Comprendre les réponses aberrantes des neutrophiles
dans le lupus pour le développement de nouvelles
approches thérapeutiques
Mémoire
Sandrine Huot
Maîtrise en microbiologie-immunologie - avec mémoire
Maître ès sciences (M. Sc.)
Comprendre les réponses aberrantes des neutrophiles
dans le lupus pour le développement de nouvelles
approches thérapeutiques
Mémoire
Sandrine Huot
Sous la direction de :
Résumé
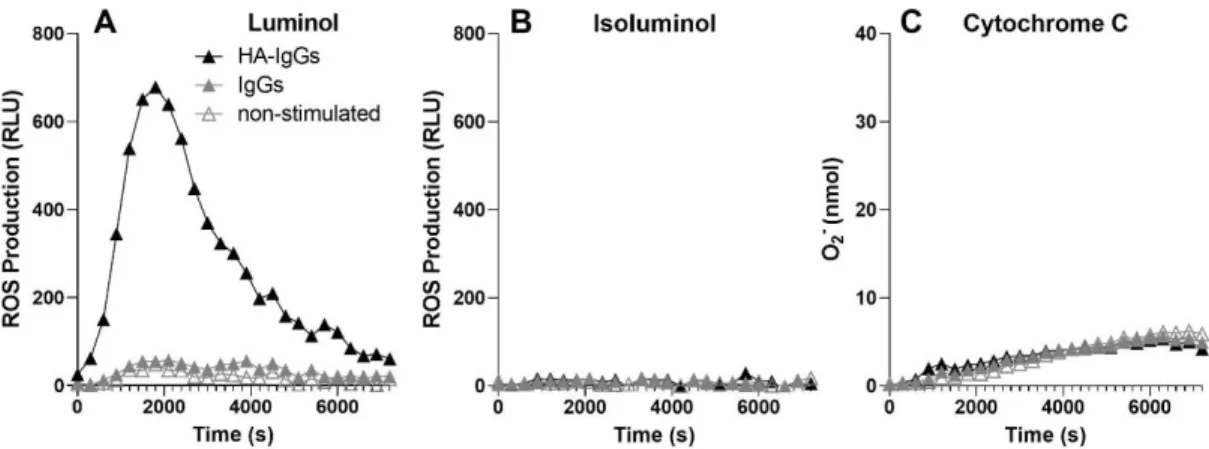
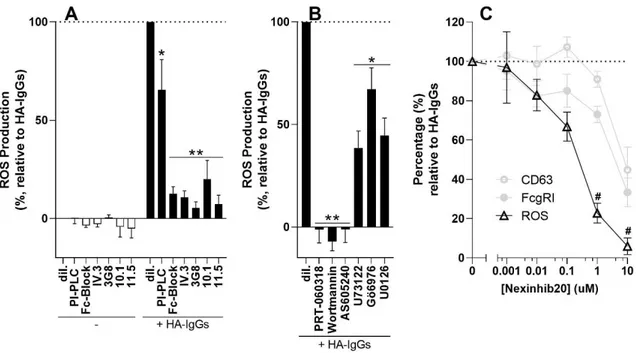
Le lupus érythémateux disséminé (LED) est une maladie auto-immune chronique à forte composante inflammatoire pour laquelle il n’existe aucun traitement curatif. Les patients qui en sont atteints présentent une pléthore d’auto-anticorps, c’est-à-dire des anticorps reconnaissant des constituants normaux du soi, que le système immunitaire considère par erreur comme étrangers. La présence de ces anticorps mène à la formation de complexes immuns, dont les taux sériques élevés et les dépôts tissulaires détectés chez les personnes lupiques corrèlent avec l’activité de la maladie. Les complexes immuns favorisent également l’activation indue des cellules immunitaires, qui peuvent à leur tour contribuer aux dommages organiques observés chez plusieurs patients. Le neutrophile, une cellule clé de l’immunité innée pour laquelle des anomalies fonctionnelles et phénotypiques ont été soulevées dans le LED, interagit avec les complexes immuns via les récepteurs Fc-gamma (FcgR). Le récepteur FcgRI, dont la présence à la surface des monocytes est positivement associée à des déterminants physiopathologiques importants du LED, est exprimé à des niveaux très faibles chez le neutrophile au repos, mais son expression est inductible en conditions d’infections sévères. La présente étude démontre que les HA-IgG, utilisés comme modèle de complexes immuns, entraînent presque instantanément une forte hausse de FcgRI à la surface des neutrophiles, et que la signalisation qui découle de ce récepteur est nécessaire à la production d’espèces réactives de l’oxygène. Ces molécules, normalement utilisées par les neutrophiles pour défendre l’organisme hôte contre les envahisseurs pathogènes, sont potentiellement dommageables pour les tissus sains. Nos travaux suggèrent que FcgRI est un contributeur important de la réponse des neutrophiles humains aux complexes immuns, et peuvent aider à expliquer le rôle délétère des neutrophiles dans les maladies auto-immunes comme le LED.
Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with a strong inflammatory component and for which there is no cure. SLE patients have a plethora of autoantibodies, that is, antibodies that recognize normal constituents of the self, which the immune system mistakenly considers foreign. The presence of these antibodies leads to the formation of immune complexes, whose elevated serum levels and tissue deposits detected in patients with lupus correlate with the disease activity. Immune complexes also promote the undue activation of immune cells, which in turn can contribute to the organic damage seen in many patients. The neutrophil, a key cell of innate immunity for which functional and phenotypic abnormalities have been raised in SLE, interacts with immune complexes via Fc-gamma receptors (FcgR). The FcgRI receptor, whose presence on the surface of monocytes is positively associated with important pathophysiological determinants of SLE, is expressed at very low levels on the neutrophil at rest, but its expression is inducible under severe infections. The present study demonstrates that HA-IgGs, used as a model of immune complexes, almost instantly up-regulate the expression of FcgRI on the surface of neutrophils, and that the signal ensuing of this receptor is necessary for the production of reactive oxygen species. These molecules, normally used by neutrophils to defend the host from pathogenic invaders, are potentially damaging to healthy tissue. Our work suggest FcgRI as an important contributor of the response of human neutrophils to immune complexes, and may help explain the deleterious role of neutrophils in autoimmune diseases like SLE.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des illustrations... vi
Liste des tableaux ... vii
Liste des figures ... viii
Liste des abréviations ... ix
Remerciements ... xii
Avant-propos ... xiii
Introduction ... 1
1. Le lupus érythémateux disséminé ... 1
1.1. Épidémiologie ... 1 1.2. Caractéristiques générales ... 1 1.3. Symptômes ... 2 1.4. Diagnostic ... 3 1.5. Mesures cliniques ... 5 1.6. Traitements ... 7 1.7. Prédispositions génétiques ... 8 1.8. Déclencheurs environnementaux ... 9 1.8.1. Les infections ... 9 1.9. Complexes immuns... 11
1.9.1. Les auto-anticorps dans le LED ... 11
1.9.2. Les sources d’auto-antigènes dans le LED ... 14
1.9.3. Les anticorps et la formation des complexes immuns ... 14
1.9.4. La clairance des complexes immuns... 16
2. Le neutrophile ... 18
2.1. Généralités ... 18
2.2. Adhésion et migration ... 19
2.3. Phagocytose ... 20
2.4. Pièges extracellulaires du neutrophile ... 21
2.5. Granules ... 21
2.6. Production d’espèces réactives de l’oxygène ... 24
2.6.1. Le complexe NADPH oxydase ... 24
3. Les récepteurs Fc ... 25
3.1. Généralités ... 25
3.2. Mode d’action et signalisation intracellulaire ... 26
3.3. Les récepteurs Fc-gamma ... 27
3.3.1. Les récepteurs Fc-gamma du neutrophile humain ... 29
3.3.2. Le récepteur FcgRI dans le LED ... 31
4. Hypothèse et objectifs ... 31
Chapitre 1 : IgG-aggregates rapidly up-regulate FcgRI expression at the surface of human neutrophils in a FcgRII-dependent fashion : A crucial role for FcgRI in the generation of reactive oxygen species ... 33
I.i. Résumé ... 33
I.ii. Abstract ... 35
I.i. Introduction ... 36
I.ii. Materials & Methods ... 38
I.iii. Results ... 41
I.v. Author’s contributions ... 47
I.vi. Acknowledgments ... 47
I.vii. References ... 48
I.viii. Figures & Legends ... 52
I.ix. Tables... 59
I.x. Supplemental Materials ... 62
Chapitre 2 : Discussion ... 67
Conclusion ... 71
Liste des illustrations
Introduction
Schéma 1. Érythème facial typique en « ailes de papillon » ... 3
Schéma 2. Représentation des différentes régions et composantes d’un anticorps (IgG) ... 15
Schéma 3. Représentation de complexes immuns de différentes tailles ... 15
Schéma 4. Voie d’activation classique du complément ... 17
Schéma 5. Mécanismes de clairance des complexes immuns ... 18
Schéma 6. Déroulement général du recrutement des neutrophiles au site inflammatoire ... 20
Schéma 7. Représentation des fonctions effectrices du neutrophile ... 21
Schéma 8. Le complexe NADPH oxydase chez le neutrophile au repos et à la suite d’un stimulus ... 25
Schéma 9. Activation et signalisation des FcgR ... 27
Schéma 10. Représentation des récepteurs Fc gamma (FcgR) humains ... 29
Chapitre 2
Schéma 11. Potentialisation des ROS par des composants bactériens ... 69Liste des tableaux
Introduction
Tableau 1. Critères de classification du LED ... 4
Tableau 2. Descripteurs du score d’activité SLEDAI ... 6
Tableau 3. Nature et prévalence de certains auto-anticorps retrouvés dans le LED ... 13
Tableau 4. Contenu des différentes granules et des vésicules sécrétoires du neutrophile humain ... 23
Chapitre 1
Table 1. Impact of heat-aggregated (HA)-IgGs on neutrophil surface markers... 59Table 2. List of compounds, their molecular target, concentrations, and diluting buffers ... 60
Liste des figures
Chapitre 1
Figure 1. HA-IgGs rapidly up-regulate FcgRI on human neutrophils, in a concentration-dependent fashion ... 52 Figure 2. HA-IgGs are a potent stimulus for FcgRI up-regulation in human neutrophils... 53 Figure 3. Signaling pathways involved in the up-regulation by HA-IgGs ... 54 Figure 4. Human neutrophils stimulated with HA-IgGs produce intracellular reactive oxygen species 55 Figure 5. FcgRI activation contributes to the production of reactive oxygen species ... 56 Figure 6. Combinations of fMLP or LPS with HA-IgGs magnify ROS production ... 57 Figure 7. Schematics: HA-IgGs rapidly up-regulate FcgRI expression at the surface of human
neutrophils in a FcgRII-dependent fashion and contribute to ROS production ... 58
Supplemental Figure S1. Description of the flow-cytometric gating strategy ... 62 Supplemental Figure S2. Impact of HA-IgGs on neutrophil viability ... 63 Supplemental Figure S3. Human neutrophils stimulated with HA-IgGs release intracellular granules . 64 Supplemental Figure S4. Absence of NET formation by HA-IgG-stimulated human neutrophils ... 65 Supplemental Figure S5. Stimulation of human neutrophils with HA-IgGs induces MCP-1 release ... 66
Liste des abréviations
7-AAD 7-aminoactinomycin D
ACR American College of Rheumatology ACTB actin beta
ADA adenosine deaminase
ADCC antibody-dependent cellular cytotoxicity ADN acide désoxyribonucléique
ADP adénosine diphosphate
ANA antinuclear antibodies
ANCA anti-neutrophil cytoplasmic antibodies AINS anti-inflammatoires non-stéroïdiens
ARN acide ribonucléique
AVC accident vasculaire cérébral BAFF B cell–activating factor
BILAG British Isles Lupus Assessment Group BLyS B lymphocyte stimulator
CCL2 C–C motif chemokine ligand 2 CCL3 C–C motif chemokine ligand 3
CI complexe immun
CMV cytomégalovirus
CR1 complement receptor 1
CXCL8 C-X-C motif chemokine ligand 8 Cytb558 flavocytochrome b558
D1 domaine 1
D2 domaine 2
D3 domaine 3
DNase désoxyribonucléase
EBNA-1 Epstein-Barr nuclear antigen 1 EBV Epstein-Barr virus
ERK extracellular signal-regulated kinase ESL-1 E-selectin ligand 1
EULAR European League Against Rheumatism FcgR Fc-gamma receptor
fMLP N-formylmethionyl-leucyl-phenylalanine G-CSF granulocyte colony stimulating factor
GM-CSF granulocyte-monocyte colony stimulating factor
GDP guanosine diphosphate
GPI glycosylphosphatidylinositol GSK glycogen-synthase-kinase GTP guanosine triphosphate H2O2 peroxyde d'hydrogène HA-IgG heat-aggregated IgG
HBSS Hank's balanced salt solution Hck hematopoietic cell kinase
hIgG immunoglobuline G (IgG) humaine HLA human leukocyte antigen
HOCl acide hypochloreux HSV Herpes Simplex Virus
IFN interféron
Ig immunoglobuline
IL interleukine
IRF5 interferon regulatory factor 5
ITAM immunoreceptor tyrosine-based activation motif ITIM immunoreceptor tyrosine-based inhibition motif L-QoL systemic lupus erythematosus quality of life LED lupus érythémateux disséminé
LPS lipopolysaccharide
LTB4 leucotriène B4
LuCIN Lupus Clinical Investigators Network MAC-1 macrophage-1 antigen
MCP monocyte chemoattractant protein
MEK mitogen-activated protein/extracellular signal-regulated kinase kinase MIP macrophage inflammatory protein
MMP-9 matrix metallopeptidase 9 (also known as gelatinase B)
MPO myéloperoxydase
NADPH reduced nicotinamide adenine dinucleotide phosphate NET neutrophil extracellular trap
O2 oxygène
O2- anion superoxyde
PAR polyarthrite rhumatoïde pDC plasmacytoid dendritic cells PGE2 prostaglandine E2
Phox phagocyte oxydase
PI3K phosphoinositide 3-kinase (also called phosphatidylinositol 3-kinase) PI-PLC phosphatidylinositol-specific phospholipase C
PKC protéine kinase C
PLC phospholipase C
PMA phorbol myristate acetate PSGL-1 P-selectin glycoprotein ligand ROS reactive oxygen species SEM standard error of the mean
SHIP-1 Src homology 2 domain containing inositol polyphosphate 5-phosphatase 1 SHIP-2 Src homology 2 domain containing inositol 5-phosphatase 2
SHP-1 Src homology region 2 domain-containing protein tyrosine phosphatase-1 SHP-2 Src homology region 2 domain-containing protein tyrosine phosphatase-2 SF-36 36-Item Short Form Survey
SLEDAI Systemic Lupus Erythematosus Disease Activity Index SLICC Systemic Lupus International Collaborating Clinics STAT4 signal transducer and activator of transcription 4 Syk spleen tyrosine kinase
TLR toll-like receptor TNF tumor necrosis factor
< À la mémoire de ma grand-mère,
Marie Jeanette Bergeron >
Remerciements
Au terme de ce travail, j’aimerais d’abord remercier sincèrement mon directeur de recherche, le Dr Marc Pouliot, qui a accepté de me prendre sous son aile pour mes études de maîtrise. Par sa disponibilité, son ouverture et son excellent mentorat, il a su alimenter mes réflexions, ma curiosité et ma motivation pour la recherche et la science. Merci pour ton amabilité et ta patience, et merci d’être un directeur aussi passionné.
Je veux également adresser un merci tout spécial à Cynthia Laflamme, qui m’a non seulement aidé quotidiennement sur le plan technique, mais qui a également été d’un précieux soutien lors des situations personnelles plus difficiles que j’ai eu à traverser durant ma maîtrise. Merci pour ta présence, ta bonne humeur et tes conseils écolos ; c’est un plaisir de travailler avec toi.
Merci à Guillaume Paré, Emmanuelle Rollet-Labelle, Isabelle Allaeys, Mihaela Robu, Rashmi Shah et Yann Becker d’avoir participé de près ou de loin à mes travaux par votre aide et vos conseils. Un merci particulier à Isabelle Dubuc et Annie Gravel pour votre assistance dans la pénible « aventure Tecan ». Gabriel Pépin, Tania Lévesque, Vanessa Colllin, Stéphane Hasse, Sofiane Berrazouane ; merci d’avoir rendu ces années plus douces et agréables. J’aimerais aussi souligner le plaisir que j’ai eu à m’occuper de la plante dont j’ai hérité, et remercier les plant ladies avec qui j’ai pu partager les plantules que j’ai réussi à faire pousser ! Aux étudiants du T-1, Romuald Babou, Audrée Laroche, Geneviève Marcoux, Étienne Doré, Andréa Murru, Alexie Doucet, Julien Vitry et à toutes les personnes que j’oublie, merci pour votre camaraderie.
Je tiens également à remercier le Dr Jacques Côté, sans qui je ne serais pas où je suis aujourd’hui. Merci d’avoir cru en moi dès mes débuts en recherche, pour ton soutien tout au long de mon parcours et pour m’avoir guidé inconsciemment vers un directeur que j’apprécie dans un domaine de recherche qui m’allume.
Finalement, merci à mon amoureux, Jimmy Gauthier, pour sa présence et ses encouragements qui m’ont permis de garder le cap et l’équilibre pendant toute cette aventure. Merci de me soutenir, de me calmer et de m’aimer comme je suis.
Avant-propos
Le chapitre 1 reprend intégralement, et sans modifications, l’article IgG-aggregates rapidly up-regulate FcgRI expression at the surface of human neutrophils in a FcgRII-dependent fashion : A crucial role for FcgRI in the generation of reactive oxygen species, publié dans la revue FASEB Journal (Federation of American Societies for Experimental Biology) le 18 septembre 2020. https://doi-org.acces.bibl.ulaval.ca/10.1096/fj.202001085R
Pour cet article, j’occupe le statut de premier auteur, accompagnée des coauteurs Cynthia Laflamme, Dr Paul R. Fortin, Dr Eric Boilard et Dr Marc Pouliot. Mes contributions dans la préparation de cet article ont été la réalisation de la plupart des expériences, l’analyse de données et la participation à l’écriture du manuscrit, notamment au montage des figures.
Introduction
1. Le lupus érythémateux disséminé
1.1. Épidémiologie
Le lupus érythémateux disséminé (LED) est une maladie auto-immune chronique à forte composante inflammatoire qui affecte principalement les femmes en âge de procréer1. Il accable environ 1 Canadien sur 2 000, et 90 % des patients atteints sont de sexe féminin2. L’Amérique du Nord signale l’incidence et la prévalence les plus élevées, mais les estimations varient considérablement en fonction de l’âge et de l’origine ethnique1. Les patients atteints de LED ont un taux de mortalité environ trois fois plus élevé que celui de la population générale et présentent des risques accrus de mortalité reliée aux maladies cardiovasculaires (plus de deux fois supérieurs), aux maladies rénales (plus de quatre fois supérieurs) et aux infections (près de cinq fois supérieurs)3.
1.2. Caractéristiques générales
En tant que pathologie auto-immune, le LED se définit comme un dysfonctionnement du système immunitaire, qui dévie de sa fonction protectrice pour devenir assaillant. L’auto-immunité, ou l’immunité dirigée contre le soi,
amène l’organisme à s’attaquer à ses constituants normaux, qu’il considère par erreur comme étrangers. Chez les patients souffrant de LED, environ 180 anticorps différents reconnaissant des composants inhérents au soi (auto-anticorps) sont rapportés4. De cette diversité immense, à peine une demi-douzaine d’auto-anticorps sont
spécifiques au LED (anticorps anti-Smith et anti ADN double brin, par exemple), tandis que la majorité d’entre eux peuvent également être détectés dans d’autres maladies4. Des anomalies sont observées dans presque
tous les aspects du système immunitaire, tant du côté inné, que du côté adaptatif. Bien que le LED soit reconnu comme « la maladie aux mille visages », ces éléments ont soulevé des doutes quant au caractère singulier de la pathologie, qui parfois, est plutôt perçue comme un regroupement de plusieurs maladies distinctes5,6.
Malgré le fait que le LED présente un large spectre de manifestations, la signature interféron (IFN), définie par une hausse de l’expression des gènes régulés par l’IFN de type I (alpha, bêta), fait partie des altérations typiques retrouvées chez les patients7. Une production excessive d’IFN-alpha par les cellules dendritiques
plasmacytoïdes (plasmacytoid dendritic cell ou pDC, en anglais) décelant du matériel nucléique endogène est aussi un phénomène décrit dans la pathogenèse du LED8 et il est traditionnellement entendu que ces effets
contribuent à la perte de tolérance au soi. Plusieurs voies extrinsèques et intrinsèques aux cellules B sont d’ailleurs rapportées dans la perte des points de contrôle de la tolérance (revues ici9). Des problèmes de
commutation de classe IgG et de différenciation des plasmocytes, les cellules B productrices d’anticorps solubles, sont notamment soulevés10.
Concernant les cellules de l’immunité innée, plusieurs revues dépeignent le rôle des neutrophiles comme un déterminant central des processus d’initiation et de perpétuation du LED11-15. En effet, des anomalies fonctionnelles et phénotypiques des neutrophiles semblent être au cœur de la pathologie et de ses manifestations. Par exemple, les neutrophiles de faible densité (low-density neutrophils ou LDN, en anglais), retrouvés en nombre plus important chez les patients atteints de LED, sont reconnus comme un sous-type de neutrophiles immatures et pro-inflammatoires ayant la capacité accrue de produire de l’IFN de type I et de libérer des pièges extracellulaires (neutrophil extracellular trap ou NET, en anglais), contribuant ainsi à la pathogenèse de la maladie. Cependant, l’existence des LDN en tant que sous-ensemble distinct demeure en pourparlers16. Outre les LDN, les aspects entourant la mort des neutrophiles dans le LED (niveaux élevés de neutrophiles apoptotiques, NETose) sont reconnus comme des éléments clés responsables du développement des lésions tissulaires et du dysfonctionnement du système immunitaire. Comme quoi l’auto-immunité, généralement considérée en tant que désordre d’étiologie principalement adaptative, peut aussi se rattacher à la branche innée de l’immunité, plus particulièrement au neutrophile et à ses altérations.
1.3. Symptômes
D’un point de vue clinique, le LED est une pathologie hautement imprévisible ; les personnes qui en sont atteintes présentent des manifestations systémiques extrêmement diverses qui apparaissent de manière cyclique, selon des périodes d’exacerbation (poussées actives) et de rémission. Les premiers symptômes du LED, impliquant généralement des douleurs articulaires, sont souvent non spécifiques, ralentissant ainsi le diagnostic17. Pendant ce temps, l’inflammation s’installe et les dommages tissulaires s’accumulent chez les patients. Dans tout le cortège des organes touchés, la peau est souvent affectée, notamment par l’érythème en papillon caractéristique qui se présente au niveau du nez et des joues (Schéma 1). En plus des symptômes visibles, le LED implique de nombreux symptômes sous-jacents pouvant représenter de réelles menaces pour la vie des patients. Des atteintes oculaires, cardiovasculaires, musculosquelettiques, neuropsychiatriques et rénales se manifestent chez les individus souffrant de LED18. La néphrite lupique constitue un élément important de la prise en charge, puisque ce type de manifestations entraîne une morbidité et une mortalité substantielles19. Compte tenu de sa nature chronique et de ses nombreuses comorbidités, le LED représente un fardeau économique important qui débute même dans les années précédant le diagnostic des patients20. Au pays, les coûts médicaux directs annuels, composés principalement des séjours hospitaliers, de la médication, des visites
de médecins spécialistes et des tests en laboratoire, peuvent s’élever jusqu’à 15 000 $ par patient souffrant de LED actif sévère21.
Érythème en papillon caractéristique présent au niveau du nez et des joues des patients souffrant de lupus. Aussi appelé éruption malaire (masque de loup).Créé avec BioRender.com
1.4. Diagnostic
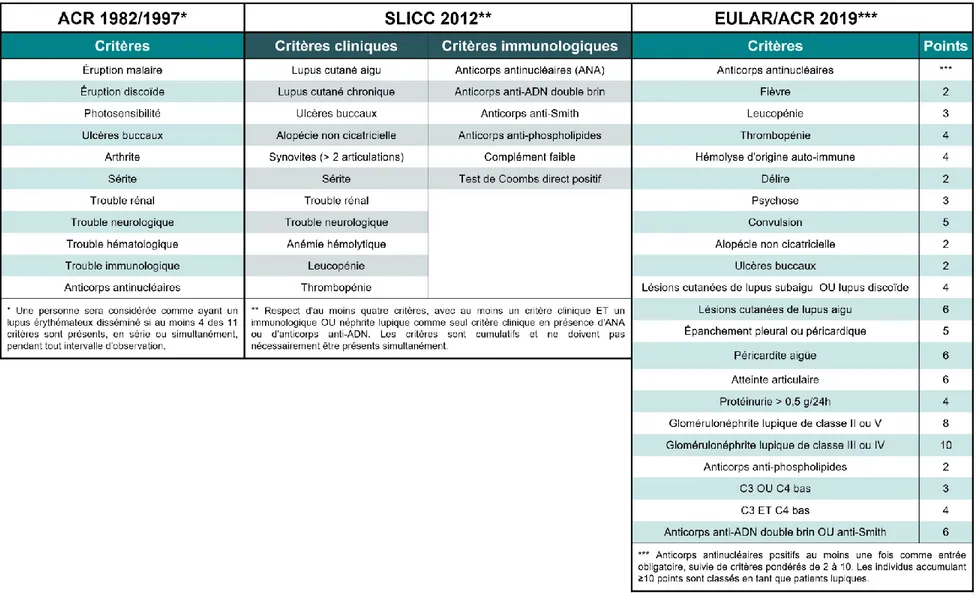
Entre l’apparition des symptômes initiaux et le diagnostic formel de LED, plusieurs années peuvent s’écouler et des diagnostics erronés peuvent être posés22,23. En raison de sa présentation clinique complexe, le LED constitue un défi diagnostique réel pour lequel il n’existe aucun test de dépistage unique. C’est plutôt un ensemble de critères évocateurs qui permet de poser le diagnostic. Puisque les différentes manifestations du LED se présentent de manière progressive, une importance particulière est accordée à l’histoire médicale des patients. En se basant sur cet historique ainsi que sur un examen physique et une sélection de tests en laboratoire, les cliniciens cherchent d’abord à exclure les diagnostics alternatifs, puis à reconnaître les combinaisons de symptômes caractéristiques du LED24. Principalement élaborés à des fins de recherche, certains critères de classification peuvent aussi servir d’outils de référence pour faciliter le diagnostic24. Plusieurs moutures de ces critères ont été mises en place au fil des ans, notamment par l’American College of Rheumatology (ACR) en 198225 et 199726, par le Systemic Lupus International Collaborating Clinics (SLICC) en 201227, et par une collaboration entre l’ACR et l’European League Against Rheumatism (EULAR) en 201928.
Cette dernière version a d’ailleurs été conçue dans le but d’améliorer la détection précoce des nouveaux cas de LED28, démontrant une fois de plus le diagnostic laborieux de cette pathologie. Les différents critères de
classifications et les conditions requises pour qu’une personne soit considérée atteinte de LED sont présentés au Tableau 1. De manière générale, un nombre minimal de critères doivent être réunis chez un même individu pour que le LED soit démontré.
1.5. Mesures cliniques
Trois domaines sont importants à considérer lors de l’évaluation complète d’un individu atteint de LED : l’activité de la maladie, les dommages aux organes et la qualité de vie (état de santé) des patients. De par l’hétérogénéité de la maladie et ses périodes d’exacerbation cycliques, l’activité du LED varie dans le temps et entre les individus. Son évaluation est cruciale pour le choix des traitements, et peut se faire selon plusieurs systèmes différents (revus ici29,30). Parmi ceux-ci, le BILAG (British Isles Lupus Assessment Group), qui donne un score spécifique pour chaque organe, et le SLEDAI (Systemic Lupus Erythematosus Disease Activity Index), qui fonctionne plutôt comme une mesure globale, se démarquent par leur degré de validation et leur fréquence d’utilisation30. Le SLEDAI se compose de 24 anomalies (descripteurs) réparties au sein de neuf systèmes organiques différents auxquels une cote d’importance distincte est accordée31,32 (Tableau 2). Le pointage maximal théorique de 105 constitue cependant un évènement impossible en pratique puisque la plupart des patients ont un maximum d’environ 10 à 15 descripteurs présents à la fois (sur les 24)31.
Dans plusieurs cas, un diagnostic de LED impliquera une accumulation de dommages permanents aux organes. Ces atteintes tissulaires peuvent être secondaires à l’activité de la pathologie, à des complications liées aux traitements ou à des maladies intercurrentes (par exemple un cancer survenant au cours du LED)33. L’index de dommage SLICC/ACR (aussi connu sous l’appellation SLICC/ACR Damage Index ou SDI, en anglais), permet d’évaluer l’accumulation des dommages chez les patients souffrant de LED, sans égard à la cause33. Il comprend l’évaluation de 12 systèmes organiques (pointage de 0 à 47) et définit le dommage comme un changement irréversible survenant après le diagnostic du LED et perdurant depuis au moins six mois33. Contrairement à l’activité de la maladie, où des périodes de stabilité complète peuvent être observées, les dommages ne sont que cumulatifs. D’ailleurs, leur importance pour le pronostic est majeure puisque la survie des patients diminue aussitôt qu’un point de dommage est atteint34.
L’activité de la maladie et les dommages aux organes ne représentent que partiellement l’état des patients atteints de lupus, qui sont également confrontés à des obstacles émotionnels importants. Au quotidien, le LED ébranle la qualité de vie physique, sociale et psychologique des personnes qui en souffrent35. Ces bouleversements peuvent être évalués à l’aide de différents questionnaires, notamment par l’enquête abrégée en 36 éléments (36-Item Short Form Survey ou SF-36, en anglais). Ce questionnaire général couvre huit dimensions de la santé qui réfèrent à la qualité de vie : (1) fonctionnement physique, (2) douleur corporelle, (3) limitations de rôle dues à des problèmes de santé physique, (4) limitations de rôle dues à des problèmes personnels ou émotionnels, (5) bien-être émotionnel, (6) fonctionnement social, (7) vitalité (énergie/fatigue) et (8) perceptions générales de la santé36,37. Deux mesures sommaires (santé physique et santé mentale) sont également disponibles38. Des outils d’évaluation spécifiques pour le LED ont aussi été mis en place dans le but
d’adapter les critères à l’imprévisibilité de la maladie39,40. Le questionnaire sur la qualité de vie LED (Systemic lupus erythematosus quality of life questionnaire ou L-QoL, en anglais) a quant à lui été conçu pour faciliter la compréhension des critères d’évaluation par les patients, le tout en se conformant aux normes psychométriques les plus strictes41.
1.6. Traitements
En raison de l’étiologie complexe du LED, aucun traitement curatif n’a été découvert jusqu’à présent. Les options thérapeutiques auxquelles les patients ont accès visent à optimiser leur qualité de vie et leur survie à long terme ainsi qu’à prévenir les périodes d’exacerbation et les dommages permanents aux organes, le tout en contrôlant l’activité de la maladie et en minimisant les comorbidités et la toxicité des médicaments42. En fonction des organes touchés, de la sévérité des symptômes et de l’activité du LED, plusieurs lignes directrices ont été établies en ce qui concerne les traitements (revues ici43). Les options palliatives disponibles comprennent, en utilisation souvent combinée, les antipaludéens (hydroxichloroquine, reconnue comme molécule clé pour le contrôle du LED44,45), les glucocorticoïdes, les immunosuppresseurs et les anti-inflammatoires non stéroïdiens (AINS)46,47. Approuvé par la Food and Drug Administration en 2011, le belimumab (BENLYSTAMC) constitue la première thérapie approuvée conçue spécifiquement pour le traitement du LED en plus de 50 ans. Ce traitement est dirigé contre le facteur d’activation des cellules B (B cell–activating factor ou BAFF, aussi connu sous le nom B lymphocyte stimulator ou BLyS, en anglais) et vise une diminution de la survie des cellules B autoréactives48. Selon des données positives de phases III (étude BLISS-LN) obtenues en décembre 2019 par GlaxoSmithKline, cet agent biologique, utilisé à l’origine chez les patients dépourvus de manifestation rénale sévère ou d’atteinte au niveau du système nerveux central48, pourrait bientôt être autorisé chez les patients souffrant de néphrite lupique active sévère49.
En août 2019, la société biopharmaceutique AstraZeneca a annoncé à travers des résultats de phase III (étude TULIP 2) que l’anticorps monoclonal anifrolumab réduisait de façon significative l’activité de la maladie chez les patients atteints de lupus50. Ce nouveau médicament potentiel pour le traitement du LED modéré à sévère agit en inhibant la signalisation dépendante des IFN de type I51. À ce sujet, le vaccin immunothérapeutique IFN-alpha kinoïde (IFN-K), qui n’a malheureusement pas atteint son critère d’évaluation principal, a en revanche démontré une forte immunogénicité polyclonale contre l’IFN-alpha et une réduction considérable de la signature IFN de type I chez les patients, se méritant ainsi une évaluation plus approfondie dans des études de phase III52. En outre, des résultats positifs de phase III (étude AURORA) ont été obtenus en décembre 2019 pour la voclosporine, un inhibiteur de la calcineurine développé par Aurinia Pharmaceuticals53. Cet immunosuppresseur
expérimental (qui vise l’inhibition de la production d’IL-254), s’inscrit comme molécule potentielle pour traiter la
maladie rénale associée au LED53.
Malgré ces avancées encourageantes, le développement de nouvelles approches thérapeutique pour le lupus n’est pas sans embuche. Ainsi, un communiqué de presse de The Janssen Pharmaceutical Companies of Johnson & Johnson annonçait, fin juin 2020, la suspension de la phase III de l’étude LOTUS en raison d’un manque d’efficacité. Ces travaux investiguaient l’utilisation de l’ustekinumab, un antagoniste humain de l’IL-12
et de l’IL-23 approuvé pour le traitement du psoriasis, chez les personnes touchées par le LED55. À l’heure actuelle, de nombreuses études cliniques de phase I, II et III menées par le Lupus Clinical Investigators Network (LuCIN) sont en cours afin d’évaluer l’innocuité et l’efficacité de nouveaux médicaments contre le LED56. Ces études cliniques, de même que les études fondamentales s’intéressant à la pathogenèse du LED, se doivent de persévérer afin d’offrir des agents thérapeutiques plus efficaces et plus sécuritaires aux patients touchés par cette maladie.
1.7. Prédispositions génétiques
Sur le plan génétique, le regroupement de maladies auto-immunes à l’intérieur des familles d’individus touchés par le LED a été démontré57,58. D’ailleurs, dans la parenté des patients (premier, deuxième et troisième degrés de parenté), environ 10 % sont touchées par le LED et près de 7 % ; par la polyarthrite rhumatoïde57. Les parents, frères/sœur et enfants des personnes lupiques ont une prévalence de LED d’environ 3 %, tandis que celle des oncles/tantes et neveux/nièces avoisine le 2 %57. Chez les jumeaux monozygotes (identiques), la concordance de la maladie entre les deux jumeaux atteint autour de 25 %59,60, soit trois fois plus que chez les jumeaux dizygotes60. Collectivement, ces observations démontrent l’existence de facteurs génétiques prédisposant au développement du LED. Ces prédispositions peuvent être polygéniques, c’est-à-dire relatives à l’altération de plusieurs gènes (multifactorielles), ou monogéniques ; occasionnées par des mutations dans un seul gène. La plupart des altérations géniques sont observées principalement dans des voies clés de l’immunité innée et adaptative, notamment la clairance des CI, les voies de l’interféron (IFN) de type I et des récepteurs de type Toll (Toll-like receptors ou TLR, en anglais) et la signalisation lymphocytaire (pour des revues, voir61-64). Du côté inné, plusieurs polymorphismes retrouvés dans les FcgR de faible affinité (FcgRII et FcgRIII) sont associés au LED et à la néphrite lupique65-67, et des carences en divers composants du complément représentent un facteur majeur de prédisposition au LED. En fait, plus de la moitié des patients atteints d’une déficience en C1q souffrent du LED68 et des anomalies génétiques dans les éléments C1q, C1r/C1s, C2 et C4 du complément prédisposent au développement de la pathologie (revues ici62). Des altérations associées au LED ont aussi été retrouvées dans différents gènes des enzymes désoxyribonucléases (DNase), responsables de la dégradation de l’ADN pouvant servir de substrat pour la formation de CI (revues ici62). De plus, des polymorphismes dans IRF5 (interferon regulatory factor 5), qui lui fait partie de la famille des facteurs de régulation de l’IFN, démontrent une forte association avec le LED, et ce, quel que soit le phénotype de la maladie69. Les variations génétiques de STAT4 (signal transducer and activator of transcription 4), un gène impliqué dans la signalisation en réponse à une activation par les cytokines IL-12 et IL-23, mais aussi activé par l’IFN de type I, sont quant à elles associées à un phénotype d’insuffisance rénale sévère69. Du côté adaptatif, plusieurs polymorphismes présents
dans le système HLA (human leukocyte antigen), responsable de la présentation antigénique, ont été rapportés comme contributeurs au développement du LED70.
1.8. Déclencheurs environnementaux
Comme exposée à la section précédente, la concordance du LED chez les jumeaux identiques représente environ 25 % des cas59,60. Autrement dit, dans 75 % des cas, le deuxième jumeau de la paire n’est pas atteint de lupus. Dès lors, ces résultats soulignent la participation de facteurs additionnels à la génétique, parmi lesquels les déclencheurs environnementaux ont été rapportés. L’utilisation d’œstrogènes exogènes (contraceptifs oraux et hormones post-ménopausiques) et la ménarche (apparition des premières règles) hâtive prédisposent au développement du LED71. Le tabagisme72 et l’exposition professionnelle à la silice73 font également partie des contributeurs environnementaux principaux. Chez les individus génétiquement prédisposés, ces facteurs peuvent donc agir comme déclencheurs de la maladie. De plus, bien que le rôle des rayons ultraviolets (UV) dans l’apparition du LED soit soupconné74, leur responsabilité dans l’éclosion des poussées inflammatoires chez les individus préalablement diagnostiqués est documentée davantage75-77. Cet aspect est d’ailleurs bien connu des patients, et la plupart d’entre eux évitent les heures d’ensoleillement auxquels les rayons sont les plus intenses78. Une exacerbation des symptômes cutanés, mais aussi des manifestations non cutanées du LED, est rapportée durant le printemps et l’été79,80.
1.8.1.
Les infections
Le rôle des agents infectieux en tant que déclencheurs environnementaux du LED est au cœur d’une littérature grandissante81-83. La présentation d’une infection au parvovirus humain B19 ressemble en plusieurs points aux manifestations du LED (revue en détails ici84). Les similarités comprennent non seulement des symptômes cliniques comme l’éruption malaire et la photosensibilité, mais aussi des ressemblances en ce qui a trait aux résultats des tests en laboratoire (ANA, anticorps anti-ADN double brin, etc.)84,85. De plus, l’infection au parvovirus B19 est une des infections virales les plus courantes chez les patients souffrant de LED86. Les mécanismes d’activation indirecte, de mimétisme moléculaire et d’infection virale persistante ont d’ailleurs été proposés pour relier l’infection du parvovirus B19 au développement du LED87, mais aucun lien de causalité n’a été clairement établi.
L’activation indirecte (bystander activation, en anglais) constitue un des mécanismes potentiellement déclencheurs de maladies auto-immunes. Elle se produit en contexte pro-inflammatoire, par exemple lors d’une
infection virale. La réponse inflammatoire de l’hôte qui est montée contre l’agent infectieux occasionne des dommages collatéraux inévitables, générant ainsi des débris tissulaires (auto-antigènes). En raison de leur présence dans l’environnement pro-inflammatoire, ces débris constituent de nouvelles cibles potentielles. Leur reconnaissance par des cellules autoréactives se trouvant à proximité favorise alors le développement d’une réponse indirecte contre les auto-antigènes, parallèlement à la réponse immunitaire initialement dirigée contre le virus. Ce mécanisme peut d’ailleurs aider à expliquer le fait que plusieurs virus différents arrivent déclencher l’auto-immunité.
L’exposition à un antigène étranger peut aussi conduire au développement subséquent de l’auto-immunité en raison de similitudes entre certaines structures de l’agent pathogène (épitopes étrangers) et les constituants de l’hôte. Ces ressemblances amènent le système immunitaire à produire des anticorps dirigés contre l’envahisseur infectieux qui vont cependant aussi reconnaître certains épitopes présents chez l’hôte (auto-épitopes), augmentant le risque d’une rupture de la tolérance au soi. Ce mécanisme, appelé mimétisme moléculaire, semble impliqué dans la pathogenèse du LED par le biais de l’infection au virus Epstein-Barr (EBV). La protéine virale EBNA-1, d’importance majeure dans le stade latent de l’infection à l’EBV, présente une réactivité croisée avec des auto-antigènes du lupus, notamment avec l’auto-antigène Smith hautement spécifique au LED (revue ici88,89). Une fois la tolérance perdue, les anticorps produits peuvent ensuite reconnaître d’autres déterminants autoantigéniques, donnant de l’ampleur à la réponse auto-immune. Ce phénomène, connu sous le nom de propagation d’épitopes (epitope spreading, en anglais), peut lui aussi résulter de l’infection à l’EBV et conduire au développement du LED88.
Tandis que les infections virales représentent un facteur environnemental majeur pouvant mener au déclenchement du LED, celles d’origine parasitaire auraient plutôt un rôle protecteur en ce qui a trait au développement de la pathologie (revues ici82,90,91). Les évidences à ce sujet reposent principalement sur des études in vivo. Par exemple, il a été montré que l’administration d’une glycoprotéine parasitaire sécrétée par le nématode filaire chez un modèle de souris lupiques diminuait la production d’anticorps antinucléaires et les dépôts d’IgG et de C3a (un composant du système du complément) dans les reins92. Une équipe a également démontré que l’infection de souris lupiques par le parasite responsable du paludisme protégeait le tissu rénal murin en diminuant le stress oxydatif 93, et réduisait les niveaux sériques d’IgG (IgG2a et IgG3) et d’anticorps anti-ADN double brin94. En revanche, des études in vitro ont démontré que les anticorps présents dans le sérum des patients atteints de LED pouvaient réagir avec Plasmodium falciparum et inhiber sa croissance95. Ces résultats reflètent donc une interaction bidirectionnelle entre le parasite responsable du paludisme et le LED, mais soulèvent également la présence d’une réactivité croisée entre les antigènes de Plasmodium et les auto-antigènes présents chez les personnes lupiques. En ajoutant à l’équation le rôle des antipaludéens dans le soulagement du lupus, la relation entre le paludisme et le LED est encore loin d’être élucidée.
Indépendamment de leur rôle dans le développement de l’auto-immunité, les agents infectieux représentent une menace pour les individus lupiques. En fait, les patients présentent un taux de mortalité par infections graves de plus de 25 %96. Ils sont plus enclins aux infections urinaires, aux pneumonies et aux bactériémies sans foyer, notamment en raison de l’utilisation de certains d’immunosuppresseurs97. Un risque plus élevé d’infections sévères au virus de l’herpès simplex (Herpes Simplex Virus ou HSV, en anglais) est aussi observé98. Comme le parvovirus B19, le cytomégalovirus (CMV) est un des agents pathogènes responsables des infections virales les plus courantes chez les patients souffrant de LED86. Le développement de la maladie à CMV chez ces patients est notamment associé à une plus longue durée du LED et à des doses de corticostéroïdes plus élevées99. Ces données mettent en évidence l’implication des microorganismes pathogènes dans la pathologie du LED, et ce même au-delà de leur rôle en tant que déclencheurs environnementaux.
1.9. Complexes immuns
D’étiologie complexe et incertaine, le LED se caractérise par des taux sériques élevés de CI circulants qui sont en corrélation l’activité de la maladie100,101 ainsi qu’avec les faibles niveaux de complément retrouvé chez les patients101. Les personnes atteintes de LED présentent d’ailleurs des problèmes de solubilisation des CI102, et leur abondance en circulation est également reliée au dépôt de CI dans les glomérules rénaux103. À ce propos, la présence de CI déposés dans le tissu rénal (membrane basale tubulaire) corrèle positivement et significativement avec la plupart des marqueurs pathologiques rénaux104. L’étude des réponses immunitaires aux CI est donc primordiale afin de développer une meilleure compréhension des processus physiopathologiques impliqués dans le LED, qui à son tour permettra la découverte de nouvelles cibles thérapeutiques prometteuses.
1.9.1.
Les auto-anticorps dans le LED
Avec 180 auto-anticorps recensés, le LED est la maladie auto-immune qui détient la collection d’auto-anticorps détectables la plus garnie (revus ici4). Parmi ceux-ci, une vingtaine d’anticorps différents sont en association avec l’activité de la maladie, une quinzaine sont reliés à la néphrite lupique et au moins une dizaine ciblent les neutrophiles, dont six sont dirigés directement contre des composants des granules (protéinase 3, myélopéroxidase, lysozyme, lactoferrine, élastase et cathepsine G)4. La présence d’auto-anticorps antinucléaires (antinuclear antibodies ou ANA, en anglais) est un critère presque universel chez les personnes atteintes de LED24 et leur association avec les sérites, soit l’inflammation des membranes séreuses qui protègent les organes du corps, a été rapportée (revue ici4). La présence d’auto-anticorps nucléaires spécifiques comme
les anti-ADN double brin et les anti-Smith est caractérisque du LED28. L’antigène Smith (Sm), nommé en l’honneur de la patiente Stephanie Smith105, a été découvert pour la première fois par Tan et Kunkel106. Il s’agit d’un regroupement de sept petites protéines nucléaires disposées en anneau et participant à l’épissage des pré-ARN messagers107. Malgré le fait que les anti-Sm ne soient détectés que chez 5 % à 30 % des patients4, ils sont hautement spécifiques au LED108. La prévalence d’auto-anticorps anti-ADN double brin se situe quant à elle dans la gamme des 40 %-80 %4 (voir le Tableau 3 pour un aperçu de la nature des auto-anticorps et de leur prévalence respective dans le LED). Malgré l’abondance et la diversité incontestable des auto-anticorps retrouvés dans le LED, leur présence converge potentiellement vers la formation de CI, puis vers les réponses immunitaires et inflammatoires qui découlent de leur interaction avec les récepteurs Fc-gamma (FcgR).
Adapté de4
1.9.2.
Les sources d’auto-antigènes dans le LED
Dans le contexte du LED, les neutrophiles sont reconnus comme source majeure de matériel autoantigénique menant à la formation de CI. En raison de leur courte durée de vie et de leur abondance significative, ils représentent au quotidien une charge importante de cellules mourantes. Étant donné les problèmes de clairance retrouvés chez les patients lupiques, ces taux élevés de neutrophiles apoptotiques sont difficilement éliminés, ce qui prédispose les neutrophiles à évoluer vers la nécrose secondaire109. Ce processus de mort, qui entraîne la libération d’éléments qui ne sont normalement pas exposés au système immunitaire, contribue alors à l’accumulation de matériel autoantigénique dans l’espace extracellulaire. Ces molécules, notamment la myélopéroxidase (MPO) et la protéinase 3, sont ensuite ciblées par les auto-anticorps dirigés contre les antigènes cytoplasmiques des neutrophiles (Anti-neutrophil cytoplasmic antibodies ou ANCA, en anglais)110, générant ainsi des CI.
De par leur libération de pièges de chromatine (neutrophil extracellular trap ou NET, en anglais), les neutrophiles génèrent également d’importantes quantités d’ADN extracellulaire qui sont, elles aussi, difficilement éliminées chez les patients lupiques, cette-fois en raison d’une plus faible activité des DNase111. Ces filets d’ADN ornementés de peptides antibactériens et de protéases représentent une matière première pour la formation de CI112 et constituent une source d’activation pour les cellules B autoréactives113.
1.9.3.
Les anticorps et la formation des complexes immuns
Les anticorps sont des protéines sécrétées lors de la réponse immunitaire humorale de l’hôte envers un antigène spécifique. Ils se présentent sous forme d’Y et sont constitués de deux chaînes lourdes identiques (50 kDa) reliées entre elles ainsi qu’à deux chaînes légères identiques (25 kDa) par des ponts disulfures (Schéma 2). Le clivage enzymatique spécifique d’un anticorps par la pepsine génère deux fragments : F(ab')2 et Fc. Le fragment F(ab')2 correspond à l’association de deux Fab (antigen-binding fragment) par une région charnière (hinge) (Schéma 2). Ces Fab, qui représentent les « bras » de l’Y, sont composés des chaînes légères et d’une portion des chaînes lourdes et assurent les interactions antigène-anticorps. Le fragment Fc (cristallisable) est composé du reste des chaînes lourdes et forme la base du Y. Il s’agit d’une portion constante effectrice qui interagit avec les récepteurs Fc (FcR) de plusieurs cellules immunitaires et avec le système du complément.
La structure de base de l’anticorps IgG humain se compose de deux chaînes lourdes identiques (bleu) composées d’une région variable (bleu pâle) et d’une région constante (bleu foncé), et de deux chaînes légères identiques (vert) composées d’une région variable (vert pâle) et d’une région constante (vert foncé). Les régions CH2 et CH3 de la région constante des chaînes lourdes composent le fragment Fc de l’anticorps. Créé avec BioRender.com
Le terme complexe immun (CI) désigne la combinaison d’un anticorps avec son antigène. En fonction du nombre d’antigènes et d’anticorps impliqués, la taille des CI est variable (Schéma 3). Leur formation est un processus dynamique généralement protecteur permettant la neutralisation des antigènes. Ils sont produits en réponse à des envahisseurs pathogènes, par exemple durant les infections, ou encore contre des antigènes étrangers libérés par des tissus lésés. Lorsque le système immunitaire fonctionne normalement, les CI sont éliminés naturellement par des processus qui sont présentés à la section suivante.
La taille des complexes immuns varie en fonction du nombre d’antigènes (noir) et d’anticorps (gris) impliqués. Créé avec BioRender.com
Schéma 2. Représentation des différentes régions et composantes d’un anticorps (IgG)
1.9.4.
La clairance des complexes immuns
Bien que la présence des CI contribue à la neutralisation et à l’élimination des envahisseurs pathogènes, un trop grand nombre de CI en circulation peut engendrer leur déposition tissulaire et entraîner des dommages inflammatoires. Pour cette raison, des mécanismes de clairance des CI impliquant les récepteurs Fc-gamma
(FcgR) et le système du complément servent à empêcher leur accumulation excessive. Le complément est un système complexe regroupant plusieurs dizaines de protéines fonctionnant pour la plupart par cascades protéolytiques. Il peut être activé de trois manières distinctes (voie classique, voie alterne ou voie des lectines), mais culmine dans tous les cas par la formation d’un complexe d’attaque membranaire (Membrane attack complex ou MAC, en anglais) qui a pour but la lyse des cellules pathogènes. C1q, le premier acteur de la voie d’activation classique, amorce la cascade en se liant aux CI (Schéma 4) par le biais d’une interaction avec une portion de l’IgG qui inclut la base de la région charnière et le haut de la région CH2114 (voir Schéma 2 pour les différentes régions de l’IgG). Ces régions chevauchent d’ailleurs celles qui permettent la liaison des IgG au FcgR (revues ici114-116).
Le système du complément participe de manière essentielle aux traitements appropriés des CI. Il permet leur solubilisation117,118, empêche leur précipitation119 et facilite aussi leur élimination, notamment via des sous-produits de la voie classique d’activation du complément, comme C3b et C4b120. En effet, la fixation covalente des protéines C3b et C4b aux CI permet une interaction avec la molécule CR1 (Complement Receptor 1, CD35) des érythrocytes. Ces derniers vont remettre les CI aux macrophages résidents du foie et de la rate pour qu’ils soient ingérés et détruits121 (Schéma 5). Le transfert des CI des érythrocytes vers les phagocytes implique une interaction avec les récepteurs Fc (FcR) phagocytaires et les récepteurs CR3 et CR4 du complément122. Ce processus de clairance médié par le CR1 érythrocytaire est unique aux primates, comme il l’est exposé dans cette revue121. Les phagocytes peuvent également internaliser les CI directement via leurs FcR sans avoir recours au complément (Schéma 5). La section Phagocytose couvre plus en détail le processus d’internalisation, de digestion et de destruction par les phagocytes.
C1q lie les portions Fc des anticorps composant le complexe immun. Cette fixation induit des changements conformationnels dans la molécule C1q, conduisant à l'activation du complexe C1, composé de C1q ainsi que de deux molécules C1r et de deux molécules C1s. Le complexe C1 clive ensuite les composants C2 et C4, dont les fragments se réassemblent pour former la C3 convertase. Cette C3 convertase clive le composant C3, dont une partie s’ajoute à la C3 convertase pour former la C5 convertase. La convertase C5 clive ensuite le composant C5, dont une partie sert à la formation du complexe d’attaque membranaire (membrane attack complex ou MAC, en anglais), composé de C5b, C6, C7, C8 et C9. Créé avec BioRender.com
À gauche) Clairance médiée par le CR1 érythrocytaire : les protéines du complément (C3b/C4b) se lient aux complexes immuns (CI), qui sont ensuite reconnus par le CR1 (récepteur 1 du complément) érythrocytaire. Une fois lié à l’érythrocyte, le CI est présenté et transféré aux cellules phagocytaires du foie et/ou de la rate. Les phagocytes lient alors le CI via les récepteurs Fc (FcR) et les récepteurs CR3 et CR4 du complément afin d’internaliser le CI, conduisant ainsi à son élimination. À droite) Clairance médiée par les récepteurs Fc de la cellule phagocytaire : les portions Fc des anticorps composant le CI sont reconnues et liées par les récepteurs Fc (FcR) d’une cellule phagocytaire, qui à son tour ingère le CI afin de le détruire. Adapté de121. Créé avec BioRender.com
2. Le neutrophile
2.1. Généralités
Chez l’humain, les neutrophiles sont les leucocytes circulants les plus abondants du sang (50-70 %). Chaque jour, ils sont produits dans la moelle osseuse au nombre d’environ 109 (soit l’équivalent du nombre de secondes contenues dans 31 ans et 259 jours) par kilogramme123. Les neutrophiles sont les cellules effectrices qui
patrouillent l’organisme à la recherche d’infections microbiennes et de lésions tissulaires naissantes. Ils sont armés de nombreux mécanismes de défense intracellulaires (phagocytose, production d’espèces réactives de l’oxygène) et extracellulaires (dégranulation, libération d’agents peptidiques et lipidiques, formation de pièges Schéma 5. Mécanismes de clairance des complexes immuns
[NET], etc.). Lorsqu’un préjudice est détecté, les neutrophiles circulants migrent rapidement vers les tissus infectés ou endommagés et s’y faufilent pour exécuter leur plan tactique à la défense de l’organisme hôte. Au terme de l’affrontement, les neutrophiles meurent par apoptose en exposant des molécules de phosphatidylsérine à leur surface, leur permettant ainsi d’être éliminés par efferocytose124. Ce processus
physiologique permet de désencombrer la zone de combat des neutrophiles mourants et met fin au recrutement additionnel de neutrophiles, contribuant ainsi à la résolution de l’inflammation125. Les neutrophiles circulants qui ne sont pas activés ni recrutés dans les tissus ont une durée de vie intrinsèquement courte, traditionnellement estimée à <24 h, mais certains résultats tendent à démontrer qu’ils pourraient vivre jusqu’à plus de 5 jours126.
En plus d’incarner la première ligne de défense dans la lutte contre les microorganismes, les neutrophiles
répondent aussi en tant que médiateurs de l’inflammation. Ils produisent une grande variété de cytokines, y compris des cytokines pro-inflammatoires, anti-inflammatoires, immunorégulatrices, chimiotactiques (chimiokines), angiogéniques et plus encore (revues ici127,128). Les neutrophiles synthétisent également des médiateurs lipidiques pro-inflammatoires, principalement la prostaglandine E2 (PGE2)129, le thromboxane A2 (TXA2)130 et le leucotriène B4 (LTB4)131-133, et participent à la synthèse de médiateurs anti-inflammatoires comme les lipoxines, les résolvines et les protectines (revus ici125). Compte tenu de leur abondance significative, les neutrophiles peuvent se servir de ces métabolites pour moduler l’activité des cellules immunitaires voisines dans une diaphonie bidirectionnelle. Ils orchestrent donc les réponses immunitaires innées, mais aussi adaptatives, notamment via leurs interactions avec les lymphocytes B et T (revues ici134,135).
2.2. Adhésion et migration
Le recrutement des leucocytes aux sites inflammatoires est un processus relativement bien documenté136,137. Tous les jours, des neutrophiles sillonnent la circulation sanguine à la cherche de dommages tissulaires ou d’intrusions microbiennes. Lors d’un préjudice, l’inflammation induit l’expression des sélectines (P- et E- sélectines) à la surface des cellules endothéliales qui bordent les vaisseaux sanguins. L’interaction de ces sélectines avec leurs ligands (PSGL-1 et ESL-1, respectivement), exprimés à la surface leucocytaire, ralentit le neutrophile et le fait rouler tranquillement sur la paroi du vaisseau (bien étudié chez la souris138). En circulant ainsi à proximité de la paroi endothéliale, le neutrophile sonde le microenvironnement local pour détecter des signaux inflammatoires supplémentaires (des cytokines, par exemple). Ces signaux secondaires induisent un changement de conformation des intégrines du neutrophile (passant de faible affinité à forte affinité) permettant l’interaction avec les ligands (ICAM-1) exprimés sur l’endothélium enflammé. Cette interaction de plus forte affinité permet l’arrêt et l’adhésion ferme du neutrophile sur la cellule endothéliale. Ce processus est suivi par la
diapédèse, soit la migration du neutrophile à travers la paroi vasculaire (Schéma 6). Une fois dans les tissus, le neutrophile activé migre par chimiotaxie au site précis de l’inflammation pour y éliminer l’agent causal.
Schéma 6. Déroulement général du recrutement des neutrophiles au site inflammatoire (Voir le texte pour les détails.) Créé avec BioRender.com
2.3. Phagocytose
Les cellules de l’organisme possèdent plusieurs stratégies distinctes pour internaliser les particules et les liquides qui se retrouvent dans leur environnement (pinocytose médiée ou non par la clathrine, macropinocytose et phagocytose, revues ici139). En tant que phagocyte professionnel, le neutrophile est spécialisé dans l’ingestion de microorganismes par le biais de la phagocytose, le mécanisme cellulaire utilisé pour internaliser les particules de taille égale ou supérieure à 0,5 micron (μm)140,141. L’interaction de ligands de cette taille (microorganismes opsonisés, débris cellulaires, CI) avec les récepteurs phagocytaires apparentés (récepteurs Fc, récepteurs du complément) déclenche le développement de projections temporaires en forme de bras (pseudopodes) qui vont entourer et internaliser la cible dans une vacuole nommée phagosome (revue ici142) (Schéma 7). Chez le neutrophile, ce processus peut s’effectuer en aussi peu qu’une vingtaine de secondes139. Une fois la cible engloutie, un sous-type de granules azurophiles du neutrophile, les azurophiles non-sécrétoires143, vont rapidement fusionner avec le phagosome pour y libérer des molécules hautement toxiques ainsi que des
enzymes catalysant la formation d’espèces réactives de l’oxygène (ROS) (revues ici 142,144). Contrairement aux cellules présentatrices d’antigènes qui traitent spécifiquement les particules ingérées pour initier des réponses immunitaires adaptatives, le neutrophile utilise son artillerie pour dégrader le contenu ingéré, permettant ainsi l’élimination de l’agent pathogène144.
Schéma 7. Représentation des fonctions effectrices du neutrophile
La phagocytose, la formation de pièges extracellulaires de chromatine (NET) et la dégranulation sont des mécanismes qui permettent aux neutrophiles de combattre les envahisseurs pathogènes (illustrés en jaune). (Voir la description plus détaillée de chaque mécanisme dans le texte.) Créé avec BioRender.com
2.4. Pièges extracellulaires du neutrophile
Les pièges extracellulaires du neutrophile (NET) correspondent à des filaments de chromatines recouverts de protéines antimicrobiennes. Bien que la composition protéique des NET soit hétérogène et variable selon les stimuli et les contextes physiopathologiques112,145, ces pièges sont expulsés dans l’espace extracellulaire dans le but d’attraper et de tuer les envahisseurs pathogènes, sans qu’ils ne soient internalisés par le neutrophile (Schéma 7). En plus des propriétés antimicrobiennes des NET, ces derniers agissent comme barrière physique en séquestrant les microorganismes, empêchant ainsi leur dissémination systémique.
2.5. Granules
Les neutrophiles appartiennent à la catégorie des granulocytes, c’est-à-dire qu’ils sont caractérisés par la présence de granules dans leur cytoplasme. Ces compartiments intracellulaires permettent d’emmagasiner une pléthore de molécules préformées afin de répondre rapidement aux microorganismes envahisseurs. Le
neutrophile utilise la dégranulation, soit la libération extracellulaire du contenu de ses granules, pour mobiliser à sa surface différents récepteurs, ou encore pour sécréter certaines cargaisons microbicides dans l’espace extracellulaire. Le neutrophile mature possède classiquement quatre types de granules qui se forment séquentiellement lors de son processus de maturation143. Chaque type diffère par son contenu, et par extension, par sa fonction effectrice. Les granules primaires (ou azurophiles : GA) constituent la principale ressource microbicide des neutrophiles. Elles présentent spécifiquement à leur surface la protéine membranaire CD63, et leur contenu regorge de protéines antimicrobiennes. Les GA contiennent l’enzyme myélopéroxidase (MPO) productrice d’oxydants, des protéases telles que la cathepsine G, l’azurocidine, l’élastase et la protéinase 3, ainsi que des défensines augmentant la perméabilité membranaire des bactéries146. Les granules secondaires (ou spécifiques : GS) sont généralement identifiables par leur contenu en lactoferrine, un effecteur antimicrobien, tandis que les granules tertiaires (ou de gélatinase : GG) contiennent la gélatinase, une métallopeptidase matricielle impliquées dans la dégradation de la matrice extracellulaire146. Les dernières granules à être formées sont appelées vésicules sécrétoires (VS) et contrairement aux autres sous-types granulaires, la production de VS est la seule qui persiste chez les neutrophiles matures en circulation143. Comme
les GA, ces vésicules contiennent de l’azurocidine147, mais aussi des protéines plasmatiques, des protéines du
cytosquelette ainsi que certains TLR (TRL2, TLR8) et FcgR (FcgRIIIb)146. Le noyau catalytique responsable de
l’explosion oxydative des neutrophiles (cytb558, discuté dans la section suivante) se retrouve également dans la membrane des VS ainsi que dans celle des GS et des GG148-150. Munis d’un tel arsenal, les neutrophiles déploient leurs cargaisons selon un processus finement régulé permettant la libération spécifique de leurs médiateurs à des moments bien précis. Par exemple, la dégranulation des VS possède le seuil de signalisation le plus bas et s’exécute en réponse à des stimuli inflammatoires faibles et/ou transitoires, suivie respectivement de la dégranulation des GG, des GS et des GA, mobilisées en réponse à des stimuli d’amplitude croissante143,151.
Cet ordre d’exocytose est précisément l’inverse de celui observé lors de la formation des granules. Une excellente revue143 couvre d’ailleurs les différents mécanismes régulant la libération des sous-ensembles granulaires. Rab27a, une petite GTPase, est un élément clé de la machinerie exocytaire des neutrophiles qui module la sécrétion extracellulaire des GS et des GG152, et d’une partie des GA153. Une liste non-exhaustive du contenu des différentes granules est présentée au Tableau 4.
Données combinées et adaptées de 146,147,150,154
2.6. Production d’espèces réactives de l’oxygène
Les espèces réactives de l’oxygène (ROS) sont définies comme des radicaux d’oxygène contenant un ou plusieurs électrons non appariés, comme l’anion superoxyde (O2-), et certains agents oxydants non radicaux, comme le peroxyde d’hydrogène (H2O2) et l’acide hypochloreux (HOCl). Bien qu’il existe de nombreuses voies enzymatiques par lesquelles les ROS peuvent être générés dans la cellule (notamment dans le cadre de la phosphorylation oxydative mitochondriale, au niveau de la chaîne de transport d'électrons), la libération de ces dérivés toxiques est un mécanisme que le neutrophile utilise pour détruire ses proies. Effectivement, lors de la phagocytose, l’O2- et l’ H2O2 sont produits et libérés à l’intérieur des phagosomes, amorçant ainsi une série de réactions menant à la mort du pathogène ingéré. En présence de MPO, apportée au phagosome grâce à une fusion phagosome-GA, l’ H2O2 sert à la production d’HOCl, un métabolite microbicide qui, à son tour, sert à la formation additionnelle de ROS155. Ces derniers attaquent alors les protéines, les lipides et l’ADN afin de détruire l’ennemi155. Lorsque les cibles du neutrophile sont trop imposantes pour être phagocytées, les ROS peuvent aussi être libérés de manière extracellulaire. Ce mécanisme, par lequel les neutrophiles sécrètent des dérivés toxiques de l’oxygène, est aussi connu sous le nom d’explosion oxydative, et occupe une place fondamentale dans la défense contre les infections. En effet, les individus atteints de la maladie granulomateuse chronique, chez qui les phagocytes sont incapables de provoquer l’explosion oxydative, souffrent d’infections bactériennes et fongiques sévères à répétition156.
2.6.1.
Le complexe NADPH oxydase
Le complexe NADPH oxydase est la principale source d’oxydants du neutrophile. Il est composé de cinq sous-unités spécifiques phox (phagocyte oxydase), soit gp91phox, p22phox, p40phox, p47phox et p67phox. Lorsque le neutrophile est au repos, les protéines gp91phox et p22phox forment un dimère transmembranaire appelé flavocytochrome b558 (cytb558), reconnu comme le noyau catalytique, tandis que p40phox, p47phox et p67phox forment le complexe régulateur, qui réside dans le cytosol (revues ici157). La protéine GTPase Rac2, qui est une sous-unité non spécifique, mais essentielle au complexe NADPH oxydase, se trouve également dans le cytosol157. Lors d’une stimulation, Rac2 est activée (échange du GDP pour un GTP) et p47phox est phosphorylée, engendrant ainsi la translocation de Rac2 et du complexe régulateur au niveau du cytb558, à la membrane (Schéma 8). Le complexe ainsi formé permet le transfert d’électrons du donneur NADPH, présent dans le cytosol, vers l’autre côté de la membrane, soit dans le milieu extracellulaire ou dans divers compartiments intracellulaires comme le phagosome (phagolysosome) ou certaines vésicules non phagosomales (revues ici 158). Ce transfert d’électrons permet la réduction de l’oxygène en O2-, entraînant par le fait même une production massive de ROS. Plusieurs agents peuvent aussi, sans induire l’activation totale du complexe NADPH oxydase,