontrées pour satisfaire l'objectif ultime de cette entreprise :
"la réalisation d'un grand 11Jârché unique des médicaments d'ici 1992. Il
11
Dans une première partie, nous parlerons de la Communauté Economique Européenne et de ses objectifs, puis nous étudierons l'harmonisation qui s'est établie entre les différents Etats membres, ainsi que le bilan de cette première ébauche d'harmonisation, et dans une troisième partie, nous envisagerons le système futur de la libre circulation des médicaments au sein de la CEE.
LE TRAITE DE ROME
INSTITUTIONS FONCTIONNEMENT
Chapitre I : CONSTRUCTION DE LA COXMUWAUTE
ECONOMIQUE EUROPEENNE
I. 1. Le principe de la mise en place de
la Communauté Européenne (72>
Des considérations politiques, mais surtout économiques ont favorisé la prise de conscience au ni veau des Gouvernements des Etats membres concernés de créer une Europe forte et unie et d'assurer une paix durable à notre continent, mettant ainsi un terme aux rivalités combien sanglantes qui avaient si longtemps déchiré les pays d'Europe, et laissé de graves séquelles,
Le 9 mai 1950, Robert SCHUMAN, alors Ministre des Affaires Etrangères, énonçait un certain nombre de propositions baptisées de
"
plan Schuman, qui peuvent être résumées en une seule phrase par la mise en coi11111un de productions de base et l'institution d'une Haute Autorité nouvelle, dont les décisions lieront la France, l'Allemagne et les pays qui y adhèreront, cette proposition réalisera les premières assises concrètes d'une Fédération européenne
,
Et c'est ainsi que trois Communautés ont été instituées Le 18 avril 1951, six Etats <République Fédérale Allemande, Belgique, France, Italie et Pays-Bas) ont fondé la première Communauté Européenne, celle du Charbon et de l'Acier
<CECA) par la signature du Traité de Paris.
Très rapidement, la Haute Autorité de la CECA a réalisé que le développement de l'énergie atomique justifiait la création d'une institution spécialisée. Par ailleurs, les promoteurs de l'idée européenne ont estimé que si l'on proposait aux Etats de généraliser
1' intégration déjà commencée et de faire un :Marché Commun à 6, il fallait faire aussi une Communauté atomique dans le même cadre.
C'est ainsi que devaient être instituées
- la Communauté Européenne de l'Energie Atomique <CEEA) connue également sous le nom d'Euratom;
- la Communauté Economique Européenne <CEE> ou Marché Commun, dont la Charte est le Traité de Rome signé le 25 mars 1957. <47)
Depuis 1958, pour des raisons d'efficacité et d'économie, les trois organisations CECA, CEE et EURATOK ont fusionné, essentiellement au niveau des exécutifs.
I. 2. 1&a modalités d • élargissement de.. la. ClŒ.
La Communauté qui, comme nous le savons, a un caractère ouvert demande deux conditions à un Etat postulant :
- il doit être reconnu comme démocratique, au sens donné à
cet objectif par les pays occidentaux
- il dai t accepter ce qui est convenu d'appeler l'acquis communautaire, c'est- à- dire les poli tiques générales , mais aussi l'ensemble des mesures en vigueur.
Bien entendu, tous les Etats membres ont toujours la possibilité, ensuite, de demander un changement au une inflexion des disposi tians communautaires, mais ils dai vent le faire de l'intérieur, dans le cadre des institutions et des procédures existantes.
I. 3. Elaboration d'un ordre juridique supra-national
La réalisation du Grand Marché unique intéresse bien évidemment le domaine pharmaceutique avec cependant ses problèmes spécifiques.
C'est dans cet esprit que tout d'abord six pays indépendants Frimee, Allemagne, Italie, Bénélux <Pays-Bas, Luxembourg et Belgique), puis le 22 janvier 1972 après Décision du Conseil des Communautés européennes, le Royaume-Uni, l'Irlande et le Danemark, en 1980 la Grèce et en 1986, l'Espagne et le Portugal ont
délégué aux institutions communautaires, une partie de leur Souveraineté Nationale dans certains domaines, pour 1' élaboration d'un ordre juridique supra-national.
Mais cet ordre juridique supra-national de la Communauté Européenne suppose la constitution d'un cadre juridique que l'on se propose d'exposer d'une manière schématique.
Chapitre II LE CADRE JURIDIQUE DE LA CEE
I
I
.
1. ~ Institutions Communautaires<47)
Les institutions communautaires créées par les traités constitutifs de laCommunauté sont au nombre de quatre
-le Conseil des Ministres
-le Parlement européen <Assemblée Parlementaire) -laCour de Justice
-laCommission des Communautés Européennes II. 1. 1.~ o o
C'est certainement le moteur de l'intégration européenne dont le siège estàBruxelles. Les membres de laCommission, nommés d'un commun accord par les Gouvernements des Etats <un ou deux suivant les pays membres) s'engagent par sermentà exercer leurs fonctions en pleine indépendance dans l'intérêt général de la communauté, et ne sollicitent ni n'acceptent d'instructions d'aucun gouvernement.
Les commissaires ont d'abord une responsabilité collégiale, dans
- l'exécution des décisions prises en vertu des pouvoirs de la Commission,
- la formulation des recommandations ou des avis sur les matières qui font l'objetdu présent traité.
Ilsdisposent d'un pouvoir de décision propre et participent
à la formation des actes du Conseil et de l'Assemblée dans les conditions prévues au présent traité.
La Commission a à sa disposition des services et peut instituer tous comités d'études nécessairesà 1' accomplissement de sa mission :
, Groupe de travail "Produits Pharma"... .... 1960 . Comité pharmaceutique... 1975 . Comité des Spécialités pharmaceutiques... 1975
II. 1. 2.kaConseil~
Le Conseil des Ministres est formé par lesreprésentants des Etats membres. Chaque gouvernementydélègue un de ses membres. La Présidence est exercéeàtour de rôle par chaque membre du Conseil pour une durée de six mois.
Le Conseil participeàla fois au pouvoir exécutif puisqu'il est le stade le plus élevé de décision, et au pouvoir législatif. En principe, aux termes du Traité, ilse prononceàla majorité simple ouàla majorité qualifiée.
A côté de ce Conseil, s'est développée une Institution dont le Traité de Rome n'avait pas prévu 1' importance le Cami té des Représentants Permanents <COREPER> composé d'agents diplomatiques représentants permanents des Etats membres, et qui est l'organe des relations entre les gouvernements-n:at-i-orraux-et-1-es-communautés.
II. 1. 3. L'Assemblée ou "Parlement Européen"
L'Assemblée, plus connue sous le nom de Parlement européen -son secrétariat est à Luxembourg- est composée de représentants des peuples des Etats membres, élus au suffrage universel direct, tous les cinq ans.
Le Parlement a des pouvoirs de délibération et de contrôle, mais n'exerce pas de pouvoir législatif au sens habituel du terme.
Il émet donc un avis sur les propositions de la Commission. Il peut également formuler des recommandations sur des sujets dont il s'est lui-même saisi.
II. 1. 4. La cour de Justice
La Cour de Justice siège, en séance plénière, à Luxembourg et est composée de juges assistés d'avocats généraux. Elle exerce le pouvoir judiciaire et assure le respect du droit dans l'interprétation et l'application du traité.
institutions de la Communauté,
Eru:. exemple
Si la Commission estime qu'un Etat membre manque à· une de ses obligations en vertu du Traité, elle lui demande ses observations et si elle n'en est pas satisfaite, elle émet un avis motivé. Si l'Etat ne se conforme pas à l'avis dans le délai fixé, la commission peut saisir la Cour.
Chapitre Ill FOBCTIONBEXEBT DE LA COKMUBAUTE
1 II. 1. !&fi sources d& di:ID.t. communautaire
C'est bien entendu le Traité de Rome qui constitue la source principale de ce droit communautaire. Ce dernier se manifeste par des actes administratifs qui sont soit des règlements, soit des directives, ou encore des décisions.
Le règlement a une portée générale. Il est obligatoire dans tous ses éléments et directement applicable dans tout Etat membre .
. La directive fournit aux Etats membres un but à atteindre dans un certain délai, tout en laissant à ses Etats le choix des moyens pour y parvenir.
. La décision est obligatoire en tous ses éléments pour les destinataires qu'elle désigne.
Ces mécanismes permettent la réalisation des objectifs du Traité qu'il est utile de rappeler.
III. 2. Les fonctions de la Communauté
Les objectifs du Traité de Rome sont à la fois généraux et simples. Il s'agit d'arriver à l'instauration d'un grand espace sans frontière dans lequel les personnes, les marchandises, les services et les capitaux circuleront dans les mêmes conditions qu'à l'intérieur d'un Etat .
. La libre circulation des marchandises
Elle sera assurée par la suppression des droits de douane et la suppression des barrières techniques.
La Cour de justice européenne prévoit que toute marchandise, légalement fabriquée et commercialisée dans un Etat membre, doit pouvoir être vendue dans un autre Etat membre.
Il faut que d'ici 1992, il y ait harmonisation des normes et réglementations .
. La libre circulation des personnes
Elle concerne la libre circulation des personnes dans la communauté, sans barrière douanière, ainsi que la possibilité d'exercer son métier.
La liberté de s'établir n'est garantie que dans la mesure où le candidat remplit les mêmes conditions que celles exigées des citoyens du pays d'accueil.
. La libre circulation des services
Elle concerne l'information, le marketing et l'audiovisuel. Il est prévu, en ce qui concerne la publicité et le marketing de l'industrie pharmaceutique européenne, une harmonisation de 1' information des médecins et des malades, en vue de l'usage rationnel du médicament .
. La libre circulation des capitaux
Sont concernés l'absorption, le rachat et donc les concentrations des firmes pharmaceutiques.
En ce qui concerne l'Industrie en France, le simple démantèlement douanier, aussi complet qu'il ait été, n'a pas amélioré les échanges intra-communautaires, encore moins abouti à la libre circulation.
Ce constat ayant été fait dès la parution des premières statistiques en 1960-1961, le recours au rapprochement des législations appelé harmonisation était inéluctable. Il importait dans un premier temps de réduire les divergences de point de vue des Etats membres sur un certain nombre de questions et d'harmoniser dans toute la mesure
différentes, tant du possible, au plan du pharmaceutiques pharmaceutiques. qu'à celui du
des législations parfois fort Statut des Etablissements régime des Spécialités
Reoorques :
Dans ce travail, seul le premier point sera évoqué
la libre circulation des oorchandises et plus particulièrement les autorisations administratives de mise en circulation des Spécialités Pharmaceutiques.
Avant d'évoquer les fondements juridiques de cette harmonisation des législations, nous allons étudier l'influence du système français sur l'élaboration de la législation communautaire.
Chapitre IV INFLUENCE DU SYSTEME FRANCAIS SUR L'ELABORATION DE LA LEGISLATION PHARMACEUTIQUE COXKUHAUTAIRE
IV. 1. Historique (56) <57)
Il y a 50 ans, il n'y avait dans la plupart des pays aucune réglementation d'ensemble de la mise sur le marché des médicaments qui était sous la seule responsabilité du producteur ou du pharmacien.
Le développement de la recherche pharmaceutique, le passage à la fabrication industrielle, et plusieurs accidents iatrogènes graves (affaire du Stalinon, de la poudre Baumol, de la Thalidomide) ont rendu nécessaire l'intervention des pouvoirs publics.
C'est ainsi que furent promulgués en France, en 1941 pour la première fois dans le cadre de la législation pharmaceutique, des textes législatifs et réglementaires concernant la préparation industrielle des médicaments et instituant notamment sous le nom de
"visa" (désigné depuis 1967 par 1' appellation "autorisation de mise
sur le marché") une autorisation ministérielle obligatoire.
Depuis, quatre étapes ont été parcourues jusqu'à l'actuelle autorisation de mise sur le Marché (55).
Un bref rappel sur la législation française peut présenter un certain intérêt, car le système français est souvent considéré comme l'inspirateur du système européen, du moins dans ses grandes options fondamentales.
IV. 2. La législation avant 1941
Le Décret du 13 juillet 1926 (1) relatif aux médicaments préparés à l'avance précisait que la mise en vente d'une telle spécialité n'était légalement soumise à aucune formalité particulière.
Aucun contrôle n'était obligatoire sur les matières premières et le produit fini.
Cet état de fait était doublement préjudiciable :
- pour la Santé Publique car il n'y avait aucune garantie ni d'efficacité, ni d'innocuité du médicament ;
- pour l'inventeur, qui pouvait voir sa spécialité imitée par n'importe quel autre fabricant sans possibilité de recours.
(1) Décret du 13 juillet 1926
IV. 3. La législation après 1941
IV. 3. 1. La loi du 11 septembre 1941 <2>
La loi du 11 septembre 1941 fixe la définition de la spécialité pharmaceutique et apporte les premiers éléments de réglementation en matière de fabrication industrielle du médicament,
à savoir :
- l'ouverture d'Etablissements Pharmaceutiques soumise à une autorisation
- la préparation des spécialités sous la responsabilité d'un pharmacien ;
- la nécessité d'une autorisation ministérielle préalable_ à
la mise sur le marché d'une spécialité pharmaceutique : le visa. Toutefois, ce visa n'est accordé au fabricant que s'il garantit l'innocuité et l'intérêt thérapeutique de son produit, et prouve son caractère de nouveauté.
De plus, l'inventeur est protégé des imitations pendant 6 ans.
(2) Loi n· 3890 du 11 septembre 1941
Bien autorisation entendu, cette sera reprise obligation comme base législations des Etats européens.
d'obtention d'harmonisation
IV. 3. 2. L'Ordonnance du 4 février 1959 (4)
d'une des
L'Ordonnance 59-250 du 4 février 1959 et le Décret 60-326 du 5 avril 1960 (5) comblent les lacunes de la précédente réglementation (3) sur les conditions de fabrication et le contrôle systématique des lots de fabrication, et apporte une réforme dans la législation des visas. Les nouveaux visas sont désignés : VISA NL
<Nouvelle Législation).
Sont également introduites les notions de :
- protection de l'originalité du médicament par la créâtion d'un brevet spécial de médicament <B.S.M.) qui donne à son titulaire un monopole d'exploitation de 20 ans ;
d'experts, qui doivent vérifier la conformité de la formule. (3) Loi n· 46-1154 du 22 mai 1946
J.o.
du 23 mai 1946, p. 4482 (4) Ordonnance n· 59-250 du 4 février 1959J.o.
du 8 février 1959,p.
1'756-1'759 (5) Décret n· 60-326 du 5 avril 1960J.o.
du '7 avril 1960,P·
3227-3232La France est alors le seul pays européen à avoir un statut d'expert.
Trois types d'expertises deviennent dès lors nécessaires l'expertise analytique, ayant pour but d'établir la conformité du produit en fonction de la formule annoncée, et de vérifier la validité des techniques de contrôle ;
~ l'expertise toxicologique et pharmacologique qui détermine
les propriétés du médicament et qui permet de vérifier l'innocuité du produit dans les conditions normales d'emploi
l'expertise clinique dont le but est de démontrer l'intérêt thérapeutique du produit et de déterminer la posologie, les conditions d'emploi, les contre-indications et les effets secondaires.
On peut constate.r que cette ordonnance, unanimement appréciée, sert encore de base à notre législation et a eu une importance et une influence considérable au niveau européen, au point de constituer une autre base solide d'harmonisation.
IV. 3. 3. L'Ordonnance du 23 septembre 1967 (7)
L'Ordonnance du 23 septembre 1967 intègre les premiers
(7) Ordonnance n· 67-827 du 23 septembre 1967 JO du 28 septembre 1967, p. 9553-9554
éléments d'une réglementation européenne communautaire la Directive 65/65/CEE du 26 janvier 1965 (6),
Cette ordonnance modifie la dénomination de "visa" en
"autorisation de mise sur le marché" <AMM) dont la durée de validité
est limitée à 5 ans, renouvelable par période quinquénale.
Cette ordonnance est sui vie par trois arrêtés fixant les protocoles déterminant les normes et les méthodes applicables à
l'expérimentation des médicaments, à savoir :
- pour l'expertise analytique : arrêté du 26 juin 1972 (10) - pour l'expertise pharmacotoxicologique : arrêté du
27 avril 1972 (8)
- pour l'expertise clinique arrêté du 16 mai 1972 (9). IV. 3. 4. Le Décret du 20 septembre 1978 <12)
Ce décret modifie certaines dispositions relatives à l'établissement pharmaceutique et à 1' autorisation de mise sur le marché, en application des Directives Européennes 75/318 et 75/319 du 20 mai 1975.
Par ailleurs, ce décret confirme la distinction faite entre le "fabricant", "le responsable de la mise sur le marché", et le
"titulaire de l'AHH'.
- le fabricant assure la préparation et le contrôle des produits pharmaceutiques ;
- le responsable de la mise sur le marché est la personne qui introduit la demande, et réunit les éléments constitutifs du dossier soumis au :Ministre de la Santé en vue de l'octroi de mise sur le marché i
-si cette autorisation est accordée, le responsable devient alors titulairede l'A:M:M.
Aujourd'hui, le titulaire de l'A:M:M: · n'est plus obligatoirement une personne qualifiée, c'est-à-dire un médecin ou un pharmacien, ilpeut s'agir de personnes n'ayant aucun rapport avec la pharmacie.
(6) Directive du Conseil 65/65/CEE du 26 janvier 1965 JOCE n· 22 du 9 février 1965, p. 369-373 (8) Arrêté du 27 avril 1972 JO du 28 mai 1972, p. 5400-5401 (9) Arrêté du 16 mai 1972 JO du 11 juin 1972, p. 5902 <10) Arrêté du 26 juin 1972 JO du 13 juillet1972, p. 7395-7397 (12)D ~ 78-988 du 20 septembre 1978 JO du 6 octobre 1978, p. 3486-3488
T A B L E A U
RE C A P I T U L A T I F
REGLEMENTATION FRANCAISE DE
L'AMK
AB/VISAAvant 1941
Enregistrement des formules auprès du Laboratoire Natio-nal de Contrôle des Médica-ments. EXPERTS/EXPERTISES Non obligatoire
---·---Après 1941 loi du 11 sept, 1941 - Institution du ~- Absence de dangers pour l'homme
- Intérêt thérapeutique - Caractère de nouveauté
.
Faire preuve des 3 conditions imposées par des cliniciens
Experts non obligatoires
---:---. Ordonnance du 4 féy, 1959 - VISA NL
- Création d'un BSK
- Experts choisis par le fabricant expertise analytique expertise pharmaco-t_oxicologique expertise clinique
---:---. Ordonnance du 23 sept---:---. 1967 - Harmonisation de la législationfrançaise sur les bases de la Directive 65/65/CEE
. Décret du 20 sept. 1978 - Harmonisation de la législation
française sur les bases de la Directive Européenne
(75/319/CEE-75/318/CEE>
. loin· 87-588 du 30 iuil. 1987:
3 arrêtés fixant les proto-coles normes et méthodes applicables à l'expéri-mentation des médicaments
Nouvel alignement avec les normes européennes. Agrément des experts n'est plus obligatoire
Chapitre V LISTRUJIEIT ET FOJDEJIEJIT JURIDIQUE DU RAPPROCHEJIE!TT DES LEGISLATIO!TS JlATIO!TALES DES PAYS DE LA CEE
v.
1. Intérêtde l'barDQnisation des dispositions législatives, réglementaires et arlpinistratives <5?>Dans tous développement ou
les pays, quels leur système
que soient politique
leur niveau de et économique, l'introduction de médicaments sur le marché est soumise à une réglementation de plus en plus rigoureuse dont l'objet est de vérifier la qualité, l'efficacité et l'innocuité des substances ainsi mises àla disposition des médecins et des malades.
Chaque pays possède des normes qui ~ aux autorités
nationales de vérifier que lesproduits importés sont bien conformes à leur propre législation. La difficulté majeure du rapprochement provient donc essentiellement des écarts d'exigences en matière d'autorisation de mise sur le marché entre les pays.
Il s'agit donc d'éliminer entre les pays européens les "entraves techniques'' aux échanges appliqués aux spécialités pharmaceutiques, et il faut donc que 1' effort porte sur l'harmonisation des contrôles, afin de développer un esprit communautaire de la pharmacie et du médicament, et d'éliminer les conséquences défavorables aussi bien pour les autorités sanitaires
que pour les fabricants et la Santé Publique en général <retenir les mêmes critères et utiliser les mêmes moyens pour accorder la mise en circulation des médicaments),
C'est ainsi qu'en application de l'article 100 du Traité de Rome,
"Le Conseil, statuant à l'unanimité sur proposition de la
Commission, arrête les Directives pour le rapprochement des dispositions législatives, réglementaires et administratives des Etats :membres qui ont une incidence directe sur l'établissement ou le fonctionnement du Xarché Commun",
(et selon la procédure prévue par celui-ci, c'est-à-dire après avis des assemblées européennes, qui ont été signées par le Conseil des Communautés <avec un écart de 10 années) des directives qui forment la base essentielle de la législation européenne en matière de médicament. L'objectif principal de ces Directives étant de protéger la Santé Publique tout en assurant le libre développement de l'industrie pharmaceutique et du commerce des médicaments.
v.
2. La Directive, instruuent du rapprochementV. 2. 1. Définition <47)
"La Directive lie tout Etat membre destinataire quant au résultat à atteindre, tout en laissant aux instances nationales le compétence quant à la forme et aux mayen$'.
V. 2. 2. Elaboration d'une Directive <72)
La Commission, constatant que des disparités existent entre les réglementations des Etats membres et que ces disparités entrainent des distorsions, va d'abord demander aux différents Etats une documentation sur leurs réglementations dans un domaine déterminé.
Après une première consul tatien, la Commission va soumettre un projet aux experts désignés par les Etats, qui forment le Groupe de Travail Produits Pharma de la Commission.
- Les discussions se poursuivent au sein de ce Groupe de Travail jusqu'à ce qu'un concensus apparaisse.
- Quand la commission estime que la proposition de Directive est au point, elle la soumet au Conseil, et en même temps le Parlement Européen <PE> et le Comité Economique et Social <CES> en sont saisis.
La propos! tian est alors publiée au Journal officiel des Communautés Européennes (JOCE>.
- Après adoption des avis de ces différents organismes, la Commission peut, toujours dans l'exercice de son droit d'initiative, soumettre au Conseil une proposition modifiée.
- Le Conseil renvoie la proposition et les avis au COREPER qui en safsi t son groupe de Travail.
Quand le COREPER s'est mis d'accord, la décision du conseil est entérinée -la proposition est devenue Directive-.
Au cours de cette dernière étape, la Commission peut, à tout moment, retirer la proposition.
La Directive entre en vigueur dans un délai fixé spécifiquement dans chaque cas <18 mois) à partir de sa publication au Journal Officiel.
Ce délai est mis à profit par les Etats pour mettre leur législation en conformité avec les prescriptions communautaires.
V. 2. 3. Valeur des Directives au regard des Etats membres Les Directives arrêtées par le Conseil des Ministres ont valeur de lois communautaires, lois supranationales qui sont contraignantes pour chaque Etat signataire dès qu'elles sont adoptées par le Conseil des Ministres de la Communauté Européenne.
Elles obligent les Etats membres à s'y conformer dans les délais prévus à dater du jour de leur signification officielle dans chaque Etat.
Les instances nationales ont compétence quant à la forme et aux moyens à mettre en oeuvre, quant à l'application des dispositions arrêtées.
ELABQRATIOI D'UIB DIRECTIVE
Etats membres
Documentation
PROJET
COJDHSSIO:I
-=@--4) 1re G TDB
consultation ProduitsDIRECTIVE
Phar:œaCES
DB
COISEIL
~
1 ÙOREPBR~~
PROPOS Il lOI
DIRECTIVES GT ComitésPUBLICATIOI
DécisionsAU
JOCE prises par lesDB LA
X Ill SIRES38
Les Directives doivent être transposées dans le Droit national de l'Etat membre, aussi chaque Etat doit communiquer à la Commission les textes des dispositions essentielles du Droit interne pris en application des Directives.
V. 2. 4. Obiectifs de ces Directives
Les objectifs de ces Directives sont nettement définis dans le préambule de la première Directive publiée en 1965 :
"Considérant que toute réglementation en :matière de production et de distribution des spécialités pharmaceutiques doit avoir comme objectif essentiel la sauvegarde de la Santé Publique ; considérant toutefois que ce but doit être atteint par des moyens qui ne puissent pas freiner le développement de l'Industrie pharmaceutique et les échanges de produits pharmaceutiques au sein de la Communauté . .. "
De ce préambule, il ressort trois buts essentiels - l'intérêt de la Santé Publique,
-le développement de l'Industrie pharmaceutique,
la libre circulation des médicaments au sein de la CEE (objectif essentiel).
v.
3. Enoncé des différentes Directives visant les spécialités phanaaceutiquesLe bilan de plus de vingt années d'harmonisation des réglementations pharmaceutiques en Europe comprend actuellement, pour les médicaments à usage humain, 9 Directives importantes, que nous aurons l'occasion de détailler ultérieurement, et
2 Recommandations du Conseil.
V. 3. 1.Les Directives principales(50)
DIRECTIVE DU CONSEIL 65/65/CEE du 26 janvier 1965 concernant le
réglementaires
rapprochement des et administratives,
dispositions législatives, relatives aux spécialités pharmaceutiques. (JO n • 22 du 9 février 1965).
- DIRECTIVE DU CONSEIL 75/318/CEE du 20 mai 1975 relative au rapprochement des législations des Etats membres concernant les normes et protocoles analytiques, toxicopharmacologiques et cliniques en matière d'essais de spécialités pharmaceutiques.
- DIRECTIVE DU CONSEIL 75/319/CEE du 20 mai 1975 concernant le rapprochement des disposi tians législatives, réglementaires et administratives, relatives aux spécialités pharmaceutiques.
<JO L 147 du 9 juin 1975).
Elle introduit un système transitoire de reconnaissance mutuelle des enregistrements, et définit le rôle des experts.
DIRECTIVE DU CONSEIL 83/570/CEE du 26 octobre 1983 modifiant les Directives 65/65/CEE, 75/318/CEE et 75/319/CEE relatives aux spécialités pharmaceutiques.
<JO L 332 du 28 novembre 1983).
Elle modifie, sur la base de l'expérience acquise la procédure de demande d'autorisation dite du CSP, et définit le rôle et les limites d'intervention du CSP.
DIRECTIVE DU CONSEIL 87/19/CEE du 22 décembre 1986 modifiant la Directive 75/318/CEE relative au rapprochement des législations des Etats membres concernant les normes et protocoles.
<JO L 15 du 17 janvier 1987).
Cette Directive vise à introduire une procédure simplifiée d'adaptation au progrès technique des Directives techniques existantes.
modifiant DIRECTIVE DU la Directive 41 CONSEIL 87/21/CEE du 65/65/CEE relative pharmaceutiques. (JO 1 15 du 17 janvier 1987).
22 décembre 1986 aux spécialités Cette Directive donne une protection de 10 ans, pour les médicaments de haute technologie ou issus des biotechnologies ayant suivi la procédure de consultation préalable, contre tout second demandeur qui ferait une demande relative à un produit semblable.
- DIRECTIVE DU CONSEIL 87/22/CEE du 22 décembre 1986 portant rapprochement des mesures nationales relatives à la mise sur le marché des médicaments de haute technologie, notamment ceux issus de la biotechnologie. <JO L 15 du 17 janvier 1987).
Cette Directive introduit l'exigence de saisine obligatoire du CSP pour avis en ce qui concerne les produits de la biotechnologie.
Ce processus est décrit sous le nom de "concertation
préalable".
V. 3. 2. Autres Directives
DIRECTIVE DU CONSEIL 78/25/CEE du 12 décembre 1977 relative au rapprochement des législations des Etats membres concernant les matières pouvant être ajoutées aux médicaments en vue de leur coloration (JO 1 11 du 14 janvier 1978).
DIRECTIVE DU CONSEIL 87/18/CEE du 18 décembre 1986 relative à l'application des principes de bonnes pratiques de laboratoire et au contrôle de leur application pour les essais sur les substances chimiques. (JO L 15 du 17 janvier 1987).
V. 3. 3. Propositions de Directives
- Proposition de Directive sur les médicaments exclus.
La comndssion des Communautés Européennes présente des propositions de Directives relatives à trois des quatre catégories de médicaments exclus jusqu'à présent de l'harmonisation européenne.
Ces propositions
appliquent aux sérums et vaccins, produits radiopharmaceutiques et aux dérivés du sang humain les dispositions des Directives existant pour les autres médicaments
. les complètent sur certains points spécifiques puisqu'il s'agit de médicaments très particuliers ;
. précisent certaines définitions.
Les textes de ces propositions sont joints en annexe I. - Proposition de Directive 88/C36/02 pour modifier les
Directives 65/65, 75/318 et 75/319.
Une première modification, très générale introduit la notion de médicament préfabriqué.
v
...
43
Les autres modifications concernent . l'élargissement du contrôle, . lanotice,
. l'exportation,
. lesbonnes pratiques de fabrication, . le retraitd'un produit.
Le texte de ces propositions est jointen annexe II.
R o D ~ d o ou •GuidelineS'
Les Guidelines sont des notes explicatives qui ont pour objet de codifier et d'harmoniser lesnormes et protocoles d'essais.
Contrairement aux Directives, ces Guidelines ne lient pas lesEtats membres.
- RECOUANDATION DU CONSEIL 83/571/CEE du 26 octobre 1983 concernant les essais en vue de la mise sur le marché des spécialités pharmaceutiqu€s(JQL 332 du 28 novembre 1983).
- RECOMMANDATION DU CONSEIL 87/176/CEE du 9 février 198'7 concernant les essais en vue de la mise sur le marché des spécialités pharmaceutiques (JO L '73 du 16 mars 198'7).
Cette recommandation complète la recommandation du Conseil 83/5'71/CEE et exprime dans quatorze notes explicatives ~ principes
LA MISE EN PLACE DE LA
RECONNAISSANCE MUTUELLE DES
A M M :
HARMONISATION DES LEGISLATIONS
INTERNES PAR VOIE DE DIRECTIVES
Chapitre 1 LE PASSE
1. 1 Autorisation supranationale
Avant la proposition de Directives, la Communauté Européenne a tenté il y a 25 ans de réduire les obstacles à la libre circulation des spécialités pharmaceutiques en proposant
"1 'autorisation supranationale".
Celle-ci envisageait la mise en place d'une haute autorité au sein de la Communauté Economique Européenne, qui aurait été responsable de l'autorisation de mise sur le marché des médicaments de la Communauté et qui aurait supplanté les autorités nationales.
D'une part, ces dernières ont refusé rle déléguer leur responsabilité à une haute autorité. D'autre part, des Etats européens n'ont pas voulu courir le risque de se voir déjugés dans l'intérêt qu'ils portent à la qualité de la vie et de l'environnement. Enfin, la difficulté pratique à établir une "F.D.A. européenne", a conduit à l'abandon de ce concept.
On a toutefois pensé à une époque à une AMM communautaire pour les "grosses molécules et des AMM nationales pour les autres, mais inacceptables dans le concept même de la CEE.
La CEE a préféré, à l'autorisation supranationale, une harmonisation des critères nationaux élaborée par des Directives Européennes.
I. 2. Harmonisation des AIX des différents Etats Européens<55)(75> L' initiative de la mise en place de la première Directive remonte à 1962, elle a été adoptée par le Conseil le 26 janvier 1965.
- Pirective 65/65/CEE du 26 janvier 1965 dite première Directive (6)
Cette première Directive a été publiée 3 ans après le drame de la Thalidomide, et peut être considérée comme la loi fondamentale de toutes les règles communautaires dans le domaine des médicaments.
Rédigée par les seuls "si:1t', elle est de prétention modeste puisque dans ses considérants, elle précise que
"ce rapprochement ne peut être réalisé que progressivement et qu'il importe en premier lieu d' éli:m:iner les disparités qui peuvent le plus affecter le fonctionnement du Harché Co111111un".
L'article 1er de cette Directive précise ce qu'il faut entendre par spécialités pharmaceutiques, médicament et substance.
La défini tien du médicament, qui est la plus importante de ces trois définitions, est très proche de la définition française.
Cette définition est très importante car elle sert de base à
l'ensemble de la réglementation pharmaceutique communautaire.
En France, le Code de la Santé Publique <56) définit ce terme <art. 511) : on entend par médicament :
"Toute substance ou compost tion présentée comme possédant des propriétés curatives ou préventivesà 1 'égard des maladies humaines ou animales, ainsi que tout produit pouvant êtreadministré
àl'homme ouàl'animal, en vue d'établir un diagnostic médical, ou de restaurer,corriger ou od ~ leurs fonctionso ~'.
La Directive européenne cependant ne s'applique pas aux médicaments que le pharmacien prépare lui-même dans son officine pour les dispenser au détail, sans publicité, ni aux médicaments délivrés par des médecins, des vétérinaires ou des"praticiens de 1 'artdentaire".
Elle impose, dans tous les Etats membres, le principe de 1'autorisation préalableà la mise sur le marché d'une spécialité pharmaceutique, précise la documentationàsoumettre aux autorités afin d'obtenir une autorisation de mise sur le marché, et formule lesraisons exhaustives pour lesquelles une autorisation de mise sur le marché peut être refusée, retirée, ou suspendue.
Chaque Etat membre se voit contraint d'adapter son droit interne de façonà n'autoriser un médicament qu'après examen d'un dossier scientifique et technique.
Il est tenu de refuser cette autorisation si,àl'analyse du dossier, il apparaît que la spécialité est nocive dans les conditions normales d'emploi, inactive ou non conformeà laformule déclarée.
Pour certains, l'harmonisation des législations devait conduire à une reconnaissance réciproque et automatique des autorisations nationales. C'était notamment le point de vue des Allemands : le fait d'obtenir une AMM dans un seul pays entraînait le droit d'introduire le médicament dans l'ensemble du territoire de la Communauté.
En effet, dans sa première proposition d'introduire un Gystème de reconnaissance mutuelle que la Commission a soumise au Conseil en 1967, la reconnaissance devait se faire de façon automatique dès que le premier Etat communiquait à tous les Etats membres désignés par le responsable de la mise sur le marché une copie de cette autorisation.
Dès que l'Etat membre désigné publiait le nom de la spécialité dans son JO, l'autorisation était validée dans ce pays.
Ce système de reconnaissance mutuelle signifiait en fait que l'Etat auquel le fabricant s'était adressé en premier lieu, examine la demande d'une autorisation de mise sur le marché pour le compte de tous les autres Etats membres, sur la base des critères d'une législation harmonisée.
Après de longues discussions au sein du Conseil, il était toutefois clairement apparu que les Etats n'étaient pas prêts pour accepter la proposi tian de la commission pour introduire un tel système.
C'est ainsi, qu'en 1975, est finalement apparu un accord sur une deuxième Directive pharmaceutique 75/319/CEE <14) concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives aux spécialités pharmaceutiques, prévoyant 1' élaboration des principes repris dans la Directive 65/65/CEE, et sur une Directive spécifique, relative au rapprochement des législations des Etats membres concernant les normes et protocoles analytiques, toxico-pharmacologiques et cliniques en matière d'essais de spécialités pharmaceutiques : Directive 75/318/CEE (13).
1. 3. Directive 75/318/CEE du 20 aai 1975 dite Directive normes et protocoles
Cette Directive complète la première Directive de 1965 en détaillant longuement dans trois annexes, le contenu des dossiers analytique et technique, pharmacologique et toxicologique, clinique.
Avec cette Directive, nous entrons vraiment dans la phase de la normalisation précise :
<13) Directive 75/318/CEE du 20 mai 1975 JOCE n• L 147 du 9 juin 1975, p. 1-12 <14) Directive 75/319/CEE du 20 mai 1975
- Détails des contrôles à pratiquer systématiquement sur les formes pharmaceutiques ;
- Conditions des essais de toxicité
Présentation des documents dans le dossier des expérimentations toxicologiques et pharmacologiques
- Libellé des fiches cliniques.
Une fois de plus, la CEE utilise l'harmonisation des exigences comme instrument de suppression des obstacles à la libre circulation et met l'accent sur la qualité des essais, leur importance pour la sauvegarde de la Santé, et sur la nécessité d'adapter régulièrement ces normes et protocoles au progrès de la science.
I. 4. Directive 75/319/CEE du 20 mai 1975 dite deuxième Directive
Le choix du régime final de libre circulation, après maintes discussions étant reporté vers le futur, un système transi taire était introduit avec la Directive 75/319/CEE (61).
Cette Directive qui, à côté de dispositions très importantes concernant les conditions de fabrication et d'importation des médicaments, poursuivait deux buts principaux
- imposer une autorisation préalable pour la fabrication industrielle des spécialités pharmaceutiques ;
De même, elle définit le rôle des experts qui doivent procéder aux travaux relevant de leur discipline, ou justifier le recours éventuelà la documentation bibliographique ainsi que la façon dont les autorités compétentes des Etats membres doivent instruirece dossier.
Dans ce système transitaire, la Communauté fut amenée à
introduire la notion d'un enregistrement communautaire en facilitant la procédure d'introduction d'un dossier déjà accepté par une
..
a.aY.u t.l<.!.!.or.._._i . . . ... . .. ...~oo . ... . ' .. . <. d.. ' ' .. .. ... o .. ' ' ~ ' ' .... nwq4-.!<!a""u-"'t"-r .... e""'s'--'E"'-t>tla""-tl<Jsii!--""m!liie"""m"'b.._r_..e=s;
Chacune des autorités nationales restant libre de sa décision. Toutefois, un Comité des Spécialités pharmaceutiques était mis en place.
I. 4. 1. La notion d'expert en France et en Europe (78) I. 4. 1. 1. Fondements juridiques
Comme nous l'avons vu dans le Chapitre IV. 3. 2. de la première partie, c'est la France qui a le privilège de l'antériorité en matière, avec l'Ordonnance du 4 février 1959 qui a établi, pour tous les médicaments à usage humain, une réglementation unique destinéeàassurer laprotection de laSanté Publique.
Désormais, le fabricant était tenu d'assortir toute demande de visa d'un dossier scientifique d'expérimentation, garanti par l'intervention d'experts agréés par les autorités administratives.
Au fur et à mesure de l'introduction des Directives européennes dans le Droit interne, les modalités d'octroi du visa ainsi que la notion d'experts ont évolué.
I. 4. 1. 2. Selon la Directive 75/319/CEE
La Directive 75/319/CEE en son article 1 prévoit que toute demande d'AD d'un médicament doit être établie et vérifiée par des experts possédant les "qualifications techniques et
professionnelles nécessaires''. Ces documents et renseignements sont
signés par ces experts.
Cette définition étant donnée, l'article 2 de la même Directive précise le rôle de l'expert :
"Selon leurs qualifications, le rôle des experts est :
- de procéder aux travaux relevant de leur discipline et de
décrire objectivement les résultats obtenus ;
de décrire les constatations qu'ils ont faites conformément à la Directive 751318/CEE concernant les normes et protocoles analytiques, et de dire, pour l'analyste, si le produit est conforme à la compost ti on dé cl a rée . .. "
Le même article ajoute que les rapports détaillés des experts font partie du dossier que le demandeur présente aux autorités compétentes.
.
I. 4. 1. 3. Les textes en vigueur en France
Le Décret du 21 novembre 1972 <11), modifiant l'Ordonnance du 4 février 1959 a fixé le statut des experts agréés au sens de l'article L 605 du Code de la Santé. A nouveau modifié en 1979, il reprend la défini ti on de l'expert telle qu'elle figure dans la Direqtive 75/319/CEE.
Toutefois, les autorités administratives françaises n'ont pas renoué au principe de l'agrément préalable des experts, alors même que la Directive européenne pose pour suffisante l'existence d'une
11 qualification technique ou professionnelle".
L'agrément est accordé par le Ministre chargé de la Santé, sa durée est de cinq années renouvelables.
<11 Décret n· 72-1062 du 21 novembre 1972 JO du 30 novembre 1972, p. 12405-12408
Depuis 1979, il est possible pour les collaborateurs salariés d'une entreprise d'obtenir l'agrément et d'effectuer des expertises pour le compte de leur employeur, à partir du moment où ils n'ont "aucun intérêt financier direct ou indirect" <art. R 5119 du Cade de la Santé Publique) dans l'exploitation des médicaments expertisés.
Bien que prise tardivement, cette mesure témoigne d'un effort d'harmonisation avec la Directive 75/319/CEE qui n'exige à aucun moment le recours à des spécialistes extérieurs à la firme.
De plus, les autorités administratives françaises acceptent l'agrément d'experts étrangers pour permettre aux firmes nationales et internationales d'utiliser les travaux réalisés dans d'autres pays que la France à l'appui des demandes d'autorisation de mise sur le marché.
Dans ces candi tians, l'agrément ne représente pas, en soi, une distorsion majeure par rapport au Droit européen.
Depuis le 30 juillet 1987, la loi n· 87-588 <26) a rendu l'agrément des experts en France non obligataire (38).
I. 4. 2. Le Comité des Spécialités Pharmaceutiques
Le Cami té des Spécialités Pharmaceutiques ou CSP institué par l'article 8 de la deuxième Directive est apparu en 1976.
(26) Loi n· 87-588 du 30 juillet 1987 JO du 31 juillet 1987
I. 4. 2. 1. Composition
Le CSP est composé d'un représentant par Etat membre et d'un représentant de la Commission.
Son rôle est limité à une mission de coordination en cas de demande d'un enregistrement par la procédure communautaire, et à une mission d'information du Conseil sur 1' évolution de la libre circulation.
Il ne s'agit donc pas d'une assemblée supranationale prenant des décisions, mais d'un organe qui doit permettre de rapprocher les points de vue divergents des di vers Etats membres de la Communauté comme l'indique l'article 8 de la deuxième Directive :
"Ce comité a été créé en vue de faciliter l'adoption d'une attitude coiJ11!/une par les Etats membres relative aux autorisations de mise sur le marché".
I. 4. 2. 2. La procédure dite du CSP
En se tenant à la Directive 75/319/CEE, le Comité peut intervenir de trois façons différentes (69) :
Le premier cas est le plus important <voir schéma 1).
Lorsqu'un Etat a accordé une autorisation de mise sur le marché, il doit, à la demande du responsable de la mise sur le marché, transmettre l'autorisation de mise sur le marché accordée,
ainsi que le dossier présenté pour obtenir cette autorisation dans au moins cinq Etats ainsi qu'au Comité ; cette transmission valant dépôt d'une demande d'autorisation de mise sur le marché.
Le point de départ du délai d'examen devant impérativement être le même pour tous les Etats désignés, c'est au Comité à déterminer et à diffuser cette date, dès qu'il saura avec certitude que tous les Etats concernés sont effectivement en possession de la demande et du dossier complet, Des exemples ont montré qu 1 il s 1 agissait là d 1 une
difficulté importante.
Cette date ayant été communiquée, les Etats ont alors un délai de 120 jours pour étudier le dossier.
Si un Etat estime devoir octroyer l'AMM, il ne se manifeste pas auprès du Comité.
, Si, au contraire, l'Etat estime ne pas pouvoir at tri bu er 11 AMM, il dai t transmettre son opposition mati vée au Comité dans le
délai de 120 jours.
différente Etats :
A ce ni veau de la procédure, 11 intervention du Cami té est
selon qu'il y a eu ou non opposi tian d'un ou plusieurs - lorsque le Comité n'a reçu aucune opposition dans le délai de 120 jours, il en informe immédiatement les Etats qui doivent se prononcer dans un délai de 30 jours sur la demande d' AMX. De toute évidence, l'autorisation devra être accordée ;
- lorsque le Cami té a reçu une ou plusieurs opposi tiens mati vées à l'octroi de l' AMM, il dei t, à compter de l'expiration du délai de 120 jours, se réunir dans un délai de 60 jours et émettre un avis mati vé sur l'ensemble du dossier en fonction des critères de base nocivité dans les conditions normales d'emploi, effet thérapeutique, composition qualitative et quantitative de la spécialité. Cet avis motivé du Comité, ou ceux de ses membres en cas d'avis divergents, est ou sont transmis aux Etats concernés.
A compter de la réception de cet avis, les Etats ont trente jours pour prendre une décision sur la demande d'autorisation de mise sur le marché.
SCHEXA
1
LA
PROCEDURE
DITE
DU CSP
DIRECTIVE
75/3191re ETAPE -DEKAIDE
===================
OOSSIER
11---'
_ -, LAUTORISATIO:fi DE 1ETAT JŒXBRE 1A11----)DEBUT DE LA PROCE- 1 DURE XULTI-ETATIQUE 2e ETAPE -NOTIFICATIOI=======================
ETAT lŒIBRE1A1
ADRESSE AU CSP le dossier et copie de son autorisation
3e ETAPE -DECISIOI
===================
LE CSP ADRESSE ledossier, etc.à --) au DDins5Etats membres "ilLES ETATS XEJŒRES RECOIVEIT LE DOS-SIER ET DOCUMENTS
(équivalantàune demande d"autori -sation
____-7
(en pratique, lesd et autorisations on fournis par ledemaossiers t été ndeur> 1 (Délai de 120j)
t
( 1DECISION1 . _[LE~ ~R DI
ETATS lŒJIBRES1B, C, F' etc...PREDENTDECISIOI 1___0_U_I ____~ reçoit aucune •ob
LE CSP, s'ilne
<
jectionDDtivée"dans les120j IOJI
LE CSP reçoit "11op- DEBUT DE LA PROCEDURE
position :motivée"
1
-
-
-
-
7
DU CSP conformément aux Art. 10 et 11LE CSP Il -FORlŒLES
ETATS JIEJIBRES
1
LES ETATS XE:M-BRES OCTROIEIT L'AUTORISATION
PROCEDURE DU CSP EXISTANTE DANS LE CAS D'UNE OPPOSITION ARTS 10 ET 11 DE LA DIRECTIVE 75/319 ETAT <S> R ~ DELIVRE<NT) UNE "OPPOSITION :MOTIVEE" DUREE TOTALE DE LA PROCEDURE MULTI-ETATIQUE EXISTANTE : ETAPE INITIALE 120 AVIS DU CSP 60 DECISION 30 JOURS 210 CSP : RECOIT <PAS D'OBLIGATION D' INFORMER LE DEMANDEUR, PAS D'AUDITION>
"OPPOSITION MOTIVEE" <IL A DEJA LE DOSSIER
ET LE<S> DOCUMENT<S>
60 JOURS AVIS DU CSP
<DOIT ETUDIER LA CONFORJIITE DES SPECIALITES AUX GONDI-TIONS DE L'ART 5 DE 65/65) 30 JOURS
"v
DECISION FINALE DE L'ETAT<S> MEMBRE <S> AYANT FAIT OPPOSITION
61
Après avoir évoqué le premier type d • intervention de ce Comité, passons au second prévu par l'article 12 de la deuxième Directive
une autorisation de mise sur le marché a été donnée par un Etat, cette autorisation -demandée avec le même dossier- a été refusée par un autre Etat
une spécialité est commercialisée dans plusieurs Etats, l'un de ces Etats suspend ou retire cette autorisation, les autres ne le font pas.
Dans ces hypothèses, l'un des Etats concernés peut saisir le Co mi té, qui délibère et dai t émettre un avis mati vé dans un délai maximal de 120 jours. Cet avis est transmis aux Etats concernés qui doivent faire connaître, dans les 30 jours, la suite qu'ils donnent à l'avis du Comité.
Jusqu'à Octobre 1980, cette procédure n'avait été utilisée qu'une seule fois, un Etat ayant saisi le Cami té, en application de l'article 12 de la Directive, lorsque les autorités de la République Fédérale d'Allemagne ont retiré l'autorisation de mise sur le marché du Clofibrate.
En ce qui concerne la troisième possibilité d'intervention de la part du Comité, elle résulte de l'article 14 de la deuxième Directive :
"dans des cas particuliers présentant un intérêt
communautaire, un Etat membre peut saisir le Comité avant de prendre une décision sur une demande, ou de procéder à une suspension ou à un
retrait d'autorisation".
Cette procédure a été suivie par quelques Etats qui voulaient avoir des informations, avant d'octroyer une autorisation de mise sur le marché, auprès des Etats qui avaient déjà accordé cette autorisation.
Voilà les trois actions que la Directive reconnalt au Comité. A côté de ces trois interventions, le Comité a beaucoup travaillé sur des sujets divers qui lui ont été transmis pour étude par le Comité pharmaceutique.
Le principal est l'élaboration de protocoles d'essais spécifiques dans les domaines de la toxicologie et de la clinique. Pour élaborer ce projet, il est tenu compte des protocoles qui existent déjà dans ce domaine et des connaissances scientifiques actuelles.
Ce projet est ensuite discuté au sein du groupe de travail, puis est transmis au Cami té des Spécialités pharmaceutiques, chaque Etat devant alors examiner ce projet et faire connaître ses observations.
Après un délai de quatre mois en général, les Etats doivent avoir fait conna1tre leurs observations au Comité. L'ensemble des remarques est alors transmis au groupe de travail qui les examine et les intègre au projet initial.
Pendant cette phase d'élaboration, la commission recueille l'avis de la Fédération européenne de l'industrie du médicament (sigle anglais EFPIA) qui représente l'ensemble
pharmaceutiques des Etats devant la Communauté.
des établissements
I. 4. 3. Décision du Conseil du 20 mai 1975 <15)
En même temps que le Conseil adoptait les deux Directives de 1975, il décidait la création du Comité Pharmaceutique.
Ce Comité est placé auprès de la commission et est présidé par un représentant de la Direction générale du marché intérieur et des industriels. Chacun des Etats membres de la Communauté y est représenté.
Ce Comité se réunit deux ou trois fois par an, son rôle est primordial. Il doit examiner :
- toute question relative à l'application des Directives ou toutes autres questions relevant du domaine des spécialités pharmaceutiques.
La Commission consulte le Comité à l'occasion de la préparation de propositions de nouvelles directives ou de modifications des Directives relevant du domaine des spécialités pharmaceutiques.
(15) Décision du Conseil 75/320/CEE du 20 mai 1975 JOCE n· L 147 du 9 juin 1975, p. 23
I. 5. La Directive 76/25 du 12 décembre 1977<16>
Cette Directive concerne les colorants qui peuvent être ajoutés aux médicaments. Elle retient le principe de l'alignement des colorants utilisés en pharmacie sur ceux employés dans les denrées alimentaires.
Cette Directive est modifiée par la Directive 81/464/CEE <17)du 24 juin 1981. Il n'existe plus de distinction entre les matières colorantes pour la coloration dans la masse et en surface, et les matières colorantes pour la coloration en surface seulement.
(16> Directive 78/25/CEE du 12 décembre 1977 JOCE n· L 11 du 14 janvier 1978, p. 18 <17) Directive 81/464/CEE du 24 juin 1981
CHAPITRE I I : BI LAH DU FOJJCT I ODEJŒJifT DU COMITE DES SPECIALITES
Depuis l'entrée en vigueur du traité CEE, la Commdssion s'est attachée à créer un marché commun des médicaments et a entrepris d'harmoniser les conditions de leur mise sur le marché.
Comme nous l'avons suivi précédemment, quatre Directives ont été adoptées à ce sujet.
La procédure communautaire instaurée à titre d'al te rna ti ve, à côté de la procédure nationale par la Directive 75/319/CEE du 20 mai 1975 n'a pas entrainé une adhésion massive des industriels.
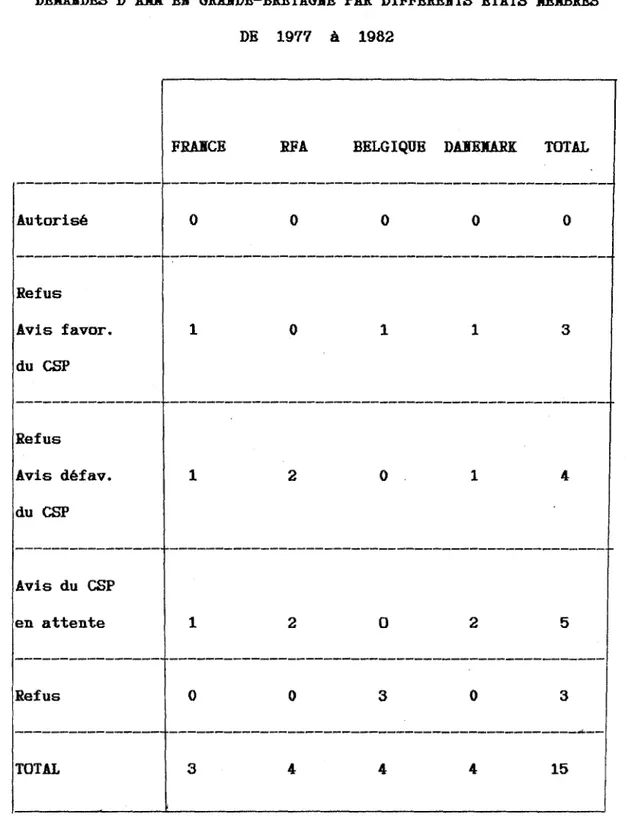
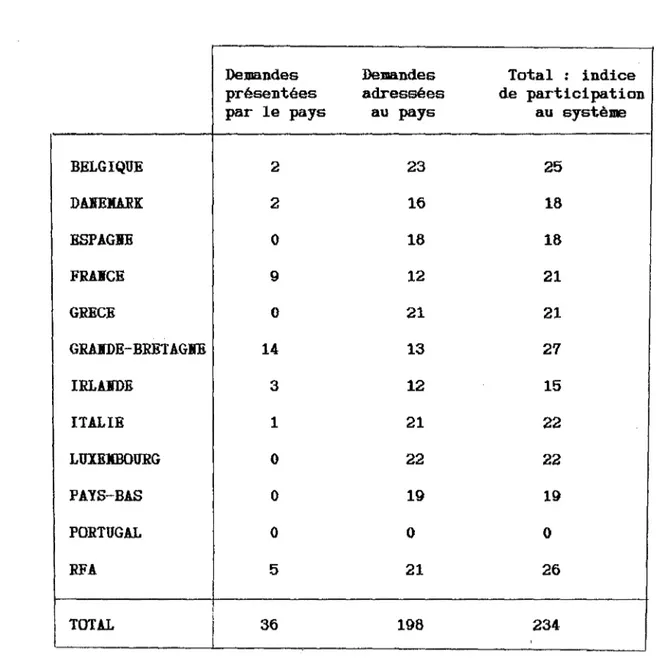
Ainsi que le montre le bilan fait après huit ans d'expérience 41 demandes ont été introduites selon cette procédure, ce qui correspond à 248 demandes adressées à des Etats membres autres que l'Etat membre d'origine (76).
II. 1. Exploitation statistique des résultats
Le Cami té des Spécialités Pharmaceutiques a été saisi de 41 demandes d'autorisation de mise sur le marché depuis son instauration.