Développement d'une méthode robuste de
caractérisation du microbiote de tissus pulmonaires
tumoraux
Mémoire
Nathan Dumont-Leblond
Maîtrise en microbiologie - avec mémoire
Maître ès sciences (M. Sc.)
Développement d’une méthode robuste de
caractérisation du microbiote de tissus
pulmonaires tumoraux
Mémoire
Dumont-Leblond, Nathan
Sous la direction de :
Duchaine, Caroline, PhD
Joubert, Philippe, M.D., PhD
Résumé
Bien que longtemps considérés stériles, il est maintenant confirmé que les poumons sont peuplés d’une communauté microbienne diversifiée. De plus, il a récemment été observé que les microorganismes peuplant le corps humain peuvent influencer le développement du cancer et son traitement dans l’intestin et le pancréas. Cependant, peu d’études ont tenté de caractériser le microbiote pulmonaire et son impact sur le cancer du poumon. Davantage d’efforts se doivent d’y être consacrés. L’absence de standardisation méthodologique et de considération pour les contraintes de l’environnement pulmonaire diminuent par contre la validité des résultats obtenus et des conclusions qui peuvent être faites actuellement. Le présent projet propose donc le développement d’une démarche expérimentale appropriée à la caractérisation du microbiote tumoral pulmonaire. Nous avons mis sur pied une marche à suivre complète, allant du recrutement de patients au traitement des données bio-informatiques. Nous avons minimisé l’incorporation de microorganismes contaminants et avons établi des points de contrôle afin de les détecter et de les retirer des échantillons. La combinaison de méthodes d’homogénéisation enzymatiques et mécaniques et d’une trousse d’extraction d’ADN commerciale a permis une détection rapide et fiable du microbiote. L’utilisation des communautés microbiennes de composition connue, autant de cellules entières («whole») que de génomes, a permis de confirmer que notre capacité de récupération bactérienne est adéquate et que l’abondance d’ADN humain dans les extraits pulmonaires n’a pas d’influence significative sur notre capacité de détection. La signature bactérienne des tissus cancéreux et sains de cinq patients a pu être différenciée de celles des contrôles méthodologiques. Nos travaux approfondissent les connaissances nécessaires à l’étude d’environnements microbiens faiblement peuplés. Ils sont un point de départ vers la standardisation méthodologique et l’implémentation de précautions d’échantillonnage appropriées pour l’étude du microbiote pulmonaire.
Abstract
Although historically considered sterile, the lung is now known to harbor a diverse microbial community. It has also been recently observed that microorganisms that populate the human body can influence the development of cancer and its treatment in the intestine and pancreas. However, very few studies have attempted to characterize the composition of the pulmonary microbiota to correlate it to lung cancer, a deeper look into this possible interaction seems mandatory. To this day, the absence of methodological standardization and considerations for the constraint of the pulmonary environment reduces the validity of the results collected and the conclusions drawn. This project aims at developing an appropriate experimental approach for the characterization of the pulmonary microbiota. We developed a complete pipeline from patient enrollment to bioinformatics analyses. We minimized the incorporation of contaminating microorganisms and established controls to detect and account for them in lung samples. A combination of enzymatic and mechanical homogenization technic with adapted commercially available DNA extraction kit allowed for a fast and reliable extraction of bacterial DNA. The use of bacterial communities of known composition, whole cells or genomic, allowed us the confirm our ability to recover bacterial DNA adequately and that the abundance of human DNA in sample does not influence our detection efficiency. The bacterial signature of the cancerous and healthy lung tissue of five patients could also be distinguished from methodological controls. Our work expands our understanding on the analysis of low microbial burden environments. It is to be a starting point in the standardization of methods used in pulmonary microbiota study and the implementation of appropriates sampling considerations.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures, tableaux, illustrations ... vii
Liste des abréviations, sigles, acronymes ... viii
Remerciements ... ix
Avant-propos ... x
Introduction ... 1
Le cancer du poumon ... 1
Les adénocarcinomes pulmonaires ... 1
Les carcinomes épidermoïdes pulmonaires ... 2
Le microbiote pulmonaire ... 2
La composition bactérienne du microbiote pulmonaire ... 3
Écologie microbienne pulmonaire ... 3
Le modèle de l’île adaptée ... 5
Homéostasie nutritionnelle ... 5
Signalisation interdomaine ... 7
Le microbiote et le cancer ... 7
Nature des interactions entre le microbiote et le cancer ... 9
Méthodologies et limites de l’étude du microbiote pulmonaire ... 10
Abondance du microbiote pulmonaire et gestion des biais ... 10
Recrutement de patients ... 10
Échantillonnage ... 14
Contexte et réalité clinique ... 15
Méthodes de détection et de quantification ... 16
Gestions des contaminants ... 19
Objectifs et mise en contexte ... 19
Chapitre 1 : Développement d’un protocole robuste pour la caractérisation du microbiote pulmonaire ... 20
1.1 Résumé ... 20
1.2 Abstract ... 20
1.4 Materials and Methods ... 22
1.4.1 Patient selection ... 22
1.4.2 Sampling ... 22
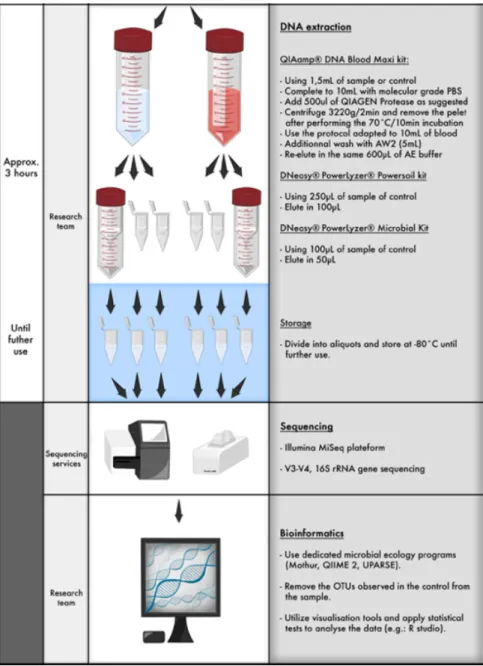
1.4.3 DNA extraction process ... 23
1.4.4.1 Enzymatic digestion ... 23
1.4.4.2 Mechanical Homogenization ... 23
1.4.4.3 DNA Extraction ... 23
1.4.5 Spectrophotometric Quantification of DNA ... 24
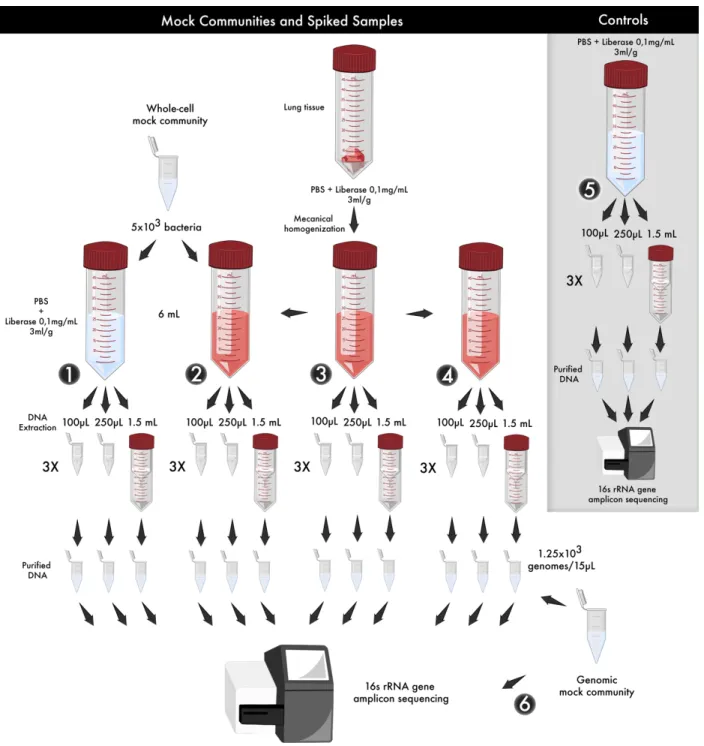
1.4.6 Detection of Spiked Bacterial Mock Community ... 24
1.4.7 Sequencing ... 25
1.4.8 Data Analysis and Selection of Techniques ... 25
1.4.9 Methodological Core Microbiota ... 25
1.4.10 Bioinformatics Management of Controls ... 26
1.5 Results ... 26
1.5.1 Detection of Spiked Bacterial Community ... 26
1.5.2 DNA yields and purity obtained with the different extraction kits ... 27
1.5.3 Alpha diversity ... 27
1.5.4 Correlation to Controls and Removal of Contaminating OTUs ... 30
1.5.5 Methological Core Microbiota ... 30
1.6 Discussion ... 34
1.6.1 Acknowledging the limits and precautions required for the study of the lung microbiota ... 34
1.6.1.1 Patient selection ... 34
1.6.1.2 Sampling Precautions ... 34
1.6.1.3 Enzymatic and Mechanical Homogenization ... 35
1.6.1.4 DNA Extraction Protocols ... 36
1.6.1.5 Sequencing Platform and Variable Region Selection ... 37
1.6.1.6 Contamination Management ... 38
1.6.2 Methological Core Microbiota ... 39
1.6.3 Methods Limitations ... 40
1.6.4 Perspectives ... 40
1.7 Conclusion ... 40
1.8.1 Competing interests ... 41 1.8.2 Data availability ... 41 1.8.3 Code availability ... 41 1.9 Supplementary material ... 44 Conclusion ... 53 Bibliographie ... 55
Liste des figures, tableaux, illustrations
Figure I-1 : Schéma récapitulatif des facteurs influençant la composition de la
communauté microbienne pulmonaire ... 6
Tableau I-1 : Facteurs concomitants pouvant modifier la composition du microbiote pulmonaire. ... 11
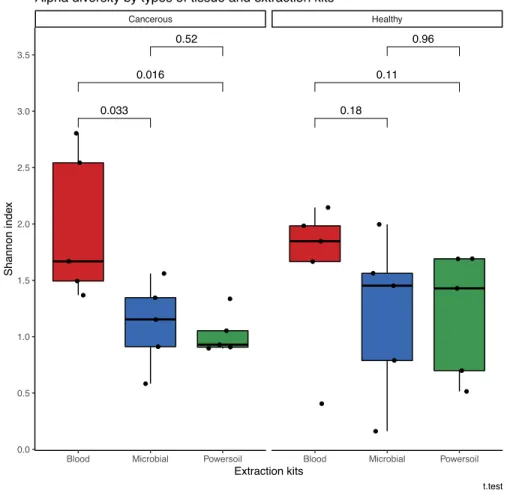
Figure 1.1.1 : Shannon’s alpha diversity of the tissue samples by type of tissue and extraction kit. ... 28
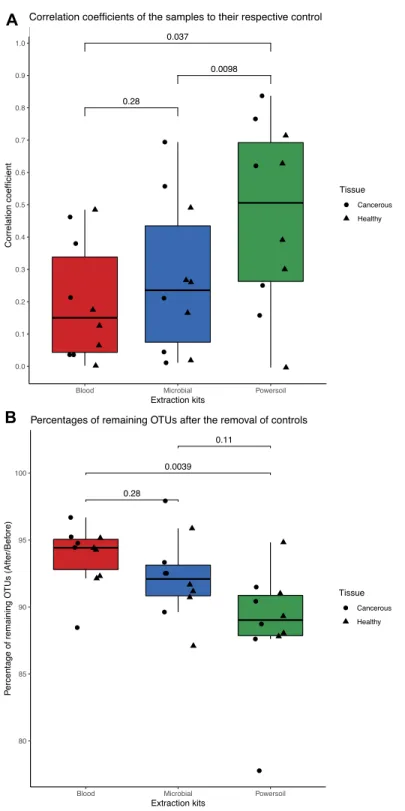
Figure 1.1.2 : A. Pearsons’s correlation coefficient between tissue samples and corresponding control. B. Percentage of remaining OTUs after the removal of controls. ... 29
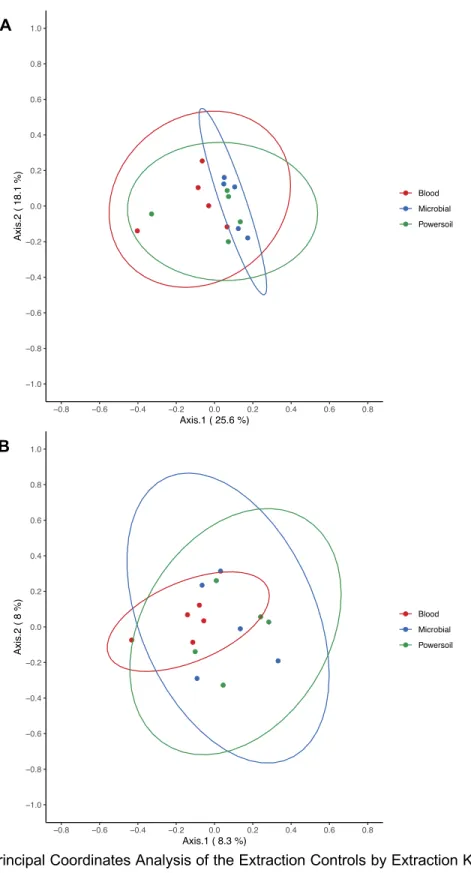
Figure 1.1.3 : Principal Coordinates Analysis of the Extraction Controls by Extraction Kits Based on Weighted (A) and Unweighted Bray-Curtis Distances (B). ... 31
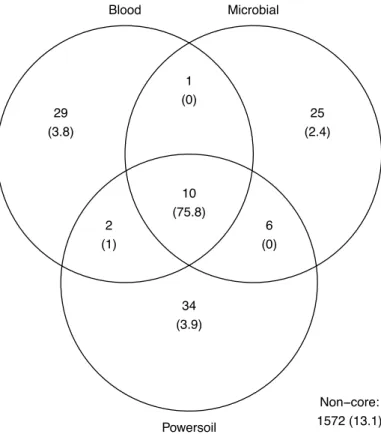
Figure 1.1.4 : Venn diagram of the core microbiota in the methodological controls. ... 32
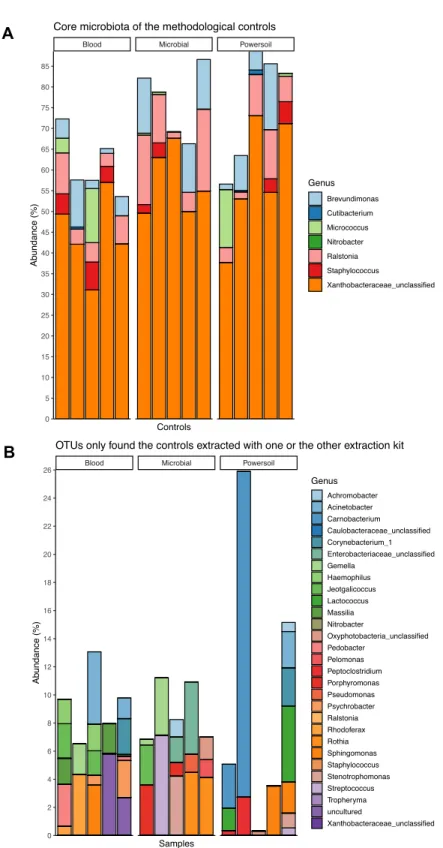
Figure 1.1.5 : Distribution of the relative abundance and taxonomic identification of OTUs found in the controls of every version of the pipeline ... 33
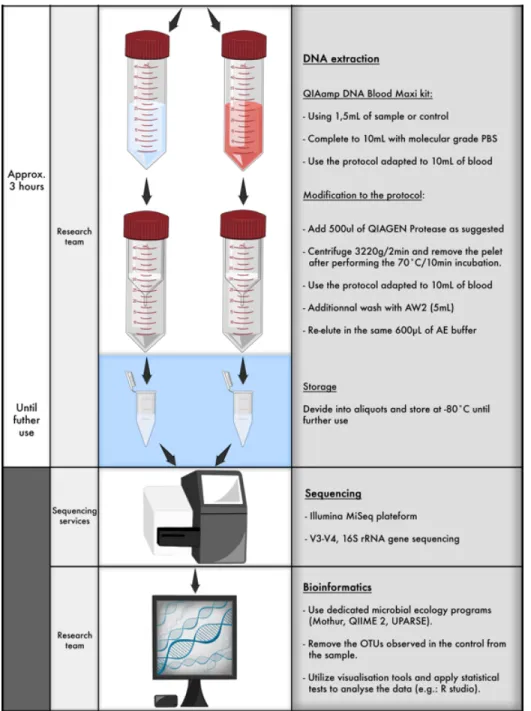
Figure 1.1.6 : Final bacterial DNA extraction and sequencing pipeline tailored to the needs and constrains of pulmonary microbiota ... 43
Figure 1.2.1 : Microbial DNA extraction optimization pipeline tailored to the needs and constrains of pulmonary microbiota. ... 45
Figure 1.2.2 : Detailed experimental design using microbial mock-communities. ... 46
Table 1.1 Patient’s clinical data ... 47
Figure 1.2.3 : Pulmonary regions defined from the origin of the lobe. ... 47
Table 1.2: 16S V3-V4 primer sequences ... 48
Table 1.3: Bacterial members of the mock communities ... 48
Figure 1.2.3 : Quantity of extracted DNA by amount of tissue treated. ... 49
Figure 1.2.4 : DNA purity (280 nm). ... 50
Liste des abréviations, sigles, acronymes
IUCPQ = Institut Universitaire de Cardiologie de Pneumologie de Québec
ITS = Internal Transcribed Spacer (Espaceur internet transcrit) LBA = Lavage bronchoalvéolaire
OTU = Operational Taxonomic Unit (Unité taxonomique Opérationnelle)
qPCR = Quantitative Polymerase Chain Reaction (Réaction en chaîne par polymérase quantitative)
Remerciements
Je tiens à remercier l’ensemble des organismes subventionnaires qui ont rendu possible la réalisation de ces travaux de maîtrise. Le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et les fonds de recherche du Québec – Santé (FRQS) pour les bourses d’étude, ainsi que le Réseau de recherche en Santé Respiratoire du Québec (RSR) et le CRSNG (Michael-Smith) pour un supplément de stage à l’étranger.
Merci aux membres de mon comité d’encadrement pour leur engagement au sein de ce processus, à Hernan Lorenzi pour m’avoir accueilli au sein de son laboratoire du J. Craig Venter Institute et à tous les membres du laboratoire Duchaine et de la biobanque de l’IUCPQ. Philippe Joubert, Marc Veillette, Christine Racine pour leur support, leur disponibilité et leur contribution essentielle à ces travaux.
Un merci spécial à Caroline Duchaine pour m’avoir montré à faire de la recherche humaine en valorisant l’esprit d’équipe, l’humilité et l’entraide. Pour m’avoir poussé à profiter de la vie, des opportunités qui s’offrent à nous et à répandre autour de nous une atmosphère positive, saine et bienfaisante. Mon passage dans ton laboratoire a consolidé mon amour pour la recherche et la manière dont je veux la pratiquer.
Je remercie également ma famille qui croit en moi depuis le début, qui me supporte sans relâche et qui me donne les moyens de mes ambitions. Merci à mes amis qui m’ont soutenu tout au long de ce parcours, qui m’ont changé les idées quand j’en avais besoin et qui l’ont rendu plus agréable. Je n’y serais jamais arrivé sans vous tous.
À toutes ces personnes qui ont parsemé mon parcours de près ou de loin et qui ne sont pas individuellement mentionnées dans ces remerciements, un grand merci. Votre contribution, bien que peut-être momentanée, à modifié ma trajectoire, m’a remis en question ou m’a permis d’aller plus loin.
Avant-propos
L’article inclus à ce mémoire a été soumis au journal Biology Communications en date du 4 juin 2020. La version présentée comprend les corrections demandées à la suite du processus de révision par les paires et a été transmise au journal le 21 septembre 2020. Aucune réponse supplémentaire de l’éditeur n’a été reçue au moment de présenter le document. Nathan Dumont-Leblond est le premier auteur de cet article et a été le contributeur principal à l’écriture, à l’analyse de données et à la production d’illustrations et de figures. Les coauteurs sont Marc Veillette, MSc et Christine Racine, MSc du Centre de recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ), Docteur Philippe Joubert, également affilié au Département de Biologie moléculaire, biochimie médicale et pathologie de l’Université Laval, ainsi que Caroline Duchaine, PhD affilié à l’IUCPQ et au Département de biochimie, de microbiologie et de bio-informatique de l’Université Laval.
Introduction
Le cancer du poumon
Le cancer du poumon est le plus répandu au Canada. Il est prévu qu’environ 29 800 Canadiens en recevront le diagnostic en 2020 et qu’environ un homme sur 14 et une femme sur 15 en seront diagnostiqués au cours de leur vie1. Les coûts associés au suivi médical et
au traitement de cette maladie («cost-of-illness») étaient estimés à plus de 75 000$ par patient en Ontario entre 2010 et 20152. De plus, 218 527 nouveaux cas de cancer du
poumon ou des bronches ont été répertoriés en 2015 aux États-Unis3. L’importance
épidémiologique et économique du cancer du poumon est donc majeure. Une compréhension des facteurs qui l’influencent est essentielle.
Le cancer du poumon se divise en deux grands groupes selon son apparence microscopique: le cancer du poumon à petites cellules et celui non à petites cellules. Ce dernier représente environ 80 à 85 % de l’ensemble des tumeurs pulmonaires malignes4.
Les adénocarcinomes, les carcinomes épidermoïdes et les carcinomes à grandes cellules sont les principaux membres du grand groupe des cancers non à petites cellules5.
Les adénocarcinomes pulmonaires
Les adénocarcinomes représentent le sous-type le plus souvent retrouvé de cancer pulmonaire (40 %)4,6. Ces cancers prennent naissance dans les cellules glandulaires
situées dans la partie distale des voies respiratoires5. Ainsi, les adénocarcinomes tendent à
apparaître en périphérie des poumons, bien qu’ils puissent également croître dans la partie centrale et tirer leur origine de l’épithélium bronchique central7. La grande majorité des
adénocarcinomes pulmonaires sont des tumeurs hétérogènes qui comportent plusieurs patrons architecturaux (lépidique, acinaire, papillaire, micropapillaire et solide), ce qui peut affecter leurs comportements biologiques et leurs réponses aux traitements8.
D’autre part, les adénocarcinomes présentent l’un des taux de mutations génétiques les plus élevés de l’ensemble des tumeurs solides humaines. Elles sont cependant légèrement moins mutées que les carcinomes épidermoïdes pulmonaires7. Ces perturbations du code
génétique modifient grandement le transcriptome des cellules cancéreuses de même que la nature et la quantité des récepteurs qu’elles expriment. Elles semblent ainsi acquérir un phénotype ressemblant davantage aux cellules épithéliales intestinales qu’aux cellules pulmonaires saines9.
Les carcinomes épidermoïdes pulmonaires
Les carcinomes épidermoïdes représentent également l’un des types de cancer du poumon les plus fréquents (25-30 %)4. Ces tumeurs prennent naissance dans les cellules
squameuses qui tapissent les bronches à la suite d’une irritation par des stimuli externes, dont la fumée de cigarette. Elles sont donc plus souvent retrouvées en région centrale et ont une légère prépondérance pour le poumon droit et les lobes supérieurs. La présence de nécrose et d’hémorragies à l’intérieur de la tumeur est également fréquente. De plus, ces tumeurs infiltrent souvent les tissus adjacents, tels l’œsophage, les vaisseaux sanguins majeurs (aorte, artère pulmonaire) ou le cœur7.
Les carcinomes épidermoïdes présentent un très haut niveau de mutations génétiques, ce qui affecte également leur transcriptome et leur morphologie. En effet, ils semblent présenter un phénotype ressemblant à celles des cellules tapissant les voies supérieures du tractus alimentaire, comme l’œsophage9.
Le microbiote pulmonaire
Le concept du microbiote humain a été introduit en 2001 par Dr Joshua Lederberg pour décrire la communauté microbienne commensale, symbiotique et pathogénique associée au corps humain10. La quasi-totalité des surfaces du corps humain est peuplée par un
consortium microbien diversifié, incluant le tractus digestif, respiratoire et la peau11–13.
Des efforts marqués ont été entrepris pour caractériser cette entité, entre autres, par la mise en place du Human Microbiome Project, une initiative concertée établie en 2008, visant à analyser le rôle du microbiote dans la santé humaine14,15. Celle-ci a été motivée par des
avancées importantes dans les technologies de séquençage de nouvelles générations, mais aussi par la réalisation commune que le métagénome bactérien participe activement au développement et au maintien de fonctions importantes, tel le système immunitaire16. Cinq
régions corporelles étaient initialement analysées dans le cadre de ce projet: les voies respiratoires supérieures (cavité nasale), la peau, la bouche, le tractus intestinal et la cavité vaginale16. Les voies respiratoires inférieures, incluant les poumons, n’y étaient pas
incluses17. En effet, celles-ci ont longtemps été considérées comme complètement stériles.
Ce dogme a cependant rapidement été révisé à la suite de l’analyse d’échantillons pulmonaires par séquençage. Il est maintenant confirmé que les poumons sont peuplés d’une communauté microbienne diversifiée, même en l’absence de maladies pulmonaires apparentes18. Par contre, de nouvelles preuves suggèrent que le microbiote est modifié
lorsque le poumon est affligé d’une pathologie, telle la maladie pulmonaire obstructive chronique19,20, l’asthme21, la fibrose pulmonaire idiopathique ou la fibrose kystique22. L’étude
du microbiote pulmonaire s’avère donc essentielle, bien qu’elle demeure un domaine en émergence23.
La composition bactérienne du microbiote pulmonaire
Les voies respiratoires humaines arborent environ 2,2 x 103 génomes bactériens par cm2 21.
Les bactéries qui les colonisent majoritairement appartiennent aux phylums Bacteroidetes,
Firmicutes, Proteobacteria et Actinobacteria24–27. De plus, Erb-Downward et coll. ont
observé, en utilisant des lavages bronchoalvéolaires (LBA), que le microbiote de la majorité (75 %) des poumons comprend les genres bactériens Pseudomonas, Streptococcus,
Prevotella et Fusobacterium. De plus, Haemophilus, Veillonella et Porphyromonas sont
retrouvés dans 50% de ces lavages20. Moris et coll. ont également observé une
prédominance des genres Streptococcus, Prevotella et Veillonella dans les échantillons de LBAs d’individus sains28. D’autre part, la population bactérienne retrouvée dans le poumon
varie fortement en fonction de sa profondeur dans le tractus respiratoire20. Une tendance
significative permettant de prédir la composition du microbiote en fonction de la localisation ne semble pas avoir pu être établie jusqu’à présent.
D’autre part, un core microbiota-ou microbiote de base, comprenant des espèces bactériennes constamment présentes dans le poumon, n’a pas été identifié chez l’humain29.
Il n’est pas non plus établi si les bactéries pulmonaires exécutent des fonctions métaboliques qui pourraient permettre la formation d’un microbiote de base, comme la dégradation de la mucine ou des carbohydrates dans le microbiote intestinal30,31. Bref, la
population bactérienne peuplant les poumons demeure peu connue. Un effort supplémentaire se doit donc d’être déployé pour en approfondir sa caractérisation.
Écologie microbienne pulmonaire
La composition du microbiote pulmonaire est déterminée selon trois facteurs majeurs : l’immigration, l’élimination et le taux de croissance des microorganismes dans les voies respiratoires. La modification du microbiote sain est donc causée par un changement d’au moins un de ces trois facteurs32–34. Au contraire, l’homéostasie pulmonaire est définie par
l’état de stabilité de l’environnement pulmonaire au moyen de mécanismes autorégulateurs34. L’homéostasie de la communauté microbienne pulmonaire s’établit donc
L’immigration bactérienne peut être initiée par l’inhalation directe de microorganismes présents dans l’air s’impactant sur les surfaces pulmonaires et migrant dans les voies respiratoires par l’intermédiaire des muqueuses. De plus, elle peut provenir de la microaspiration de sécrétions, sous forme d’aérosols, présents dans la bouche et le pharynx33. Le microbiote pulmonaire humain présente plusieurs homologies avec le
microbiote buccal, ce qui porte à croire que cette voie d’immigration est la plus importante35.
L’élimination des microorganismes peut, quant à elle, se produire par le transport mucociliaire, par la toux ou par l’effet du système immunitaire de l’hôte (immunité innée ou adaptative)33. D’autre part, un nombre important de facteurs physico-chimiques, dépendant
de la localisation dans le tractus respiratoire, influence les taux de croissance bactériens au sein du poumon. En effet, des variations importantes des conditions environnementales, tels la tension en oxygène, le pH, l’apport relatif en sang, la ventilation alvéolaire relative, la température, la structure épithéliale, la déposition de particules inhalées, ainsi que la concentration et le comportement des cellules immunitaires, sont observables d’un emplacement à l’autre32. La réaction inflammatoire médiée par le système immunitaire joue
également un rôle important dans la modification des facteurs écologiques qui gouvernent la population microbienne pulmonaire, entre autres, en favorisant l’apport de nutriments dans les poumons et en y modulant la virulence microbienne33,36–38.
L’immigration (dispersion) et l’élimination (dérive taxonomique) sont les forces écologiques majeures influençant la composition du microbiote pulmonaire sain. La multiplication microbienne a un impact limité sur le maintien de l’homéostasie dans le poumon en santé. Au contraire, en cas de pathologies pulmonaires, la reproduction microbienne, influencée par les conditions physico-chimiques locales, dicte la composition du microbiote34.
Les modèles de l’île adaptée, de l’homéostasie nutritionnelle et de la signalisation interdomaine ont été développés pour conceptualiser les forces induisant la colonisation, le maintien (homéostasie) et le débalancement (dysbiose) des communautés microbiennes qui résident dans les voies respiratoires34. Il est cependant à noter que celles-ci ne
considèrent majoritairement que les forces de sélection (déterministes), au détriment des forces neutres (non-déterministes) comme la dérive taxonomique et la dispersion passive. Sous une forte pression de sélection, l’utilisation du modèle d’assemblage des communautés de Sloan a démontré que seule une partie du microbiote est régi par les forces de sélection39,40. Une plus grande considération des forces neutres comme
contributeur au microbiote pulmonaire pourrait être nécessaire dans le développement d’un modèle global des dynamiques écologiques du microbiote pulmonaire.
Le modèle de l’île adaptée
Le modèle de l’île adaptée s’inspire de celui de MacArthur et Wilson41, basé sur l’observation
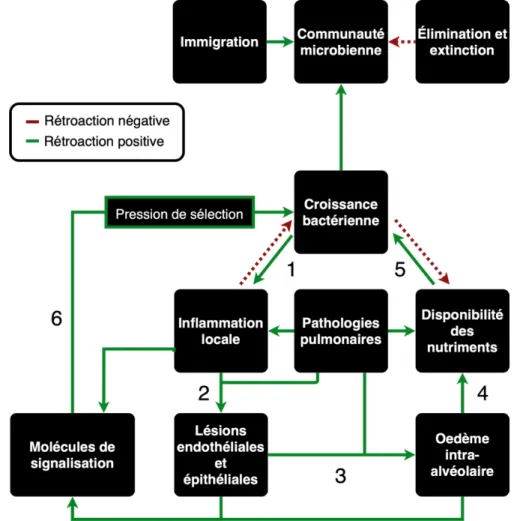
de la richesse biologique d’îles océaniques, et s’appuie sur les principes d’immigration et d’élimination/extinction préalablement mentionnés. La proximité à la source d’immigration, dans ce cas la bouche et les voies respiratoires supérieures, est le paramètre principal qui influence le ratio d’immigration. Plus l’emplacement étudié est près de cette source, plus la richesse microbienne qu’on y observe est élevée, puisque les microorganismes y sont plus facilement transportés. Moins les mécanismes d’élimination comme la toux, la clairance mucociliaire ou l’influence de l’immunité sont efficaces, plus il y aura accumulation de microorganismes (Figure I-1). Par principe de compétition et en fonction des capacités d’accueil de l’environnement pulmonaire, on s’attend également à ce qu’il y ait une plus grande chance qu’une espèce soit éliminée et remplacée si la richesse microbienne est plus élevée en condition d’homéostasie (un plus grand taux d’extinction). C’est la combinaison de ces facteurs qui détermine le niveau à l’équilibre de la population, et donc, le nombre d’espèces microbiennes (richesse) qui peut être continuellement observées (Figure I-1)34.
Homéostasie nutritionnelle
Bien que le modèle de l’île adaptée explique élégamment l’impact de l’immigration et de l’élimination microbienne et décrit les facteurs déterminants en contexte pulmonaire sain, il ne prend pas en considération l’effet de la reproduction microbienne; un déterminant crucial de dysbiose en condition pathologique. Ainsi, le modèle d’homéostasie nutritionnelle conceptualise l’influence de la présence de nutriments et de leurs origines sur la croissance microbienne.
Selon ce modèle, les bactéries qui colonisent le tractus respiratoire sont chimiohétérotrophes34. Elles utilisent les composés organiques présents dans le poumon
comme source d’énergie et de carbone. La faible abondance de nutriments dans la lumière pulmonaire en absence de pathologie ne permet pas leur prolifération soutenue. Cependant, lors de maladies pulmonaires, comme la fibrose kystique, les bronchites chroniques, l’asthme ou la maladie pulmonaire obstructive chronique (MPOC), un mucus dense et protéique s’accumule dans les poumons. Ainsi, le passage à l’état pathologique s’accompagne par l’augmentation de la quantité de nutriments disponibles dans l’espace
pulmonaire et de la croissance bactérienne (Figure I-1). Cette forte présence de bactéries se traduit par une augmentation de l’inflammation locale et de la réponse immunitaire. Cette dernière peut limiter la croissance bactérienne et rétablir l’homéostasie (Figure I-1, 1) ou, lorsque le système immunitaire est surchargé, provoquer des lésions endothéliales et épithéliales (Figure I-1, 2) et de l’œdème intra-alvéolaire (Figure I-1, 3). Ces lésions et œdème libèrent alors davantage de nutriments dans la lumière pulmonaire et favorisent la croissance bactérienne (Figure I-1, 4-5). En contrepartie, les bactéries en croissance utilisent les nutriments du milieu, ce qui diminue leur quantité disponible et régule négativement leur vitesse de croissance (Figure I-1, 5)34.
Figure I-1 : Schéma récapitulatif des facteurs influençant la composition de la communauté microbienne pulmonaire
Signalisation interdomaine
Le modèle de signalisation interdomaine prend également en considération les molécules de signalisations humaines tels les hormones, les neurotransmetteurs et les cytokines qui peuvent influencer la croissance bactérienne des membres du microbiote pulmonaire. Plusieurs publications ont démontré des impacts variés de ces différentes molécules sur les espèces Streptococcus pneumoniae42, Pseudomonas aeruginosa43, Staphylococcus
aureus44, Klebsiella pneumoniae (Freestone et al. 1999) et Escherichia coli46.
Le modèle de signalisation interdomaine est basé principalement sur l’interaction entre P.
aeruginosa, cause majeure des pneumonies nosocomiales, et les catécholamines,
médiateurs principaux de la réponse aux stress. Puisque la réponse aux catécholamines est variable d’une espèce bactérienne à l’autre et qu’elle favorise la croissance de P.
aeruginosa, leur présence dans le poumon peut mener au débalancement du microbiote
pulmonaire46–48. L’augmentation de l’inflammation pulmonaire, pouvant être causée par la
croissance microbienne elle-même, induit la production de ces catécholamines par les macrophages et neutrophiles37. Ce phénomène crée une boucle de rétroactivation positive
menant à la croissance bactérienne sélective de P. aeruginosa (Figure I-1, 6).
Les catécholamines ne sont qu’un exemple de molécules de signalisation qui pourraient indure ou faciliter le débalancement de la communauté microbienne résidente des poumons. L’influence d’autres molécules est à explorer34.
Sommairement, le corps humain module la composition du microbiote pulmonaire, entre autres, par les processus de migration, d’élimination/extinction et de croissance microbienne. À l’inverse, le microbiote pulmonaire influence les réponses physiologiques du corps et profite du débalancement nutritionnel de l’environnement pulmonaire.
Le microbiote et le cancer
L’année 2017 a été marquée par une avancée importante dans la compréhension des relations s’établissant entre les cellules cancéreuses et les microorganismes qui colonisent le corps humain. En effet, à la manière des poumons, longtemps perçus comme stériles, les tumeurs ont été observées comme étant également capables de développer des associations avec des espèces bactériennes. Celles-ci pourraient favoriser leur croissance, leur résistance aux traitements contre le cancer et leur migration (processus métastatique)49,50. Ces observations ont été faites pour le développement du cancer
colorectal et pancréatique50,51. Cependant, un profil bactérien différent a aussi été observé
entre les tissus cancéreux et les contrôles dans plusieurs autres organes, dont le sein52 et
la vessie53, ce qui laisse présager que ce genre d’association tumeur-microorganisme est
répandue chez l’humain.
Un nombre très limité d’études ont tenté de démontrer cette modification de la flore bactérienne en fonction du type de tissu (cancéreux vs sain) dans le cancer pulmonaire. Une d’entre elles, menée par Liu et coll., a utilisé le séquençage du gène codant pour l’ARNr 16S pour identifier les bactéries totales de brossages bronchoscopiques de 24 patients cancéreux54. Ils ont observé une diminution significative de la diversité alpha (nombre
d’espèces présentes) des échantillons de brossages tumoraux par rapport aux contrôles sains. De plus, les bactéries du genre Streptococcus y sont plus abondantes, alors que celles du genre Staphylococcus y sont moins présentes qu’à la surface de tissus sains. Ces résultats sont cependant basés sur un faible nombre d’échantillons et peu de considération semble avoir été portée aux possibles contaminants de la méthode. Il n’y a, en aucun cas, mention de contrôles d’échantillonnage, d’extraction ou de séquençage.
En revanche, une étude s’est attardée à l’analyse du microbiote intratumoral pulmonaire chez l’humain. Celle-ci, entreprise par Yu et coll., compare 31 spécimens de tissus cancéreux dont l’ADN a été extrait et dont le gène codant pour l’ARNr 16S a été amplifié et séquencé26. Une attention particulière a été apportée aux contrôles dans cette étude. Elle a
également observé une diminution de la diversité alpha des échantillons cancéreux. De plus, les tissus cancéreux de type adénocarcinome y présentent une plus grande diversité phylogénétique, une abondance relative plus élevée des bactéries du genre Thermus et une plus faible du genre Ralstonia que ceux de type carcinome épidermoïde. Aucune différence significative n’a cependant été observée en fonction de la localisation de la tumeur. Des analyses subséquentes ont également relevé une abondance élevée des bactéries du genre
Legionella dans les cas métastatiques, suggérant qu’elles pourraient avoir un rôle à jouer
dans la cancérogenèse. Une fois de plus, des investigations supplémentaires sont nécessaires pour confirmer ces résultats qui ont été obtenus avec un nombre relativement faible de spécimens et un protocole d’analyse nouvellement appliqué à l’étude du microbiote pulmonaire. En bref, peu d’informations sur l’importance du microbiote pulmonaire dans le développement du cancer sont présentement disponibles, bien qu’il s’agisse d’une avenue prometteuse.
Nature des interactions entre le microbiote et le cancer
La nature des interactions entre le microbiote et le cancer est diverse et complexe. Peu d’études se sont penchées directement sur le microbiote tumoral pulmonaire. Cependant, les phénomènes microbiens en enjeux dans d’autres types de cancer pourraient potentiellement s’y produire. Un état d’inflammation récurrent, la production de toxines génotoxiques, la modification de métabolismes hormonaux et la dysbiose générale du microbiote en sont les principaux.
Les bactéries composant le microbiote peuvent induire un état d’inflammation chronique qui favorise la cancérogenèse. La production de toxine pro-inflammatoire par Bacteroides
fragilis55,56 ou de dérivés réactifs de l’oxygène57, ainsi que la modulation des routes de
signalisation métabolique (Fusobacterium nucleatum) participent à la création de cet environnement inflammatoire et cancérogène58. D’autre part, les bactéries Salmonella
Typhi59 et Helicobacter spp.60 sont en mesure d’induire directement le cancer de la vésicule
biliaire et Helicobacter pylori le cancer de l’estomac chez l’humain. Bien que la cancérogenèse est souvent considérée comme un effet secondaire de l’inflammation locale chronique, ces bactéries peuvent avoir un effet génotoxique direct et perturber les voies métaboliques régulant la prolifération des cellules mucosales12,61. La production de toxines
génotoxiques, comme la colibactine chez Escherichia coli et la toxine cytholétale distendante chez Campylobacter jejuni62,63, a également été identifiée comme pouvant
mener à la formation de cancers colorectal chez la souris. La modification des profils hormonaux par les bactéries, tels ceux de l’estrogène et des phytoestrogènes, est aussi potentiellement impliquée dans l’apparition du cancer du sein64. Au contraire, un
débalancement plus global du microbiote (dysbiose) ou l’absence de certaines espèces bactériennes pourrait mener au développement du cancer, comme lors de l’utilisation répétée d’antibiotiques65,66. Les mécanismes entrainant ces phénomènes sont peu compris,
bien que considérablement sous étude à l’heure actuelle.
L’impact visiblement important du microbiote sur le cancer ne s’arrête pas à son développement, mais également à son traitement. Le microbiote bactérien peut modifier la structure chimique des agents chimiothérapeutiques, ainsi que leur concentration locale dans la tumeur67–69. Il en influence ainsi leur efficacité. Le microbiote intestinal module
également la réponse au traitement immunothérapique de type inhibiteur de point de contrôle (Checkpoint blockade)70. Ainsi, plusieurs études tentent de démystifier le
nouvelles avenues thérapeutiques. Des efforts de recherche sont actuellement déployés pour déterminer la composition du microbiote optimale pour l’obtention de réponses thérapeutiques efficaces12. Notamment, dans l’optimisation des traitements
immunothérapique ciblant le couple PD-1/PD-L170–72.
Méthodologies et limites de l’étude du microbiote pulmonaire
Plusieurs facteurs restreignent notre capacité à étudier le microbiote pulmonaire. Les défis méthodologiques de l’analyse de ce microenvironnement dépassent ou, du moins, diffèrent grandement de ceux rencontrés lors de l’étude de populations plus volumineuses comme le microbiote intestinal. La nature et l’abondance du microbiote pulmonaire, les limitations cliniques, le recrutement de patients et l’accès aux échantillons, ainsi que les limites des méthodes de détection et de quantification en sont les principaux.
Abondance du microbiote pulmonaire et gestion des biais
Le microbiote pulmonaire contient une charge microbienne grandement inférieure à d’autres microenvironnement du corps humain, tel le tractus digestif73. La description des
communautés microbiennes de faibles densités est plus sujette à être biaisée par l’introduction de contaminants ou par des choix méthodologiques74. En raison des limites
importantes dans l’échantillonnage du poumon et de l’absence de méthode d’analyse standardisée, une grande variabilité entre les résultats obtenus par les différentes études est observable. En effet, la disparité entre les méthodes analytiques utilisées rend la comparaison d’études difficile, puisqu’elles sont susceptibles d’introduire des biais distincts75. Ainsi, de plus grandes précautions doivent être prises pour s’assurer que les
microorganismes détectés sont bel et bien présents dans le tissu pulmonaire, et non pas introduits par la méthode76, et que la composition détectée est représentative de la réalité.
Une stratégie stricte de gestion des contaminations doit être mise en place à chaque étape du protocole77 et, les biais méthodologiques, pris en considération dans le devis
expérimental.
Recrutement de patients
Bien que des facteurs méthodologiques puissent avoir un impact important sur la composition détectée du microbiote, il est primordial de s’assurer que la communauté microbienne de l’individu sélectionné est adéquate pour répondre aux questionnements scientifiques. En ce sens, l’individu ou la cohorte dont on étudie le microbiote se doit d’être sélectionné avec soins afin que les facteurs connus pour influencer la composition du
microbiote, au-delà des variables de l’étude, soient pris en considération. Sans ces précautions, la variabilité interindividuelle du microbiote pourrait surpasser celle causée par les conditions expérimentales établies.
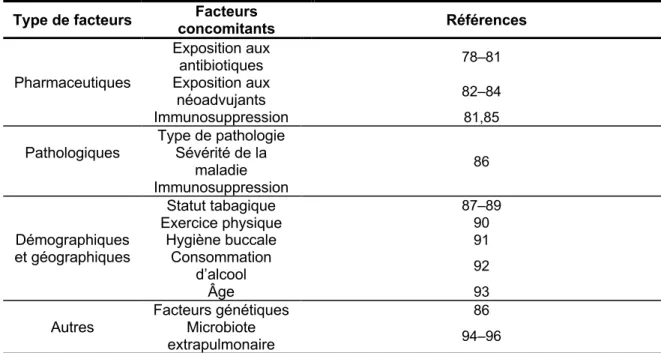
Le microbiote pulmonaire étant toujours peu décrit, les déterminants cliniques de sa composition ne sont pas bien compris. Il est cependant possible d’inférer leurs effets sur le microbiote pulmonaire en les comparant à d’autres microenvironnements humains comme le tractus digestif et à partir de notre compréhension des acteurs écologiques en jeu. Parmi ceux-ci, on compte, entre autres, des facteurs pharmaceutiques, pathologiques, démographiques et géographiques, ainsi que génétiques et associés au microbiote extrapulmonaire (Tableau I-1).
Tableau I-1 : Facteurs concomitants qui pourrait modifier la composition du microbiote pulmonaire.
Type de facteurs concomitants Facteurs Références
Pharmaceutiques Exposition aux antibiotiques 78–81 Exposition aux néoadvujants 82–84 Immunosuppression 81,85
Pathologiques Type de pathologie Sévérité de la 86
maladie Immunosuppression Démographiques et géographiques Statut tabagique 87–89 Exercice physique 90 Hygiène buccale 91 Consommation d’alcool 92 Âge 93 Autres Facteurs génétiques 86 Microbiote extrapulmonaire 94–96
Les facteurs pharmaceutiques incluent la prise d’antibiotiques, de traitements oncologiques néoadvujants et d’immunosuppresseurs. Les antibiotiques comme la ciprofloxacine, la clindamycine, l’amoxicilline et la minocycline sont connus pour profondément modifier la composition du microbiote intestinal78–80. Administrée oralement, l’amoxicilline induit une
modification rapide (24 h) de la composition du microbiote de l’intestin. Après 60 jours, sa composition tend à retrouver son état original, sans toutefois y parvenir totalement (89 % de similarité), laissant présager un effet perturbateur profond et partiellement réversible. Ces effets étaient également variables d’un patient à l’autre puisque certains patients semblaient revenir plus difficilement à leur état d’origine97. L’impact de l’administration d’antibiotiques
sur le microbiote semble cependant variable d’une région corporelle à l’autre. En effet, les communautés microbiennes peuplant la salive et la gorge leur paraissent plus résistantes78,80. Comme ces environnements sont présumés être les sources principales
d’immigration microbienne vers les voies respiratoires inférieures98,99, une résistance
similaire du microbiote qui peuple ces dernières est plausible. En absence de preuves tangibles sur la résistance ou la sensibilité aux antibiotiques du microbiote pulmonaire, user de prudence et exclure les patients ayant suivi un traitement antibiotique semble approprié. Parallèlement, l’utilisation de traitements néoadjuvants (irinotécan, 5-Fluorouracil (5-FU), capécitabine, méthotrexate, chlorambucil) a été identifiée comme pouvant modifier la composition du microbiote intestinal et causer des effets secondaires indésirables telle que la diarrhée82. Plusieurs études ont corrélé le taux de succès de ces traitements à la
composition du microbiote intestinal et exploré l’impact de sa modification sur l’efficacité des traitements83,84. Un effet similaire n’a cependant pas encore été décrit dans le cas du cancer
pulmonaire, bien que probable. Retirer ces patients d’études n’abordant pas directement l’utilisation de traitements néoadjuvants est préférable.
Les facteurs pathologiques englobent les caractéristiques intrinsèques des pathologies pulmonaires, ainsi que leur stade de progression. Les différents types de pathologies pulmonaires peuvent concrètement modifier la composition de microbiote, en favorisant plus ou moins l’inflammation et l’apport de nutriments vers la lumière et/ou le parenchyme pulmonaire. Les différents stades de progression de ces maladies peuvent aussi influencer ces mêmes variables écologiques du poumon86. De plus, la présence de comorbidités est
susceptible d’exacerber les symptômes de la maladie primaire ou de mener à d’autres dérèglements systémiques influençant le microbiote. Une sélection stricte des pathologies pulmonaires et de leurs comorbidités dans une cohorte à l’étude est cruciale.
Les facteurs démographiques et géographiques incluent le mode de vie et les habitudes de vie d’un individu, tels le statut tabagique, l’exercice physique, l’hygiène buccale, la consommation d’alcool et l’âge. La consommation de produits du tabac chez la souris accroit significativement la proportion de bactéries potentiellement pathogènes et qui induisent de l’inflammation87–89. Chez l’humain, le tabagisme pourrait avoir un impact direct
sur la composition du microbiote respiratoire, entre autres, par l’altération de la réponse immunologique pulmonaire100. Cependant, bien qu’une modification du microbiote des voies
respiratoires supérieures de fumeurs ait été quantifiée, aucune différence significative entre le microbiote pulmonaire d’individus fumeurs et non-fumeurs n’a pu être observée33,99,101. Il
est cependant possible que le tabagisme influence les populations microbiennes de façons plus subtiles dans les voies inférieures que supérieures et que ces effets soient ainsi plus difficiles à détecter99. La pratique de l’activité physique a aussi été observée comme pouvant
modifier la composition du microbiote intestinal90. Ceci laisse présager qu’un patient actif
pourrait présenter une signature bactérienne pulmonaire différente d’un individu sédentaire. De plus, l’hygiène buccale et la consommation d’alcool modifient la composition du microbiote oral91,92. Il pourrait, à son tour, modifier le microbiote des voies respiratoires
inférieures puisqu’il en est considéré comme la source primaire de la colonisation98,99.
Les facteurs génétiques, comme le polymorphisme de régions régulant la défense immunitaire et la clairance mucociliaire (TOLLIP, MUC5B, CFTR, etc.) peuvent également être à l’origine ou contributeurs au développement, ou exacerber certaines pathologies pulmonaires. Sans pour autant causer un état pathologique, ces polymorphismes peuvent aussi causer une variation interindividuelle de l’état d’homéostasie microbienne dans le poumon86.
La présence et la composition du microbiote qui colonise d’autres régions corporelles peuvent également influencer celle du poumon. Il existe, entre autres, une voie de communication bidirectionnelle entre le microbiote intestinal et pulmonaire; l’axe « intestin-poumon ». Celle-ci est médiée par la production de métabolites microbiens et leur diffusion dans le corps humain par le système circulatoire102, ainsi que par la modulation du système
immunitaire94–96. La santé pulmonaire peut modifier la composition microbienne intestinale
et, à l’inverse, le microbiote intestinal influence la santé pulmonaire 103. Bien que difficilement
contrôlable sans la mise en place d’échantillons fécaux, il s’agit d’une considération pertinente.
Suite aux avancements de la recherche sur les associations entre les microorganismes et le corps humain, il est plus que probable que d’autres facteurs influençant la composition des communautés microbiennes commensales soient identifiés. Cependant, les technologies actuellement employées pour leur étude ne permettent pas la discrimination d’un si grand éventail de facteurs. Ainsi, dans la mesure du possible, effectuer une sélection approfondie des patients en fonction de ces critères est essentiel afin de limiter la variabilité interindividuelle induite par d’autres variables que celles à l’étude. À défaut de pouvoir contrôler tous ces facteurs, leur prise en considération dans l’analyse des résultats et dans l’élaboration de conclusions est essentielle.
Échantillonnage
La capacité d’échantillonnage du microbiote pulmonaire est un frein majeur à l’expansion de ce domaine de recherche. En effet, en comparaison avec le microbiote intestinal, aucune biomasse microbienne n’est excrétée par le corps. Il est donc nécessaire d’utiliser des méthodes d’échantillonnage plus invasives et complexes afin de prélever des échantillons du parenchyme pulmonaire.
Naturellement, il n’est pas éthiquement acceptable de retirer à un individu sain un fragment de poumon afin d’en analyser la composition bactérienne. Plusieurs autres alternatives moins invasives ont donc dû être développées, dont l’utilisation d’expectorations, de lavages broncho-alvéolaires (LBA) et de brossages bronchoscopiques29,75. Les LBAs consistent à
injecter et récupérer une saline stérile dans les voies respiratoires distales et dans le compartiment alvéolaire d’un patient adéquatement anesthésié. Le liquide transitant dans les lumières bronchiolaires et dans les alvéoles capture des microorganismes à leurs surfaces ou présents dans le mucus des voies respiratoires. Les brossages bronchoscopiques, tel que leur nom l’indique, correspondent au brossage de la surface des voies respiratoires à l’aide d’une petite brosse de plastique et à l’élution de ce qui y a adhéré. Ces types de prélèvement sont cependant très sensibles aux contaminations par le système respiratoire supérieur, ce qui pourrait apporter un biais sur la population bactérienne observée. En effet, les appareillages de prélèvement se doivent de transiter par la bouche et le pharynx avant d’accéder aux poumons. De plus, l’utilisation de LBA et de brossages broncoscopiques nécessitent une procédure médicale comportant un risque pour le patient29. Cette difficulté d’échantillonnage limite donc le nombre de spécimens qu’il est
possible de récolter et rend l’obtention de résultats statistiquement valables plus ardue20,75.
L’utilisation de biopsies de poumon pourrait cependant limiter la contamination orale des échantillons75. De plus, l’utilisation et l’analyse de tissus pulmonaires pourraient permettre
la détection de genres bactériens fermement adhérés à la surface pulmonaire. Puisque les bactéries ont été observées comme pouvant migrer dans le tissu pancréatique cancéreux51,
un phénomène similaire survient probablement dans le cancer pulmonaire. Échantillonner le tissu en entier présente l’avantage d’analyser à la fois le microbiote à la surface, mais aussi potentiellement dans le tissu.
L’excision d’une partie ou d’un poumon entier (lobectomies simples/multiples ou pneumonectomie) est un traitement largement utilisé pour les cas de cancers pulmonaires. Ainsi, bien que l’utilisation de biopsies puisse sembler plus invasive que les techniques
préalablement mentionnées, un fragment de l’organe retiré chez un individu peut être échantillonné sans nécessiter une intervention médicale supplémentaire chez le patient. Bref, l’utilisation de tissus pulmonaires complets issus de résections pulmonaires semble une alternative réaliste et avantageuse.
Contexte et réalité clinique
Les milieux cliniques, incluant les salles opératoires et les départements de pathologie, ne sont pas conçus et n’implémentent pas d’emblée les précautions nécessaires pour l’étude du microbiote humain. Naturellement, les objectifs principaux sont la sécurité et le bien-être du patient, ainsi que l’exactitude diagnostique. Dans la majorité des cas, les caractéristiques microbiologiques des spécimens chirurgicaux ne sont pas considérées, puisqu’on s’intéresse plutôt aux détails histologiques.
Dans une optique de prévention des infections, les salles opératoires et leur personnel mettent cependant en place des procédures rigoureuses de stérilité qui limitent l’introduction de microorganismes exogènes chez le patient. Ces dispositions restreignent ainsi la possibilité de contamination des tissus jusqu’à leur manipulation à l’extérieur du corps. De plus, des protocoles et régulations strictes régissent le fonctionnement des salles opératoires. Le personnel y est coordonné et fortement affairé. Le matériel qui y est utilisé est spécifique et ne peut être facilement modifié. Ainsi, le contrôle sur le déroulement des procédures chirurgicales de toute équipe de recherche voulant récolter des tissus humains est limité. Les protocoles de prélèvements se doivent d’être conçus en fonction de ces contraintes.
Les laboratoires de pathologies sont, entre autres, en charge de la gestion des tissus qui sont retirés des patients, par exemple, un lobe pulmonaire, un segment d’intestin ou une biopsie mammaire, de leur dissection et de leur préparation à l’analyse pour confirmation de diagnostic. Puisqu’aucun patient n’est directement en contact avec ce laboratoire ou son personnel, très peu de précautions microbiologiques y sont prises. De plus, il peut facilement s’agir d’un environnement fortement contaminé, puisque des échantillons d’origine intestinale y sont manipulés. Cependant, bien que contraintes par le temps, les procédures pathologiques sont plus flexibles et peuvent permettre l’implémentation de précautions microbiologiques nécessaires dans le cadre d’études du microbiote.
En outre, le personnel hospitalier impliqué dans les chirurgies et la manipulation/préparation des tissus au département de pathologie (chirurgiens/ennes, infirmiers/ères, techniciens et pathologistes), travaille sur des horaires en rotations. S’assurer que les procédures établies soient appliquées conformément peut donc devenir un défi de taille, puisque plusieurs acteurs différents interviennent dans une même étape du protocole expérimental. Établir une instance de coordination, tel le laboratoire de pathologie, la biobanque de l’établissement ou un acteur externe impliqué dans la recherche clinique, permet d’assurer un suivi de la conformité des manipulations. Celles-ci se doivent cependant de demeurer simples et réalistes afin de perturber le processus clinique le moins possible et d’augmenter les probabilités que le personnel adhère aux directives.
Méthodes de détection et de quantification
L’étude du microbiote pulmonaire a été fortement influencée par le développement de nouvelles méthodes permettant la détection et la quantification des microorganismes, passant des méthodes de culture traditionnelles aux méthodes moléculaires de haut-débit aujourd’hui employées.
Les méthodes de culture microbiologique dites « traditionnelles» consistent à mettre les échantillons contenant les microorganismes en contact avec un milieu nutritif et en condition de croissance optimale pour permettre leur multiplication (température, taux d’oxygène, luminosité, etc.). L’étude du microbiote pulmonaire par les méthodes de culture a été longtemps de faible intérêt, puisque, comme pour la plupart des régions corporelles, les microorganismes colonisant le corps humain étaient perçus comme bénins et sans effets majeurs sur la santé104. Quelques études se sont cependant penchées sur la détection et
l’identification des agents bactériens responsables de dérèglements pulmonaires comme les pneumonies105. L’efficacité des méthodes de culture est cependant limitée par les
besoins nutritionnels et environnementaux des microorganismes. En effet, il est extrêmement difficile de répondre à l’ensemble de ces exigences au sein d’un même protocole expérimental, entre autres, par manque de temps et/ou de ressources matérielles et humaines. Les méthodes de culture tendent donc à sous-estimer la diversité réelle des environnements analysés au profit de genres microbiens peu exigeants. Elles peuvent cependant détecter efficacement les genres bactériens présents en très faible proportion dans une communauté106.
Les méthodes moléculaires, quant à elles, amplifient et séquencent les acides nucléiques des microorganismes de manière à les dénombrer ou les identifier. Elles nécessitent cependant l’extraction et la purification de ces acides. Les méthodes moléculaires ont l’avantage important de permettre la détection de microorganismes ne pouvant être cultivés puisqu’elles sont basées sur l’amplification de leurs acides nucléiques par des réactions biochimiques contrôlées, plutôt que par leur multiplication cellulaire. L’étude du microbiote pulmonaire a pris rapidement de l’ampleur et de l’intérêt avec l’avènement de ces méthodes moléculaires et, plus précisément, du séquençage de nouvelle génération. Celles-ci ont permis de mettre en évidence l’abondance et la diversité des microbiotes. De plus, l’importance du microbiote sur la santé humaine dans plusieurs autres régions corporelles a contribué au changement des paradigmes de la stérilité pulmonaire76. Par contre, la faible
abondance microbienne du microbiote pulmonaire rend ces méthodes plus difficilement applicables, puisque leur grand pouvoir de détection est encore plus sujet à l’incorporation de biais et de contaminants, principalement lors des étapes de traitement des échantillons et d’extraction d’ADN.
Les méthodes d’extraction et de purification de l’ADN bactérien utilisées ont été répertoriées comme ayant un impact significatif sur la communauté microbienne détectée dans divers environnements107–109. La présence de types bactériens plus difficile à lyser, comme les
bactéries à Gram positif110, peut aussi diminuer notre capacité à les récupérer et favoriser
la détection de genres plus facilement lysables. De plus, la présence d’une variété d’inhibiteurs (ions, polysaccharides, etc.) dans les échantillons peut limiter la capacité de récupération d’ADN bactérien. Chaque protocole se soit donc d’être adapté à la matrice des échantillons étudiés111. L’extraction d’ADN bactérien des tissus pulmonaires pose
cependant un important défi, puisque sa matrice tissulaire qui renferme les bactéries est difficilement détruite. Elle est constituée de 5-10 % d’élastine et de 10-20 % de collagène, ce qui la rend hautement élastique112. Les trousses commerciales d’extraction d’ADN
régulièrement utilisées dans le contexte du microbiote humain renferment également plusieurs contaminants microbiens qui peuvent artificiellement augmenter la diversité des échantillons113. Puisque l’étude de communautés de faible abondance est plus sujette à être
biaisée par ces contaminants, une attention particulière doit être prise pour limiter leur incorporation lors de l’étude du microbiote pulmonaire76. Le contrôle systématique des
Les méthodes de séquençage de nouvelles générations sont diverses et évoluent rapidement. La plateforme Illumina Miseq combinée à l’analyse d’amplicons du gène codant pour l’ARNr 16S est cependant l’une des plus utilisées dans le cadre d’études d’écologie microbienne114. Son faible taux d’erreur, sa relativement grande capacité de production de
séquences (de grandes longueurs, 300-350nt), sa facilité d’utilisation et son faible coût d’utilisation, lorsque combinés à une stratégie de double indexage, sont ses principaux avantages115,116. Plusieurs études s’étant déjà intéressées au microbiote pulmonaire ont
utilisé cette plateforme26,54,117. Au contraire, les méthodes métagénomiques s’appliquent
difficilement à l’étude du microbiote pulmonaire. L’abondance d’ADN de l’hôte provenant des cellules pulmonaires des échantillons biopsiques ajoute une pression supplémentaire sur l’efficacité et la sensibilité de la méthode de détection puisque l’ADN de l’hôte compétitionne avec l’ADN bactérien pour son hybridation aux amorces utilisées dans les réactions de séquençage. Les méthodes métagénomiques sont également plus couteuses118.
Les méthodes moléculaires détectent et séquencent souvent une seule partie du génome microbien; un marqueur. Pour l’analyse bactérienne de communautés microbiennes, le gène codant pour l’ARNr 16S est le plus utilisé. Cependant, en raison des longueurs maximales de séquençage des différentes plateformes, il n’est pas possible de le séquencer dans sa totalité. Il est donc nécessaire de sélectionner une ou plusieurs régions adjacentes de ce gène. Elles sont nommées : régions variables (V). Il n’y a présentement pas de consensus concernant la région variable à analyser dans la cadre d’analyse du microbiote111,119. Elle se doit cependant d’être constante entre les différentes études afin de
permettre leur comparaison et devrait être sélectionnée avec grande attention.
Les données issues des méthodes de séquençage à haut débit sont extrêmement volumineuses et se doivent d’être traitées par des logiciels spécialisés tels que Mothur120.
Elles sont d’abord organisées en fonction de leur échantillon d’origine et subissent plusieurs étapes de filtration afin que seules les séquences jugées suffisamment de bonne qualité soient conservées. Les séquences sont ensuite regroupées en fonction de leur similarité de séquence (seuil= 97%, 99% ou 100%) en unités taxonomiques opérationnelles (OTU) ou en variant de séquence d’amplicon (ASV). Ces regroupements peuvent être alors comparés directement en fonction des variables expérimentales associées aux échantillons, ou préalablement combinés en fonction de leur identification taxonomique121.
Gestions des contaminants
L’incorporation d’échantillons contrôles («No-template control)» est une pratique fortement encouragée dans le cadre d’études de diversité microbienne111. Elle permet habituellement
d’estimer la nature et la teneur des contaminants qui sont introduits dans les échantillons par la méthode expérimentale. Il n’y a cependant pas de consensus quant à savoir que faire lorsque des contaminants sont détectés dans ces contrôles111. Le retrait total de tous les
OTUs présent dans les contrôles des échantillons ne semble pas toujours approprié, puisque ces OTUs peuvent être également présents naturellement chez ces derniers113.
Ainsi, certains groupes de recherche ont tenté d’utiliser un modèle statistique neutre (Venkataraman et al. 2015), ou même de prendre en considération des données issues d’analyse de réaction en chaîne par polymérase quantitative (qPCR), pour rectifier proportionnellement les résultats obtenus dans les échantillons de microbiote122. Ces
méthodes semblent cependant présenter des lacunes importantes. Le développement de méthodes permettant la gestion des contaminants demeure nécessaire.
Objectifs et mise en contexte
Le présent projet s’inspirera des études précédentes afin d’établir des bases solides pour la poursuite de l’étude du microbiote pulmonaire et l’approfondissement des connaissances sur le microbiote tumoral. Un protocole expérimental permettant l’évaluation de la diversité et de l’abondance relative du microbiote des tissus tumoraux par séquençage visant le gène codant pour l’ARNr 16S sera mis sur pieds. Celui-ci sera adapté aux contraintes de ce type d’environnement et une attention particulière sera portée aux contrôles des sources de contaminations.
Chapitre 1 : Développement d’un protocole robuste
pour la caractérisation du microbiote
pulmonaire
1.1 Résumé
L’absence de standardisation méthodologique et de considérations pour les contraintes de l’environnement pulmonaire diminue la validité des résultats obtenus et des conclusions qui en sont tirées. Ainsi, nous désirons valider une marche à suivre complète de séquençage d’amplicons du gène de l’ARNr 16S, allant du recrutement de patients aux analyses bio-informatiques. Nous avons minimisé l’incorporation de microorganismes contaminants et avons établi des points de contrôle afin de les détecter et de les retirer. Une combinaison de méthodes d’homogénéisation enzymatiques et mécaniques et d’une trousse d’extraction d’ADN commerciale a permis une détection rapide et fiable du microbiote. La signature bactérienne des tissus cancéreux et sains de cinq patients pouvait être différenciée de celles des contrôles méthodologiques. Nos travaux approfondissent les connaissances nécessaires à l’étude d’environnements microbiens faiblement peuplés. Ils sont un point de départ vers une standardisation méthodologique et l’implémentation de procédures d’échantillonnage appropriées dans le cadre d’étude du microbiote pulmonaire.
1.2 Abstract
The lack of methodological standardization diminishes the validity of results obtained and the conclusions drawn when studying the lung microbiota. Therefore, we aim to validate a complete 16S rARN gene amplicon sequencing workflow, from patient recruitment to bioinformatics, tailored to the constrains of the pulmonary environment. We minimized the impact of contaminants and established negative controls to track and account for them at every step. Enzymatic and mechanical homogenization combined to commercially available extraction kits allowed fast and reliable extraction of bacterial DNA. The bacterial signatures of extracted cancerous and healthy human tissues from 5 patients could be distinguished from methodological controls. The DNA extraction kits used significantly impacted the bacterial composition of the controls. Our work expands our understanding of low microbial burdened environments analysis. This paper is to be a starting point towards methodological standardization and the implementation of proper sampling procedures in the study of lung microbiota.
1.3 Introduction
The modification of the lung microbiota has been linked to many pulmonary pathologies, such as the chronic obstructive pulmonary disease (COPD)19,28, asthma21, idiopathic
pulmonary fibrosis (IPF), and cystic fibrosis (CF)123. This pulmonary microbiota shift could
also have a significant impact on human health considering its influence in other body regions11,124. Therefore, the characterization and study of the pulmonary microbiota is of high
importance.
Bacteria can impact carcinogenesis, the evolution of cancer and the outcome of treatments of pancreatic and bowel cancers in mice50,51,125. A few studies aimed at analyzing a similar
effect of lung microbiota on pulmonary cancer26,54,126,127. Recently, Jet al. observed a
promoting effect of commensal bacteria on lung cancer development in mice117. Such a
distinct effect as yet to be confirmed in humans. Until now, human pulmonary microbiota studies integrating next-generation sequencing methodologies (NGS) have all used different protocols for tissue collection, nucleic acids extraction and bioinformatics analyses, which limit the conclusions that can be drawn from the current literature. Therefore, the development of an accurate and standardized method for the characterization of the lung microbiota seems mandatory.
DNA extraction methods have a noticeable impact on the microbial community detected107– 109. Since a wide variety of inhibitors (ions, polysaccharides, etc.) can reduce the efficiency
of extraction, protocols should be optimized to the sample matrix111. Besides, the resistance
to lysis of some bacteria, such as Gram-positive bacteria110, may reduce our ability to detect
them, creating a bias toward more easily lysed genera. The microbial biomass in the lung is low compared to what is found in the digestive system73. Bacterial profiling of such
low-density community is more prone to biases induced by contaminants and method selection74. In addition, commercial DNA extraction kits may carry a significant number of
bacterial contaminants113. Precautions are required to ensure that detected microorganisms
are not incorporated by the experimental method76. The implementation of strict contaminant
management strategy is necessary77.
The goal of our study is to validate multiple key aspects of a complete pipeline, from sampling to result analysis, that is designed and adapted to the study of the intratumoral lung microbiota. It is intended to be a first step towards achieving methodology standardization in lung microbiota studies.