FACULTE DES SCIENCES
THESE PRESENTEE
A L'ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
pour L'OBTENTION
du
GRADE MAITRE ES SCIENCES
par ALAIN BELANGER Bachelier ès Sciences Université Laval SYNTHESE D'a-PYRONES Novembre 1971
11
REMERCIEMENTS
Je voudrais témoigner ma plus vive reconnaissance à mon directeur de thèse, Monsieur le Professeur Paul Brassard, pour l'aide inestimable qu'il a apporté à la réalisation de ce travail. Ses attitudes certaines de chercheur et son esprit scientifique me servent de modèle. Qu'il veuille bien accepter le témoignage de ma profonde gratitude.
Je tiens à remercier sincèrement le Dr. Marcel Soucy qui m'a aidé de ses généreux conseils. Je suis également reconnaissant à mes condis ciples pour leur coopération et le climat de cordiale amitié qu'ils ont su créer dans le laboratoire.
J'adresse également mes remerciements à Mlle Diane Thibault et à M. Réjean Dulude pour l'aide inlassable qu'ils m'ont toujours apporté.
Je tiens à remercier enfin le Conseil National de Recherche pour la bourse qui m'a été octroyée durant l'année 1971.
TABLE DES MATIERES
Page
AVANT-PROPOS ... v
PARTIE THEORIQUE
INTRODUCTION: Les synthèses d'ct-pyrones... 1
Section I: Etude de la cycloaddition-1,4 de l'acétal méthylique
du chlorocétène à un carbonyle a,$-insaturé. ... 6 A. Action de l'acétal méthylique du chlorocétène sur un
carbonyle a,3~insaturé ... 8 B. Spectro s copie... 13 C. Insuccès de la cycloaddition de l'acétal méthylique du
chlorocétène ... 18
Section II: Hydrolyse des chloro-3 diméthoxy-2,2 dihydro-3,4
pyr annes... 20 Action d'une solution acide sur les chloro-3
diméthoxy-2,2 dihydro-3,4 pyrannes ... 22
Section III: Déshydrohalogénation ... 25 Action d'une base aqueuse sur les benzoylesters ... 28 Section IV: Synthèse de l'a-benzal y-phényl crotonolactone . . 32
IV
Section VI: Synthèse d ' ct-pyrones substituées...38
PARTIE EXPERIMENTALE Remarques générales... 43
Section I: Etude de la cycloaddition-1,4 de l'acétal méthylique du chlorocétène à un carbonyle a,$-insaturé ... 44
Section II: Hydrolyse des chloro-3 dihydro-3,4 diméthoxy-2,2 pyr annes...49
Section III : Déshydrohalogénation... 52
Section IV: Synthèse de l'a-benzal y-phényl crotonolactone ... 56
Section V: Hydrolyse du cétoester a-halogéné ... 58
Section VI: Synthèse d'a-pyrones substituées ... 60
BIBLIOGRAPHIE ... 64
AVANT-PROPOS
Un nombre important de substances possédant des activités physiolo giques contiennent un noyau a-pyrone dans leur molécule. Selon la nature des substituants, on les divise en deux classes: les stéroïdes et les aromatiques. Dans le premier cas, on retrouve des glucosides d'origine animale et végétale dont le radical stéroîdique est rattaché à 1'a-pyrone en position 5. Les propriétés cardiotoniques de ces stéroïdes, comme la bufotaline (1) par exemple, a fait l'objet d'études approfondies. Les dé rivés aromatiques se caractérisent par leur activité antibiotique et fon gicide. Les trois principaux représentants de ce dernier groupe, sont la paracotoïne (11), la yangonine (111) et l'hyspidine (IV).
OAc
OH HO HI
II
HO OMe OHIII
IV
VI
Plusieurs synthèses du noyau a-pyrone substitué ont été effectuées et elles se classent en deux groupes. Dans le premier, la substance de départ est facilement accessible mais par suite d'étapes nombreuses, les synthèses ne se réalisent qu'avec de très faibles rendements. Dans le deuxième cas, les transformations sont faciles mais requièrent des struc tures difficiles d'accès; ce qui ne permet pas alors d'en tirer des métho des pratiques et générales.
Un. projet de synthèse tirant partie des propriétés des acétals de cétène a été élaboré. Il devait comporter cinq étapes simples qui géné ralement s'effectuent avec de bons rendements. Applicable aux carbonyles a,$-insaturés, cette méthode s'avérerait en principe une synthèse générale d'a-pyrones.
Une réévaluation de la procédure a permis de simplifier considéra blement le mode opératoire en éliminant trois des étapes. Il en résulte donc une synthèse inédite d'a-pyrones à la fois simple, pratique et proba blement générale.
INTRODUCTION
Les synthèses d'a-pyrones
Un grand nombre de méthodes ont jusqu'ici été élaborées en vue de 1'obtention d'a-pyrones; sauf quelques exceptions, elles ne possèdent pas d'application générale. Nous donnerons dans cette première partie du tra vail quelques exemples importants de synthèses d'a-pyrones substituées ci tées en littérature. 0 0
2 CH
3-C-CH
2-C-OR
V
ROOC^, ÇH3 CH3/
0
CH(%r 0VI
v ÇH3R- C2H5
VII
2
OH
HOOC-CH-OH2-OOOH
^ 2 HOOO-CH2-CHO
HOOC^^ H 0VIII
2
Plusieurs auteurs (1) ont effectué la synthèse de l'acide isodéhydro acétique (VI) par condensation de l'acétoacétate d'éthyle (V) en milieu acide ; par décarboxylation, ils ont obtenu 1’a-pyrone (VII). Une méthode analogue permet d'isoler l’acide coumalique (VIII) par condensation d'un 3-formylacide.
Une méthode importante de synthèse d'a-pyrones consiste en une cycli sationde ô-cétoacide a,(3-insaturé (IX) (1). Mais très souvent on n'isole pas ce dernier produit mais plutôt 1'a-pyrone (X) correspondante vu les conditions acides de 1'hydrolyse.
C2H5OOC-OOOC2H5
4
-CH3-CH=CH-C00C2Hs
-> CgH^OOC OH HOOC v,,, HOOCIX
X
Les essais de synthèse avec un y-formylacide a,g-insaturé ont été négatifs, cependant Panizzi et Nicolletti (1) ont employé l'éther d'énol
3 R-C-C5C-C00CH3 U
CHOCHs
H- R L> H+ COOCH3 H OCH3XI
XII
La condensation du 3,3-dichloroacroléine (XIII) avec une cétone ali phatique (R = alkyle, R' = aryle), avec l'acétoacétate d'éthyle (R - CH?, R' = CO^C^Hg) et avec l'acide pyruvique (R = CO^H, R' = H) a été étudié en détail pour 1'obtention d'a-pyrones (XIV) (1). Elle présente un certain degré de généralité quoique les a-pyrones substituées en 3 et 4 demeurent
inaccessibles. 0 R-G-CH2-R' + CH0CH=CCl2
XIII
-> Q R'R-C-C=CH-CH=CC
(2v
R’ R %XIV
4
Qn emploie aussi le 3,3~dichloroacroléine (XIII) dans la conden sation avec un acétylure métallique (XV) et selon les conditions expé rimentales, on isole l'a-pyrone (XVI) ou l'hydroxy-4 a-pyrone (XVII). Mais cette méthode entraîne de nouveau les inconvénients cités précédem ment vu le substrat dihalogéné.
CCi2=CHCH0 4-
XII
VCR=CR'
XV
GO^CHCOOEGR <— CC^CH-CHCnGR —> CC^CH-CH^CHCOR
OH
XVII
XVI
R' = M
L'incorporation du noyau a-pyrone dans un stéroïde en position 17 a fait l'objet de plusieurs études récentes de synthèse (2, 3, 4). Meme si elles semblent assez générales, elles se caractérisent par des rendements peu élevés vu le grand nombre d'étapes nécessaires.
5
Cette thèse présente une synthèse inédite d'a-pyrones substituées. Le nombre des étapes [2] et le caractère probablement général de celles- ci en font une méthode des plus prometteuses. La réaction initiale de cette synthèse fait intervenir la condensation du carbonyle a,B-insaturê et de l'acétal inéthylique du chlorocétène (XVII).
OCH3
H Cl
Section I
Etude de la cycloaddition-1,4 de l'acétal méthylique du chlorocétène à un carbonyle a,g-insaturé.
7
I - Etude de la cycloaddition-1,4 de 1'acetal méthylique du chlorocetène à un carbonyle a3g-insaturé.
Les propriétés chimiques des acétals de cétène (XIX) s'expliquent par leur caractère de réactif dipolaire-1,3 (XIXa, XlXb, XIXc) qui pro vient de la conjugaison des groupements alkoxyles avec la liaison double
(5). Le méthylène devient le centre de réactivité anionique de la molé cule. OR CHn= C OR XIX OR OR OR CHg-C, OR <3--- CHg-C OR
xix o
XIX b XIX c8
Les acêtals de cétène (XIX) réagissent avec les carbonyles a,g-in-saturés comme 1'anhydride maléique, les cetones ou les aldéhydes a,g-in- saturés et les quinones (5). McElvain et ses coll. (6) ont décrit la syn thèse des diméthoxy-2,2 dihydro-3,4 pyrannes (XX, XXI, XXII) obtenus à partir de la condensation de 1'acetal méthylique du cétène (XXIII) avec 1'acroléine, le cinnamaldéhyde et de la benz al acetophenone.
CHR=CH-COR' + CH
2
=
0
(
00
H
3)2
XXIII
XXII:R=R'=06H5»
0 OMe RXXI :R=
06
Hs
r
’=
h
XX :R=R'=H
A. Action de 1'acétal méthylique du chlorocétène sur un carbonyle a,B-insature.
Les réactions de 1'acétal du chlorocétène ne semblent pas avoir été exploitées en synthèse. Suivant les règles de cycloaddition-1,4,
9
Tableau I
Cycloaddition de 1'acétal methylique du chlorocétène à un carbonyle g,g-insaturé OMe \J-CI /\ "H R' R Temps de réaction Température Rendements (%) R=R’=R- H 3 h 125° 56 R=R'= H R“= C Hg 1.50 h CO 60 R=R’= H R"= CeH5 24 h 150° 66 r’= r”= h R = C6H5 3 jours 150° 43 R=R -C^Hg R' = H 3 jours 150° 36 R'=R"=C6H5 R-H 3 jours 150° 21
10
la présence d'un groupe accepteur d'électrons, le chlore par exemple, rattaché au philodiène doit favoriser la réaction (7). Par contre, l'en combrement stérique dû à de tels substituants pouvait consister un incon vénient sérieux. Notre étude porte sur l'action de l'acétal méthylique du chlorocctène (XVIII) sur divers carbonylcs a,|3-insaturés: l'acroléine, la méthylvinylcétone, le cinnamaldéhyde, la phény1vinylcétone et la
benzalacétophénone. Le Tableau I donne un résume des conditions expéri mentales et des résultats obtenus. En général les réactions furent effec tuées en tubes scellés sauf pour la méthylvinylcétone qui réagit à son point d'ôbullition.
Chloro-3 diméthoxy-2,2 dihydro-3,4 pyranne (XXIV).
Le produit obtenu à partir de l'acroléine est le chloro-3 diméthoxy-2,2 dihydro-3,4 pyranne (XXIV) et nous 1'isolons par distillation. Le rendement de 56% s'explique probablement par une polymérisation partielle de 1'acro léine et par une décomposition du dihydro-3,4 pyranne (XXIV) qui s'avère relativement peu stable. Il a tendance à s'ouvrir au contact de l'air hu mide pour donner un formylester.
Chloro-3 méthyl-6 diméthoxy-2,2 dihydro-3,4 pyranne (XXV).
La réaction s'effectue au point d'ébullition de la méthylvinyl cétone (82°C), Après une" période d'induction d'environ une heure, elle se
complète dans les 30 minutes suivantes. Une prolongation du temps de la réaction n'en améliore pas le rendement mais au contraire provoque une certaine perte du produit prévu. Nous récupérons du mélange réactionnel une partie de la méthyIvinylcétone et de l'acétal (XVIII) qui n'a pas réagi.
11
Le chloro-3 methyl-6 diméthoxy-2,2 dihydro-3,4 pyranne (XXV) obtenu par distillation est peu stable et prend une coloration jaune après quelques minutes, d'ailleurs le cycle s'ouvre facilement au contact de l'air hu mide.
Chloro-3 phényl-6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVI).
Le fractionnement du mélange réactionnel permet d'obtenir avec 66% de rendement le chloro-3 phényl-6 diméthoxy-2,2 dihydro-3,4 pyranne à partir de la phénylvinylcétone. L'efficacité de la transformation présu me l'influence du groupement phényle relié à un carbonyle lors de la cyclo
addition.
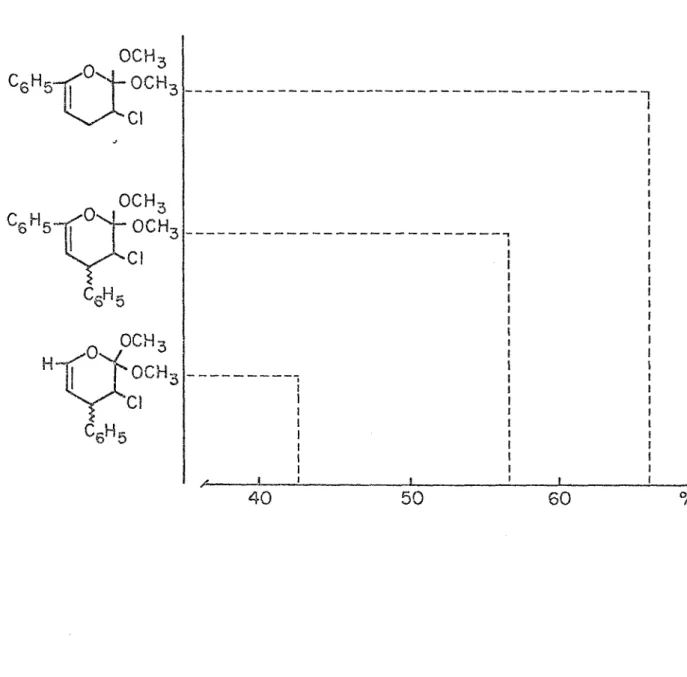
Par comparaison au rendement des substrats XXVII, XXVIII et XXIX, comme le montre le tableau II, nous observons une augmentation de rende ment lorsqu'un groupement aromatique est substitué au carbone oxygéné. Il faut suggérer ici un effet mésomère qui entraîne une concentration électro nique assez forte sur 1'oxygène favorisant directement la cycloaddition.
trans-Chloro-3 phényl-4 diméthoxy-2,2 dihydro-3,4 pyranne (XXVII).
Par distillation, nous recueillons une partie du cinnamaldéhyde qui n'a pas réagi et, avec un rendement de 43%, le trans-chloro-3 phényl-4
diméthoxy-2,2 dihydro-3,4 pyranne (XXVI). Le facteur déterminant qui abaisse le rendement est l'encombrement stérique causé par le groupement phényle. Nous notons la possibilité d'existence de deux isomères due à 1'asymétrie du carbone 4. Vu les conditions auxquelles nous soumettons le
12
Tableau II
Rendement relatif de pyrannes substitués par des groupements aromatiques
Z---!
“1
I
50
13
mélange réactionnel (3 jours à 150°), il semble que le seul diastéréo- isomère isolé soit thermodynamiquement le plus stable.
trans-Chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII) et cis-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXIX).
Les produits formés n'étant pas volatiles, ils ont été obtenus par cristallisation fractionnée. Nous recueillons ainsi 36% de trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII) et dans les deux cristallisations subséquentes, 21% de cis-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXIX). Le rendement totalise 57%,soit la moyenne des deux expériences précédentes conformément aux arguments déjà énoncés.
L'influence du groupement phényle en position 6 est de nouveau mise en évidence par la formation d'un diastéréoisomère (XXIX) relative ment moins stable. Les conditions expérimentales étant les mêmes que pré
cédemment d'où on a isolé qu'un seul isomère, il faut en conclure à un ef fet stabilisateur du groupement phényle sur le produit cinétique de la réaction.
B. Spectroscopie.
Dans le spectre infrarouge,les dialkoxy-2,2 dihydro-3,4 pyrannes se caractérisent par une bande d'absorption intense vers 1660 cm"1. Celle- ci est due à la présence de la double liaison. Rao (8) attribue les ban des intenses dans la région de 1190 à 1038 cm""1 à 1 ' absorption de l'acétal. Nous observons effectivement une série de pics dans cette même région mais légèrement déplacé (de 1150 à 1000 cm-1)probablement causé par le
grou-14
pement éther vinylique. Les spectres de résonance magnétique nucléaire des chloro-3 diméthoxy-2,2 dihydro-3,4 pyrannes se caractérisent par la présence de deux singulets dans la région de 3.50 p.p.m. Cette valeur correspond aux absorptions constatées pour les diéthoxy-2,2 dihydro-3,4 pyrannes (9).
OMe
R
Pour les substrats non-substitués en 4, les protons de cette posi tion apparaissent comme un multiplet dans la région de 2.50 p.p.m. Il s'agit d'un signal complexe englobant les couplages avec les protons en 3 et 5. Lorsque la liaison double est conjuguée à un groupement aroma tique, il y a alors glissement jusqu'à 5.50 p.p.m. du pic attribué au proton en 5. Quant au groupement chlorométhine, nous le retrouvons dans
la région de 4.30 p.p.m.
Nous tenons à préciser que la stéréochimie des produits provenant de la cycloaddition-1,4 ne constituait pas le but premier de nos travaux. Nous n'entreprenons pas une étude systématique mais nous tirerons simple ment des renseignements de la spectroscopic de résonance magnétique nucléaire.
La valeur élevé de la constante de couplage vicinal (J^ ^ - 10.5 Hz) pour 1'isomère (XXVII) indique une orientation trans-diaxiale de ces
instable en raison de 1'interaction diaxiale-1,3 et de lui attribuer la conformation [A].
XXVII(A)
XXVII(B)
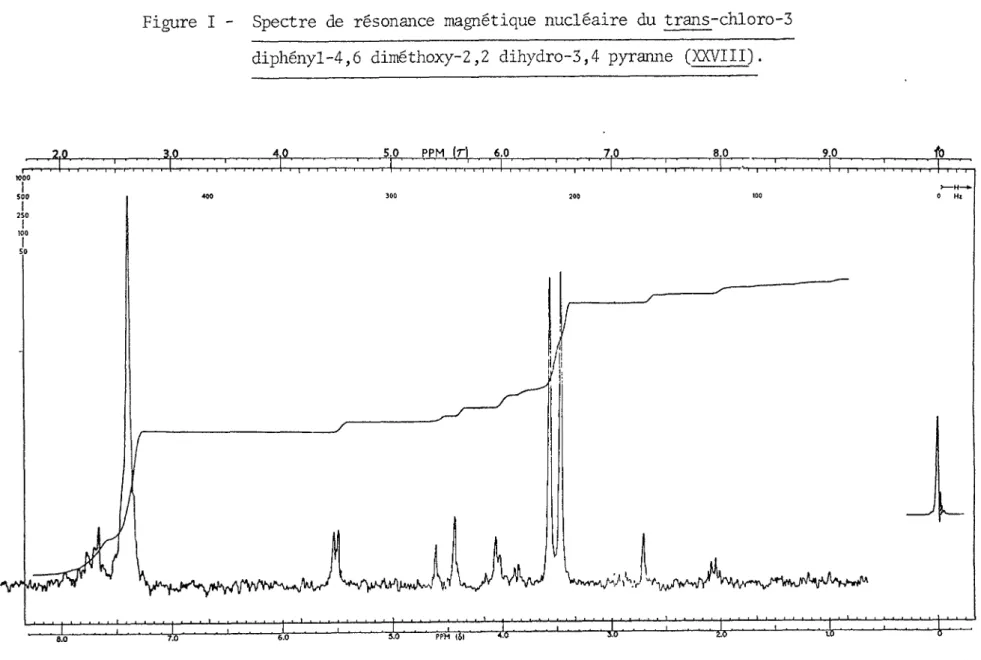
Comme l'illustre la Figure I pour l'isomère, la constante de
couplage élevé [10.5 Hz] entre les protons vicinaux 3,4 démontre une orientation trans-diaxiale de ces protons. Comme précédemment, cette conformation est stabilisée vu l'absence d'une interaction diaxiale-1,3.
OMe
Figure I - Spectre de résonance magnétique nucléaire du trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII).
Figure II - Spectre de résonance magnétique nucléaire du cis-chloro-3
diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXIX)•
18
La complexité du spectre de la Figure II est reliée à deux facteurs soit de la quasi-superposition de deux doublets dédoublés à 4.45 p.p.m. et du couplage un à un des trois protons. De la valeur faible du couplage vicinal, on en déduit la configuration des protons en 3 et 4. Le cou plage à longue distance entre les protons en 5 et 3 laisse prévoir la po
sition équatoriale de ce dernier. L'absence d'un tel couplage dans le diastéréoisomère précédent (XXVIII) est une preuve supplémentaire de 1'orientation axiale du proton en 3. Des deux conformations possibles
(XXIXA) et (XXIXB), nous ne retenons que (XXXIXA) vu 1'interaction
diaxiale-1,3 dans XXIXB et du couplage à longue distance entre les protons 3 et 5.
Cl OMe
XXIX(A)
XXIX(B)
C) Insuccès de la cycloaddition de l'acétal méthylique du chlorocétène. Nous notons jusqu'à maintenant une seule exception à la cycloaddi- tion-1,4 de l'acétal méthylique du chlorocétène. Malgré de nombreuses tentatives de condensation avec le tiglaldéhyde (XXX), nous ne parvenons
19
pas à la cyclisation désirée et nous récupérons entièrement les substrats. L'effet stérique n'est pas ici le seul facteur mais il faut inclure le fait- stabilisateur des deux groupements méthyles sur la liaison double. D'ailleurs, en littérature il ne semble pas exister de cycloaddition-1,4 entre le tiglaldéhyde et un philodiène quelconque.
G CH^ ^OCH]
+
H Cl
Section II
Hydrolyse des chloro-3 dimëthoxy-2,2 dihydro-5,4 pyrannes
21
II - Hydrolyse des chloro-3 diméthoxy-2,2 dihydro-3,4 pyrannes.
L'hydrolyse des dialkoxy-2,2 dihydro-3,4 pyrannes était tentée par McElvain (6) comme preuve de leur véritable structure. Le produit isolé était le cétoester ou le formylester correspondant (Schéma 1).
« OMe R -Tj ^ —OMe h h R-C C-OMe —>
V
RV
R Schéma 1Le mécanisme de 1'ouverture est analogue à 1'hydrolyse des acétals (9). La protonation se fait préférentiellement sur un des groupes
méthoxyles vu leur caractère plus électronégatif que l'éther vinylique; les électrons de ce dernier étant délocalisées par la liaison double
(Schéma 2). /0- OMe -OMe H Ô —Me OMe H-Q-H "OMe 0 h H-Ç 0 C-OMe <--H V OtH
b
-OMe Schéma 222
Aetion d'une solution acide sur les chloro-3 diméthoxy-2,2 dihydro-3,4 pyrannes.
Le mode opératoire généralement suivi comporte une addition d'une solution acide aux chloro-3 dihydro-3,4 diméthoxy-2,2 pyrannes. Si le substrat est liquide, 1'hydrolyse s’opère à la température ambiante et est exothermique, tandis que nous devons chauffer sur bain-marie, si la substance est solide. Comme nous le notions précédemment 1'hydrolyse peut s'effectuer au contact de l'air humide sans catalyseur acide, mais dans ce cas le temps de réaction est prolongé.
ch3 C-OMe
XXXII
L'hydrolyse fut d'abord tentée avec le chloro-2 méthyl-6 dimé- thoxy-2,2 dihydro-3,4 pyranne (XXV) et le substrat isolé fut tel que nous le prévoyions, le chloro-2 acétyl-4 butyrate de méthyle (XXXII). La bande vers 1665 cm""1, caractéristique des dihydro-3,4 pyrannes, est main tenant disparu dans le spectre infrarouge. Les deux absorptions intenses à 1720 cm-1 et 1750 cm"1 impliquent la formation d'une cétone aliphatique et d'un ester a-halogéné (10). On observe sur le spectre de résonance magnétique nucléaire la présence d'un singulet à 3.80 p.p.m. (ester méthylique), et la disparition des deux pics dus à l'acétal.
23
Le singulet qui représente le groupe méthyle de la position 6 est maintenant déplacé jusqu'à 2.15 p.p.m. région normale pour un grou pement acétyle.
L'hydrolyse du trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII) s'opère si nous le chauffons dans un mélange d'eau et
d'acide chlorhydrique. Nous recueillons le thréo-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIII).
OMe
XXVIII
XXXIII
La disparition de la bande à 1655 cm-1 dans 1 ' infrarouge implique la formation d'un nouveau produit. Les deux absorptions présentes à 1748 cm-1 et 1685 cm-1 dévoilent un groupement ester a-halogénê et un groupement benzoyle. Le signal correspondant à l'ester méthylique dans le spectre de résonance magnétique nucléaire apparaît normalement à 3.67 p.p.m.
L'érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIV) s'obtient par 1'hydrolyse du pyranne (XXIX). Nous l'identifions par la ressemblance des spectres infrarouge en comparaison avec l'isomère
pré-24
cèdent (XXXIII) et par le signal dû à l'ester méthylique dans le spectre de résonance magnétique nucléaire.
Cl
OMe
XXIX
XXXIV
L'analyse des constantes de couplage du chlorométhine des butyrates de méthyle XXVIII et XXIX fournit une preuve des configurations érythro et thréo. Le proton donne un doublet à 4.62 p.p.m. (J = 7.5 Hz) au cas de 1'isomère XXVIII et à 4.90 p.p.m. (J = 6.0 Hz) au cas de
1'isomère XXIX . Cette différence des constantes de couplage caractérise les diastérêoisomères érythro et thréo et permet de vérifier la configura tion. En effet, la constante de couplage vicinal (J\g) dans 1'isomère érythro tend à être beaucoup plus élevé que dans 1’isomère thréo (11). Ces valeurs différentes s'expliquent par les dispositions relatives des hydro gènes dans les conformations stables de chaque diastéréoisomère.
Section III
26
III - Deshydrohalogénation
Suivant le projet original de synthèse, le produit obtenu à ce stade était l'ester a,B-insaturé. Une hydrolyse simple de cet ester dans un milieu aqueux et basique amènerait probablement la formation de l'acide correspondant et une étape nous séparerait donc du but ultime (Schéma 3).
0 h R-Ç 0 C-OCH3
>
0 II R-Ç%
C-0CH3 Schéma 3Les tentatives d'opérer une élimination sur 1'érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIV) dans des conditions plus
ou moins rigoureuses (bromure de lithium dans le diméthylformamide,
nitrate d'argent dans le méthanol ou s-collidine) n'amènent pas les trans formations escomptées. Le cétoester demeure inerte à l'action du nitrate d'argent à la température ambiante tandis qu'il y a réaction à 80°C. L'examen du mélange réactionnel par chromatographie sur couche mince dé montre la formation d'un trop grand nombre de produits pour en permettre 1'identification. De plus les spectres infrarouge et de résonance magné tique nucléaire ne révèlent pas l'élimination désirée. Le bromure de lithium dans le diméthylformamide et la s-collidine, lorsque chauffés à 140° pendant deux heures, effectuent tous deux la déshydrohalogénation;
27
on isole dans le deuxième cas le chlorhydrate de collidine. Nous notons en effet la formation d'un même produit majoritaire dans les deux mélan ges réactionnels. L'analyse par spectroscopie infrarouge et de résonance magnétique nucléaire permet de supposer la formation d’un carbométhoxy-2 diphényl-3,5 dihydro-3,4 furanne (XXXV).
ii h R-C C-OCH3
XXXIV
R\z0\fC-0CH3 H' 'R R = C6"5XXXV
La déshydrohalogénation s'effectue dans une solution basique fai ble (Na2C03), mais il n'en résulte pas le produit attendu mais plutôt un cyclopropane (Schéma 4). On constate ainsi la formation d'un carbanion en a du benzoyl qui s'attaque par la suite à la position 2 suivant un mécanisme de substitution nucléophile de deuxième ordre.
0 11 R-C 9 C- OCHi ET
P
R-CP
C-OCH3 0 11 R-C 0 C-OCH 3 B y ?, 9 R-C C-OH Schéma 428
Action de bases aqueuses sur les benzoylesters.
L'action d'une base faible (Na^CO^) sur le thréo-chloro-2 phênyl-3 benzoyl-4 butyrate de méthyle (XXXIII) entraîne une déshydrohalogénation et une hydrolyse de l'ester, confirmées par 1'analyse élémentaire. L'ab sorption à 1000 cm"1 suggérait la formation du cyclopropane (12). La tentative d'interprétation du spectre de résonance magnétique nucléaire se heurte à des difficultés. Nous ne parvenons pas à attribuer les signaux aux protons du fait des faibles différences des déplacements chi- miques. Mais on observe aisément aucun déplacement qui pourrait caracté-
riser l'existence d'une liaison double. On a pu déduire la structure du diastéréoisomère par comparaison à un substrat connu. Stoemer et Schenck (13) ont obtenu en effet les quatre acides cyclopropaniques
XXVI, XXXVII, XXXVIII et XXXIX, à partir d'esters mêthyliques
C II R-C C-OH
XL
XXXVI
R R 0 II C-OHXXXVH
II 0 R29
XXXVIII
B">
XXXIX
R-c6h5correspondants (XL et XLI). Suivant les conditions de 1'hydrolyse, il y avait possibilité d'épimérisation. La structure du cyclopropane ob tenu par hydrolyse de 1'isomère (XXXIII) correspond à l'acide cis- phényl-2 brans-benzoyl-3 cyclopropanecarboxylique (XXXVI) vu les points de fusion identiques. Nc^COg C-OCH3 R=C6H5 R
XXXVI
30
Nous obtenons 1'isomère (XXXVI) en milieu faiblement basique tandis que Stoemer l'isolait par hydrolyse acide. Ainsi le milieu que nous em ployons ne favorise pas 1'épimérisation au carbone relié au groupement benzoyle. Se servant d'une base forte (NaOH) nous devions favoriser la
formation de l'épimère (XXXVII) à partir du même diastéréoisomère (XXXIII). L'expérience permet d'isoler l'acide trans-phényl-2 trans-benzoyl-3 cyclo- propanecarboxylique (XXXVII) et nous l'identifions comme précédemment.
NaOH
XXXIII
XXXVII
Nous démontrons une transformation analogue par l'obtention d'un isomère à partir de 1'érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIV). Les grandes ressemblances entre les spectres infrarouge confirment la structure cyclopropanique. L'identification précise s'ef fectue par la corrélation entre les points de fusion et nous obtenons l'acide cis-phényl-2 trans benzoyl-3 cyclopropanecarboxylique (XXXIX).
NdgCOg
XXXIV
XXXIX
Conformément à 1'argument précédent, il est normal que le cyclo propane (XXXVIII) n'est pas isolé puisque 1'épimérisation semble déjà effectué par une base faible. Mais pour vérification, nous traitons 11érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIV) dans un milieu basique fort (NaOH). Le résultat fut comme nous le prévoyions le produit isolé est 11isomère préalablement identifié comme l'acide cis-phényl-2 trans-benzoyl-3 cyclopropanecarboxy1ique (XXXIX).
NaOH
Section IV
33
IV - Synthèse de l'a-benzal y-phényl crotonolactone.
La cyclisation d'un formylacide en lactone a fait l'objet d'études récentes. Pettit et ses coll. (3) ont effectué la synthèse d'une
dihydro-3,4 a-pyrone (XLII) à l'aide de l'acide p-toluènesulfonique.
0 O
H-C C-OH /°vO
R=(CH
2
)
2
-CH
3
XLII
Puisque les acides cyclopropanecarboxyliques sont isomères des acides a,B-insaturés, ils pourraient adopter un comportement analogue. Il serait donc possible d'opérer une cyclisation et peut-être, par rearrangement, obtenir une a-pyrone (schéma 5).
R"c6H5
34
Lors du traitement de 1'isomère (XXXVI) par l'acide p-toluène-sulIo nique, nous observons l'apparition d'une coloration jaune intense dans
le mélange réactionnel. Le substrat jaune clair isolé par chromatogra phie sur gel de silice possède une formule moléculaire de dé montrant ainsi une perte d'une molécule d'eau. L'analyse du spectre
infrarouge permet d'affirmer la formation d'une lactone et d'une liaison double tout en constatant l'absence d'un cyclopropane et des fonctions acide et benzoyle. Le spectre de résonance magnétique nucléaire offre peu de renseignement car il ne présente qu'un singulet et deux multiplets, tous dans la région des aromatiques. Son point de fusion de 149-150° et son apparence physique enlèvent toute analogie avec la diphényl-4,6 a-pyrone. Nous avons déterminé la structure exacte par référence à une substance déjà
connue en littérature, l'a-benzal y-phênyl crotonolactone (XLIII) (14). La similitude du point de fusion et des absorptions ultraviolet et infrarouge 1'affirment clairement. L'hypothèse précédemment avancé s'avère fausse puisque l'hydrogène en position 1 possède un caractère beaucoup plus acide que celui en 2.
Section V
36
V - Hydrolyse du cetoester a-halogénë.
L'échec de la déshydrohalogénation au stade du cetoester nous porte à la remettre à une étape ultérieure de la synthèse. Nous formulons ainsi un plan tirant partie de la possibilité de parvenir à 1'insaturation lors que nous serions en présence du chloro-3 dihydro-3,4 ct-pyrone (XLV). Pettit et ses coll. (3) citent le cas d'une telle élimination par 1'emploi du bro mure de lithium dans le dimethylformamide (schéma 6).
R=(CH2L-CH3
Schéma 6
Pour parvenir au chloro-3 dihydro-3,4 a~pyrone (XLV) nous devons effectuer une hydrolyse de l'ester suivi d'une cyclisation qui s'opérerait selon la méthode précédemment décrite dans la section IV. Nous tentons un premier essai d'hydrolyse du cétoester a-halogéné selon une méthode employée par Engel et ses coll. (2) pour 1'hydrolyse des diesters. En traitant l'acétyl-4 chloro-3 butyrate de méthyle (XXXII) dans un mélange d'acicle acétique et d'acide chlorhydrique, nous recueillons un produit noir huileux qui n'a aucune ressemblance avec le produit attendu. Des
37
nous conduisent à 1'acide prévu (XLIV)• Par contre, 1'acide a-halogéné est instable et se transforme rapidement. Des tentatives de cyclisation avec l'acide p-toluènesulfonique immédiatement après 1'obtention du céto- acide a-halogéné (XLIV) n'ont pas permis de contourner la difficulté.
Section VI
39
VI - Synthèse d'q-pyrones substituées.
L'impossibilité d'obtenir 1'a-pyrone suivant le projet initial en traîne une réévaluation du schéma de la synthèse. Comme la faille de cette méthode se trouve au stade de l'élimination, nous tenterons de l'ef fectuer à une étape antérieure.
Des essais avec la s-collidine et le bromure de lithium dans le diméthylformamide s'avèrent infructueux et nous récoltons un grand nombre de produits impossible à identifier. Une première tentative avec le
méthylate de sodium dans le méthanol ne donne pas les résultats escomptés ; nous récupérons le substrat inchangé. L'emploi à nouveau de cette der nière base dans un solvant dipolaire aprotique, le diméthyl sulfoxyde, opère des transformations inattendues au-delà de 1'élimination prévue. Nous n'isolons pas la diphényl-4,6 diméthoxy-2,2 pyranne (XLVII) mais bien l'a-pyrone (XLVI) correspondante.'
La diphényl-4,6 a-pyrone (XLVI) s'identifie par comparaison avec un échantillon authentique obtenu par la méthode de Kohler (15). Des essais effectués de un à deux équivalents de base ne conduisent pas à une conversion complète du substrat. Par contre, quatre équivalents permettent d'isoler jusqu'à 72% de diphényl-4,6 a-pyrone (XLVI).
R
XXXVIII
R 7^ R OMe R > ;0 R-c6h5 RXLVI
Nous avons éprouvé l'efficacité de la transformation par la prépa ration d'autres a-pyrones. Le traitement du trans-chloro-3 phényl-4 diméthoxy-2,2 dihydro-3,4 pyranne, (XXVII) dans les mêmes conditions basi ques, fourni la phényl-4, a-pyrone (XLVIII) avec un rendement de 85%.
OMe —OMe ^0\_0 1 --- >
V
R R xxvnXLVIII
La chromatographie sur couche mince n'a pas révélé la présence de produits secondaires. Puisque nous avons remarqué une instabilité assez marquée de ces deux substances en solution, il s'ensuit donc que le ren dement n'est pas quantitatif. La diphényl-4, 6 a-pyrone prend rapidement
une teinte rouge en solution et il devient alors difficile d’en extraire un produit jaune pâle. Il en est de même pour la phényl-4 a-pyrone pro duit blanc et cristallin qui prend une coloration jaune.
Des essais préliminaires démontrent que la phényl-6 chloro-3
diméthoxy-2,2 dihydro-3,4 pyranne (XXVI) était réfractaire au méthylate de sodium. L'hydrogène à éliminer en position 4 n'étant plus benzylique, l'élimination nécessite une base plus forte. Le traitement par le tert- butylate de potassium permet de recueillir la phényl-6 a-pyrone (IL ), positivement identifié par les spectres infrarouge, de résonance magné tique nucléaire. De plus,le spectre ultraviolet est semblable à celui noté en littérature (16).
OMe
XXVI
IL
FrC^Hs
Ces transformations ne correspondent pas aux propriétés connues des orthoesters puisqu'on admet leur stabilité en milieu basique. Pour expliquer le cours apparemment atypique de formation d'a-pyrones, nous suggérons les deux mécanismes suivants.
1. Un premier équivalent de base effectue l'élimination. Il pourrait y avoir ensuite une attaque nucleophile avec ouverture du cycle.
42
L'hydrolyse subséquente de 1'orthoester entraîne la formation de l'a- pyrone (Schéma 7). ^0 OCH3 -OCH3 0" OCH3 <T0CH3 . B
<
B H2O Y 0 11 C-0 Schéma 72. Le deuxième mécanisme possible aborde le problème comme précé demment . Après le stade de la dêshydrohalogénation, nous pouvons considé rer une substitution concertée au niveau de 1'éther méthylique qui amè nerait la cyclisation à 1'a-pyrone (Schéma 8).
OCH3 "-OCH3 B" —> 0;CH3< B ^>r-0CH3 -> 0 /0 C-OCH3 < B" H20 Y 0 ,0- c-o-Schéma 8
PARTIE EXPERIMENTALE
Les points de fusion ont été déterminés en tube capillaire à l'aide d'un thermomètre calibré (appareil Thomas-Hoover). Les spectres infra rouge ont été réalisés sur un appareil Beckman, IR-10. Les spectres ultraviolet ont été déterminés sur un appareil Beckman, modèle DK-IA.
Les spectres de R.M.N. ont été enregistrés sur un appareil Varian A 60; les déplacements chimiques sont mesurés en <5 par rapport au tétraméthyl silane (ô=o) et ne sont pas corrigés. Les spectres de masse ont été dé terminés sur un spectrographe Varian M 66.
Les microanalyses furent effectuées par Monsieur Réjean Dulude de l'Université Laval.
Section I
Etude de la cycloaddition-1,4 de l'acëtal méthylique du chlorocétène à un carbonyle a,g-insaturé
45
I - Etude de la cycloaddition-1,4 de l'acétal mêthylique du chlorocëtène à un carbonyle a,g-insaturé.
Chloro-3 diméthoxy-2,2 dihydro-3,4 pyranne (XXIV).
Dans un tube scellé, on fait réagir de l'acétal méthylique du chlorocëtène (8.70 g) avec de l'acroléine(4.0 g). Après 3 heures de réac tion à 125°C, on distille le mélange réactionnel sous 12 mm de mercure. On récolte le chloro-3 diméthoxy-2,2 dihydro-3,4 pyranne (6.82 g) p.é. 90- 92°. Après quelques minutes, le -liquide prend une teinte jaune.
Spectre infrarouge (Film): 1665 cm-1 (double liaison), 1130-1030 cm-1 (acétal).
Spectre de résonance magnétique nucléaire (CDCl^): un multiplet centré à 2.50 p.p.m. (CHg 4), deux singulets à 3.30 et 3.32 p.p.m. (O-CH^ 2,2), un triplet centré à 4.30 p.p.m., J = 5.5 Hz (CH 3), un doublet dédoublé cen tré à 4.75 p.p.m., J = 2.0, 6.0 Hz (CH 5) et un triplet dédoublé centré à 6.25 p.p.m., J = 2.0, 6.0 Hz (CH 6).
Chloro-2 méthyl-6 diméthoxy-2,2 dihydro-3,4 pyranne (XXV).
L'acétal méthylique du chlorocëtène (10 g) est ajouté au méthyl- vinylcétone (5,7 g), préalablement distillée. Après avoir chauffé à re
flux pendant 1.5 heures, on isole par distillation un mélange de methyl- vinylcétone et d'acétal méthylique du chlorocëtène. Le chloro-2
méthyl-6 diméthoxy-2,2 dihydro-3,4 pyranne (9.49 g) est recueilli sous 12 mm. de mercure, p.é. 72-74°. Il est peu stable et se transforme en cétoester au contact de l'air.
46
Spectre infrarouge (Film): 1670 cm-1 (liaison double) et 1120-1020 cm 1 (acétal).
Spectre de résonance magnétique nucléaire (CDCl^): un multiplet centré à 1.78 p.p.m. avec un couplage à longue distance (CH^ 6), un multiplet cen tré à 2.45 p.p.m. (ŒL 4), deux singulets à 3.32 et 3.38 p.p.m. (O-CTL^ 2,2) un triplet centré à 4.12 p.p.m., J = 5.5 Hz (CH 3) et un multiplet centré à 4.48 p.p.m. (CH 5).
Chloro-3 phényl-6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVI).
Dans un tube scellé, la phénylvinylcétone (2.50 g) est addi tionné à 1'acétal mëthylique du chlorocétène (2.10 g). Après un chauf fage de 24 heures, on distille le mélange réactionnel et on récolte le chloro-3 phényl-6 diméthoxy-2,2 dihydro-3,4 pyranne (3.06 g) p.é. 110- 114° (1.5 mm).
Spectre infrarouge (Film) : 1665 cm-1 (double liaison), 1600 et 1580 cm-1 (aromatique) et 1125-1020 cm-1 (acétal).
Spectre de résonance magnétique nucléaire (CDCl^): un multiplet centré à 2.70 p.p.m. (CH-, 4), deux singulets à 3.40 et 3.45 p.p.m. (O-CH^ 2,2) ,
un triplet centré à 4.32 p.p.m., J = 5.0 Hz (CH 3), un triplet à 5.40 p.p.m. J = 4.0 Hz (CH 5) et deux multiplets centrés à 7.35 et 7.62 p.p.m. (C^H^).
trans-Chloro-3 phényl-4 diméthoxy-2,2 dihydro-3,4 pyranne (XXVII).
Dans un tube scellé, on fait réagir 1'acétal mëthylique du chloro cétène (5.1 g) et le cinnamaldéhyde (6.0 g). Le chauffage à 150°C pendant 3 jours est suivi d'une distillation du mélange réactionnel. On isole
47
ainsi le cinnamaldéhyde (3.5 g) et le trans-chloro-3 phényl-6 diméthoxy-2,2 dihydro-3,4 pyranne (4.80 g) p.é. 95-96° (1.0 mm). Le substrat cristal lise dans l'éther et on obtient le produit (XXVII), p.f. 106-107°.
Spectre infrarouge [KBr] : 1660 cm-1 (liaison double), 1610 cm-1 (aromati que) et 1120 et 1020 cm-1 (acétal).
O
Spectre de résonance magnétique nucléaire [(CD,)^ - 0]: deux singulets à 3.38 et 3.42 p.p.m. (O-CH^ 2,2), un multiplet centré à 3.92 p.p.m. (CH 4), un doublet centré à 4.22 p.p.m., J = 10.5 Hz (CH 3), un doublet dédoublé
centré à 4.88 p.p.m., J = 7.0, 2.5 Hz (CH 5)., un doublet dédoublé centré à 6.42 p.p.m., J = 7.0, 2.5 Hz (CH 6) et un singulet à 7.38 p.p.m. (C^H^).
Anal. Cale, pour C^H^ClOy
Trouvé :
C, 61.41 ; H, 5.90 C, 61.31 ; H, 5.93
trans-Chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII).
Dans un tube scellé, on chauffe à 150°C pendant 3 jours un mélange de benzalacétophénone(6.0 g) et 1'acétal mêthylique du chlorocétène (3.5 g). On laisse cristalliser le mélange réactionnel dans un mélange d'éther et d'éther de pétrole (30-60°). Les deux premières cristallisations fournis sent le trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne
(3.5 g) p.f. 125-126°. Après chaque récolte les eaux-mères sont évaporées et le résidu est repris dans un mélange des mêmes solvants.
Spectre infrarouge [KBr]: 1655 cm"1 (liaison double), 1600 cm"1 et 1580 cm-1 (aromatique), 1145-980 cm"1 (acétal).
48
et 3.57 p.p.m. (O-CHg 2,2), un doublet dédoublé centré à 3.94 p.p.m., J = 10.5, 2.5 Hz (CH 4), un doublet centré à 4.52 p.p.m., J = 10.5 Hz
(CH 3), un doublet centré à 5.50 p.p.m., J = 2.5 Hz (CH 5), un singulet à 7.40 p.p.m. et un multiplet centré à 7.72 p.p.m. (C^Hg 4,6).
Anal. Cale, pour C^gH^gClOg: C, 69.00 ; H, 5.74 Trouvé : C, 69.08 ; H, 5.71
çis-Çhloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXIX).
Les eaux-mères provenant de la cristallisation du trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (XXVIII) sont évaporées à cécité. Le résidu est repris dans un mélange d'éther et d'éther de pétrole
(30-60°). On obtient ainsi deux récoltes subséquentes constituées de cis chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (2.10 g), p.f. 79-
80°.
Spectre infrarouge [KBr]: 1660 cm-1 (liaison double), 1605 cm-1 (aroma tique) et 1115-1025 cm""1 (acétal).
Spectre de résonance magnétique nucléaire (CDClg): deux singulets à 3.45 et 3.51 p.p.m. (0-CHg 2,2), un multiplet centré à 4.42 p.p.m., J = 2.0, 1.5 Hz (CH 3 et CH 4), un doublet dédoublé centré à 5.60 p.p.m., J = 1.5, 2.0 Hz (CH 5), un singulet à 7.42 p.p.m. et un multiplet centré à 7.80 p.p.m.
(% 4,6).
Anal. Cale, pour C^gH^gClOg: C, 69.00 ; H, 5.74 Trouvé : C, 68.94 ; H, 5.59
Section II
50
II - Hydrolyse des chloro-3 dihydro-3,4 diméthoxy-2,2 pyrannes
Chloro-2 acétyl-4 butyrate de méthyle (XXXII).
On ajoute à un mélange hétérogène de chloro-2 méthyl-6 diméthoxy-2,2 dihydro-3,4 pyranne (4.13 g) et d'eau(.5 ml) quelques gouttes d'acide chlorhydrique. La température du mélange réactionnel s'élève et le milieu devient homogène. On verse dans l'eau (20 ml) et extrait à l'éther. La solution éthérée est séchée sur sulfate de magnésium et évaporée. On recueille le chloro-2 acétyl-4 butyrate de méthyle (3.68 g). Le produit est huileux et prend une coloration jaune après quelques minutes.
Spectre infrarouge (Film): 1750 cm-1(ester méthylique) et 1720 cm-1 (cétone).
Spectre de résonance magnétique nucléaire (CDC1?): un singulet à 2.15 p.p.m. 0
(CH^ ~ C -), un multiplet centré à 2.20 p.p.m. (CTL, 3), un multiplet cen tré à 2.67 p.p.m. (CTL 4), un singulet
doublet dédoublé centré à 4.42 p.p.m., J = 8.0, 6.0 Hz (CH 2).
thréo-Chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIII).
A une solution aqueuse d'acide chlorhydrique (20 ml 1.0 N), on ajou te le trans-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (.700 g) suivi d'un chauffage sur bain-marie pendant 2 heures. On verse ensuite le mélange réactionnel dans l'eau (100 ml) et extrait à l'éther. La solution éthérée est séchée sur sulfate de magnésium et évaporée. Après deux cris tallisations dans un mélange d'éther et d'éther de pétrole (30-60°), on obtient le thréo-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (.510 g), p.f. 45-46°.
51
Spectre infrarouge [KBr]: 1748 cm-1 (ester méthylique), 1685 cnr1 (benzoyle), 1600 et 1585 cm-1 (aromatique).
Spectre de résonance magnétique nucléaire (QDC1-): un doublet centré à Q
3.58 p.p.m., J = 2.5 Hz (CH^ 4), un singulet à 3.68 p.p.m. (- C - OCH^), un doublet dédoublé centré à 4.20 p.p.m., J = 6.0, 2.0 Hz (CH 3), un dou blet centré à 4.90 p.p.m., J = 6.0 Hz (CH 2), un singulet à 7.32 p.p.m. et deux multiplets à 7.55 et 8.00 p.p.m. (10 protons aromatiques).
Anal. Cale, pour C^gE^ClO^: C, 68.25 ; H, 5.37 Trouvé : C, 68.22 ; H, 5.45
erythro-Chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (XXXIV).
On porte à ébullition pendant 30 minutes un mélange de cis-chloro-3 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (.300 g), de méthanol
(5 ml), d'eau (10 ml) et d'acide chlorhydrique (1.0 ml). Après refroidis sement 1'érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (.215 g) cristallise,p.f. 99-100°.
Spectre infrarouge [KBr]: 1735 cm”1 (ester méthylique) 1682 cm”1 (benzoyle) 1600 cm”1 (aromatique).
Spectre de résonance magnétique nucléaire (CDCl^): un singulet à 3.58’p.p.m. ( - 8 - OCH3 )» un doublet centré à 3.70 p.p.m., J = 3.5 Hz (CH^ 4) un multiplet centré à 4.15 p.p.m. (CH 3), un doublet centré à 4.62 p.p.m., J = 7.5 Hz (CH 2), un singulet à 7.30 p.p.m. et deux multiplets respecti vement à 7.50 et 7.95 p.p.m. (10 protons aromatiques).
Anal. Cale, pour C^gH^ClO^: C, 68.25 ; H, 5.37
Section III
53
III - Dëshydrohalogénation
Acide trans-phênyl-2 cis-benzoyl-3 cyclopropanecarboxylique (XXXVI).
A me solution de Na^CO? (.850 g) dans l'eau (5 ml), on ajoute le thréo-chloro-2 phenyl-3 benzoyl-4 butyrate de méthyle (.800 g). Le milieu est rendu homogène par addition d'un mélange de inéthanol-tétrahydrofuranne (1:1). Après me heure d'agitation à la température ambiante, le mélange réactionnel est versé dans l'eau et la solution aqueuse est lavée deux fois à l'éther. Après acidification, on extrait trois fois à l'éther. Cette dernière solution éthérée est séchée sur sulfate de magnésium et évaporée. On isole par cristallisation dans m mélange d'éther et d'éther de pétrole (30-60°) l'acide cis-phényl-2 tram-benzoyl-3 cyclopropane
carboxylique (.590 g) p.f. 173-174°. (Litt. p.f. 174-175°) (13). Spectre infrarouge [KBr]: 1695 cm-1 (acide carboxylique), 1678 cm-1
(benzoyle), 1600 cm-1 (aromatique) et 960 cm-1 (cyclopropane).
Spectre de résonance magnétique nucléaire (CDCl^): m doublet dédoublé centré à 2.70 p.p.m. , J = 9.0, 7.0 Hz, m doublet centré à 3.28 p.p.m., J = 1.5 Hz, m doublet centré à 3.40 p.p.m., J = 2.0 Hz (3 CH 1, 2, 3), m singulet à 7.48 p.p.m. et deux multiplets centrés à 7.60 et 8.15 p.p.m.
(10 protons aromatiques) et m singulet à 10.80 p.p.m. (acide carboxyli que) .
Anal. Cale, pour C-^E^C^: C, 76.69 ; H, 5.31 Trouvé : C, 76.51 ; H, 5.29
Acide trans-phény1-2 trans-benzoyl-3 cyclopropanecarboxylique (XXXVII). Le thréo-chloro-2 phényl-3 benzoyl-4 butyrate de méthyl (.200 g)
54
est traité par le NaOH (.052 g) dans l'eau (5 ml). On isole de la façon habituelle l'acide trans-phényl-2 trans-benzoyl-3 cyclopropanecarboxylique
(.077 g), p.f. 151-152° (Litt. p.f. 152-153°) (13).
Spectre infrarouge : 3 695 cm-1 (acide) 1670 cm-1 (benzoyle) 1600 cm-1 (aromatique) 1010 cm*"1 (cyclopropane).
Spectre de résonance magnétique nucléaire (CDCl^): un multiplet centré à 3.45 p.p.m. (3 protons du cyclopropane), un s in guiet et deux multiplets centrés respectivement à 7.25, 7.45 et 8.00 p.p.m. (10 protons aromati ques), un singulet à 11.50 p.p.m. (acide carboxylique).
Anal. Cale, pour CjjH-^C^: C, 76.69 ; H, 5.31 Trouvé : C, 76.79 ; H, 5.31
Acide cis-phényl-2 trans-benzoyl-3 cyclopropanecarboxylique (XXXIX)
a) Par le Na2CC>2
A une solution de Na^CO, (.600 g) dans 5 ml d'eau, on ajoute 1'érythro-chloro-2 phényl-3 benzoyl-4 butyrate de méthyle (.200 g). On suit le mode opératoire décrit plus haut et on isole l'acide cis-phényl-2 trans-benzoyl-3 cyclopropanecarboxylique (.030 g) p.f. 133.5-134.5°,
(Litt. p.f. 136-137°) (13).
Spectre infrarouge [KBr]: 1708 cm-1 (acide), 1680 cm*"1 (benzoyle), 1600 cm"1 (aromatique) et 1015 cm"1 (cyclopropane).
Spectre de résonance magnétique nucléaire (CDC1?): un doublet dédoublé centré à 2.78 p.p.m., J = 5.0, 10.0 Hz, un doublet dédoublé centré à 3.28 p.p.m., J = 10.0, 7.0 Hz, un doublet dédoublé centré à 3.80 p.p.m.
55
J = 7.0, 5.0 Hz (3 CH 1, 2, 3), un singulet à 7.58 p.p.m. et deux multi plets à 7.60 et à 8.15 p.p.m. (10 protons aromatiques) et un singulet à 10.60 p.p.m. (acide carboxylique).
Spectre de masse: pic moléculaire à m/e 266. b) par le NaOH
Le composé précédent (XXXIV) (.190 g) est traité par une solution de NaOH (.050 g) dans l'eau (5 ml). On suit le mode opératoire décrit plus haut et on recueille l'acide cis-phényl-2 trans-benzoyl-3 cyclopropane- carboxylique p.f. 134-135°. (Litt. p.f. 135-136°) (13).
Les spectres infrarouge et de résonance magnétique nucléaire sont identiques au produit précédent.
Section IV
57
IV - Synthèse de l'a-benzal y-phênyl crotonolactone ÇXLIII).
On chauffe à reflux pendant 3 heures un mélange d'acide trans -
phényl-2 cis-benzoyl-3 cyclopropanecarboxylique (.190 g) d'acide p-toluène- sulfonique (2 mg) et Ce benzène (40 ml). On verse le mélange réactionnel dans l'eau et extrait à l'éther. La solution éthérée est lavée avec une solution de NaHCCL (5%) et séchée sur sulfate de magnésium. Après purifi cation par chromâtographie sur couche mince (gel de silice), on obtient l'a-benzal y-phényl crotonolactone, p.f. 149-150° (Litt. p.f. 155° (14).
Spectre infrarouge [CCl^J; 1790-1785'cm-1 (lactone), 1630 cm-1 (double liaison), 1600 et 1590 cm-1 (aromatique).
Spectre de résonance magnétique nucléaire (CDCl^): un singulet à 7.00 p.p.m. (CH 4) et un multiplet centré à 7.70 p.p.m. (11 protons aromatiques).
Spectre ultraviolet (EtOH): 250 mu,
Anal. Cale, pour G^ti^C^: C, 82.25 C, 82.18
log e = 1.85; 384 mp, log e = 2.94.
Trouvé
H, 4.85 H, 4.95
Section V
59
V - Hydrolyse du cétoestex 2-halogène.
Acide chloro-2 acétyl-4 butyrique (XLIV).
Ou agite pendant 24 heures à température ambiante un mélange hété rogène de chloro-3 acétyl-4 butyrate de méthyle (.500 g), de résine
Dowex 50 W-X4 (70-50 maille) et d'eau (30 ml). Après filtration on verse le mélange dans l'eau et on extrait à l'éther. La solution éthërée est sé chée sur sulfate de magnésium et évaporée. On recueille ainsi l'acide chloro-2 acétyl-4 butyrique (.397 g). Il se transforme rapidement en gou dron noir.
Spectre de résonance magnétique nucléaire (CDC1,) un s in guiet à 2.12 p.p.m.
O 5
(OH, - C -) j un multiplet vers 2.15 p.p.m. masqué par le groupement acétyle (CH^ 3), un multiplet centré à 2.70 p.p.m. (CH. 4), un doublet dédoublé centré à 4.51 p.p.m., J = 7.0, 5.5 Hz (CH 2) et un singulet à 9.66 p.p.m. (acide carboxylique).
Section VI
61
VI - Synthèse d'a-pyrones substituées.
Diphênyl-4,6 a-pyrone (XLVI).
On laisse à la température ambiante pendant 24 heures un mélange de méthylate de sodium (.288 g) de diméthyl sulfoxyde anhydre (60 ml) et de trans-chloro-2 diphényl-4,6 diméthoxy-2,2 dihydro-3,4 pyranne (.430 g). On verse le mélange réactionnel dans l'eau et extrait à l'éther. La solu tion éthérée est séchée sur sulfate de magnésium et évaporée. La diphényl-4,6 a-pyrone (.230 g) est purifiée par deux cristallisations, p.f. 136-137,
(Litt. 137-138°) (15). Le produit sous forme cristalline est stable mais prend rapidement une coloration rouge en solution.
Spectre infrarouge [KBr]: 1700 cm-1 (lactone), 1630 cm-1 et 1540 cnn1 (liaisons doubles).
Spectre de résonance magnétique nucléaire (CDCl^): un doublet centré à 6.65 p.p.m., J = 1.5 Hz (CH 3), un doublet centré à 7.35 p.p.m., J = 1.5 Hz (CH 5) et deux multiplets à 7.60 et 8.00 p.p.m. (10 protons aromatiques)
Spectre ultraviolet (EtOH): 263 m , log e = 3.90; 345 my, log e = 2.95.
Anal. Cale, pour C-^yH^Og: 82.25 ; H, 4.85 Trouvé : C, 82.34 ; H, 4.97 Phényl-4 a-pyrone (XLVIII)
On ajoute le méthylate de sodium (.487 g) à un mélange de diméthyl sulfoxyde anhydre (150 ml) et de trans-chloro-2 phényl-3 diméthoxy-2,2 dihydro-3,4 pyranne (.570 g). Suivant le mode opératoire précédemment
62
décrit, on obtient la phényl-4 a-pyrone (.330 g) p.f. 75-76°. Le produit est blanc sous forme cristalline, mais prend une teinte jaune en solution. Spectre infrarouge [KBr]: 1705 cm-1 (lactone), 1640 cm-1 et 1540 cm-1
(liaisons doubles).
Spectres de résonance magnétique nucléaire (CDCl^): un doublet centré à 6.50 p.p.m., J = 1.0 Hz (CH 3), un doublet dédoublé centré vers 6.58 p.p.m.
(masqué par le doublet à 6.50 p.p.m.), J = 2.0, ? Hz (CH 5) et un mul tiplet centré à 7.58 p.p.m. (6 protons aromatiques).
Spectre ultraviolet (EtOH): 228 mp, log e = 4.00; 272 mp, log e = 3.62; épaulement 298 mp, log e = 3.15.
Anal. Cale, pour G^jHgOg: C, 76.73 ; H, 5.70 Trouvé : C, 77.03 ; H, 4.47 Phényl-6 a-pyrone (IL).
On abandonne à la température ambiante pendant 14 heures, un mélange de tert-butyl at e de potassium (1.023 g), de dimethyl sulfoxyde anhydre
(150 ml) et de phényl-6 chloro-3 diméthoxy-2,2 dihydro-3,4 pyranne (.500 g). Le procédé habituel permet d'isoler un mélange de produits contenant la phényl-6 a-pyrone comme produit majoritaire.
Spectre infrarouge (CCl^): 1738-1720 cm-1 (lactone) 1635 cm-1 et 1552 cm-1 (liaisons doubles).
Spectre de résonance magnétique nucléaire (CDCl?): un doublet centré à 6.27 p.p.m., J = 9.5 Hz (CH 3), un doublet centré à 6.65 p.p.m., J = 7.0 Hz
63
(CH 5) et deux multiplets vers 7.50 et 7.90 p.p.m. (protons aromatiques).
BIBLIOGRAPHIE
1. N.P. Shusherina, N.D. Dmitrieva, E.À. Juk'yanets et R. Ya. Levina, Russ. Chem. Rev. 36_, 176 (1967).
2. C.R. Engel, R. Bouchard, A.F. de Krassny, L. Ruest et J. Lessard, Steroids, 14, 637 (1969).
3. G.R. Pettit, D.C. Fessier, K.D. Pauli, P. Hofer et J. Khight, J. Org. Chem. 35, 1398 (1970).
4. F. Sondheimer, W. McCrae et W.G. Salmond, J. Am. Chem. Soc. 91, 1228 (1969).
5. S.M. McElvain, Chem. Rev. 45, 453 (1949).
6. S.M. McElvain, E.R. Degginger et J.D. Dehun, J. Am. Chem. Soc. 76, 5736 (1954).
7. J.B. Hendrickson, D.J. Cram et G.S. Hammond, "Organic Chemistry, McGraw-Hill, 1970, p. 853.
8. C.N.R. Rao, "Chemical Applications of Infrared Spectroscopy", Academie Press, 1963, p. 190.
9. Vu Moc Thuy, Bull. Soc. Chim. France, 4429 (1970).
10. J. March, "Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", McGraw-Hill, 1968, p. 662.
11. L.B. Jackman et S. Sternhell, "Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry". Pergamon Press, 1969, pp. 291-
293.
L.J. Bellamy, "The Infra-red Spectra of Complex Molecules", Methuen Co., London, 1958, p. 179.
65
13. R. Stoener et Fr. Shenck, Ber., 60_, 2566 (1927).
14. F.W. Shueler et C. Hanna, J. Am. Chem. Soc., 73, 3528 (1951). 15. E.P. Kohler, J. Am. Chem. Soc., 44, 379 (1922).
16. T.L. Jacobs, D. Danbner et A. Danbner, J. Am. Chem. Soc., 80, 864 (1958).
APPENDICE A
Spectres infrarouge des chloro-3 dihyd.ro-3,4 pyrannes.
R” OMe
—OMe Cl f*'r H
Position des bandes (cm-1)
liaison double acétal
R-R -R - H 1662 1130-1090-1050-1030 r=r'=h; R"= C H3 1670 1120-1100-1040-1020 R=r'=h; 1665 1125-1095-1050-1020