Altérations de l’épissage et maladies rares
Texte intégral
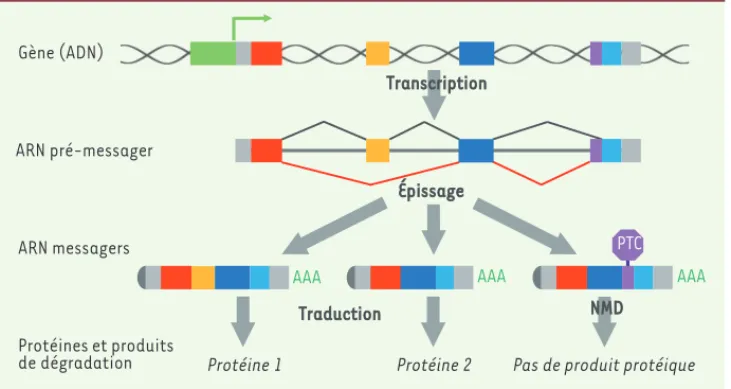
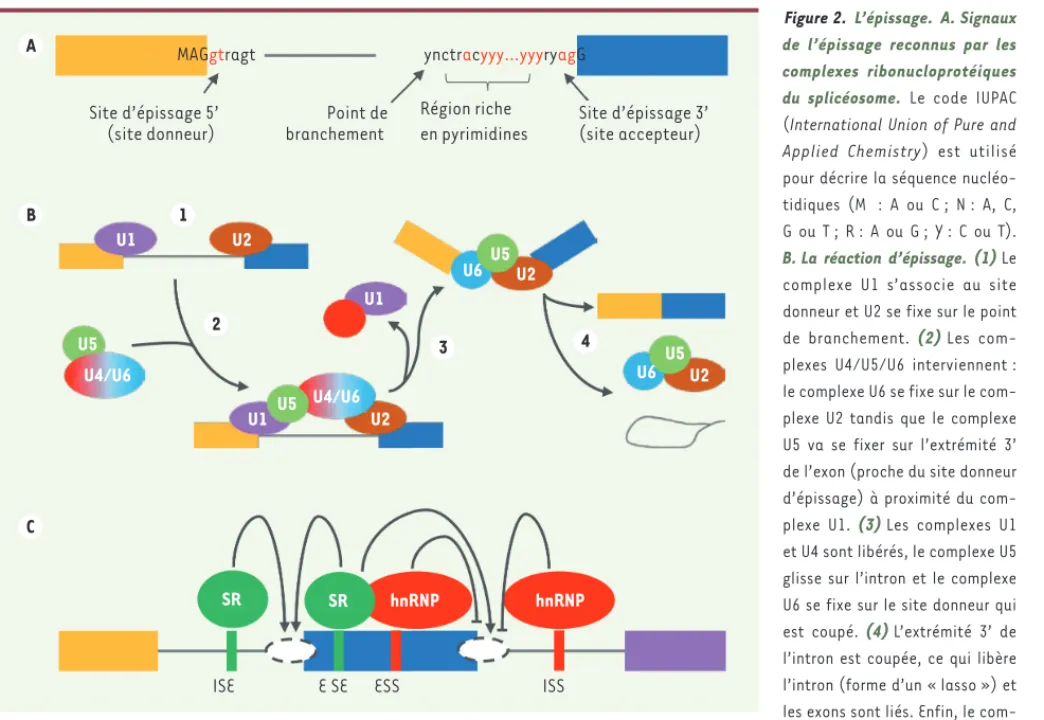
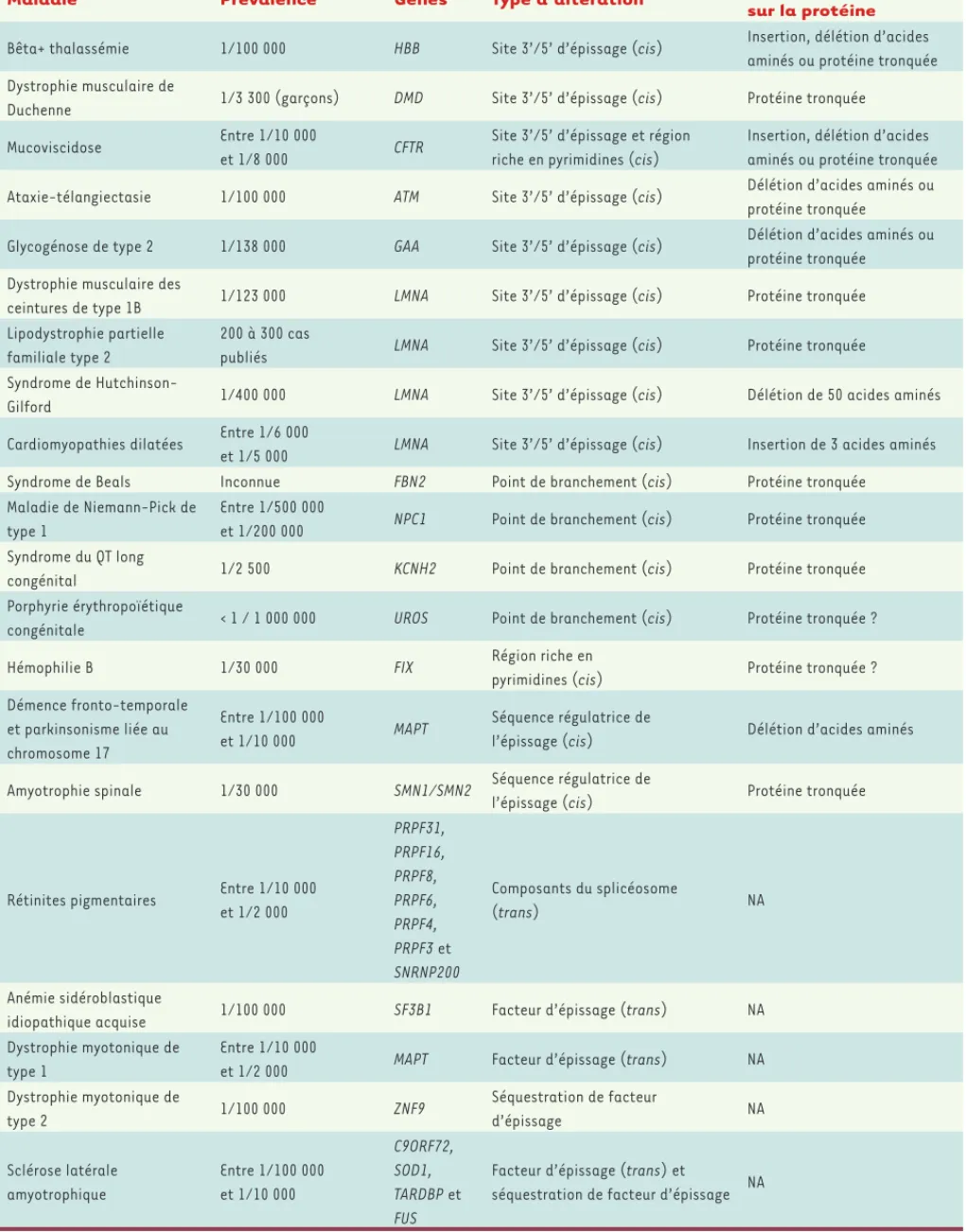
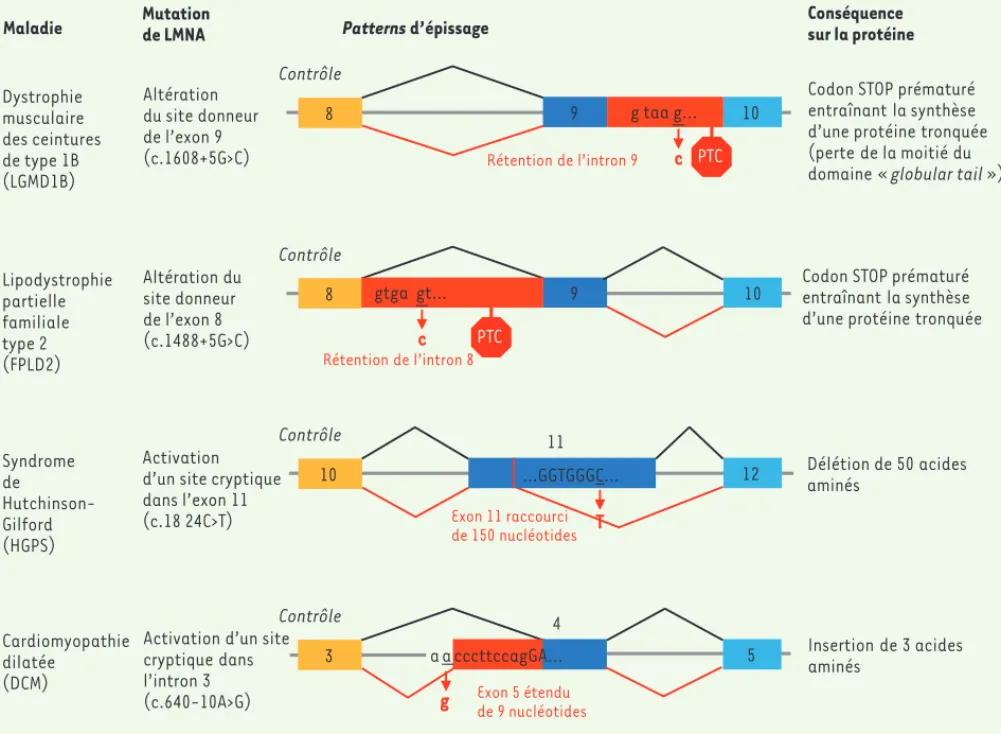
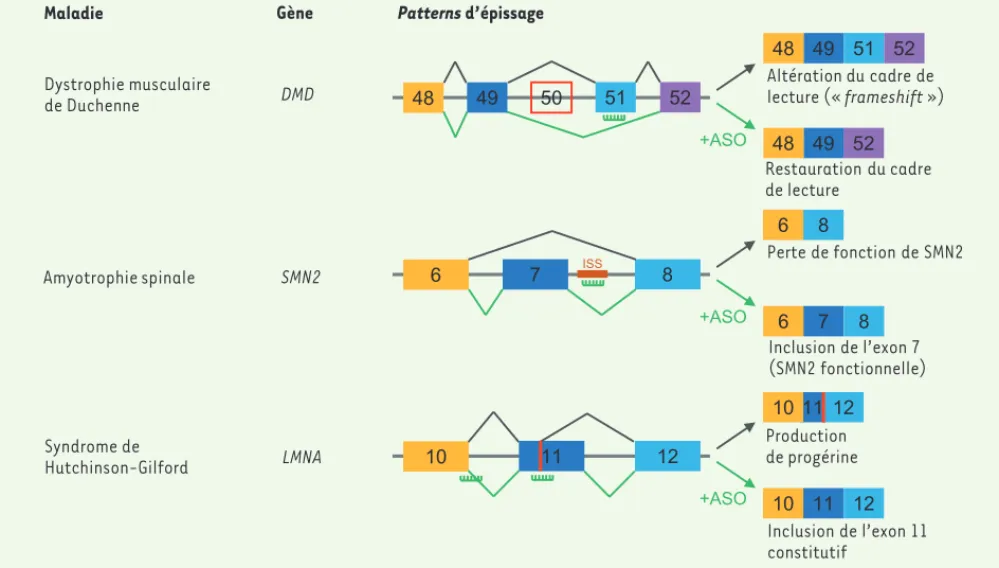
Figure
Documents relatifs
One consequence of the machinery developed in the previous section is that we can use it to provide an exact characterization of all of the Martin-L¨of random Turing degrees
Si SRSF3 recrute le complexe exosomal à ARN en l’absence d’EB2, en présence de la protéine virale, SRSF3 apparaît nécessaire à l’export nucléaire des ARNm viraux
The protons and the secondary particles penetrate into the target and specific energy deposition by one proton bunch on copper and tungsten targets along the beam direction is shown
tique dans la configuration ionique, de l’énergie de la liaison dative et de l’énergie de répulsion. La décroissance de l’énergieD*, dans la série
Cela a permis de réaliser un dosage plasmatique chez des patients atteints d'un cancer médullaire de la thyroïde (CMT) et quelques sujets témoins.. Le CCP II
Des neurones visuels ajustent le déplacement de leur champ récep teur avant la réalisation d'un mouve ment oculaire (p.. Vers le transfert de gènes dans les
En conclusion, nous avons montré que la formation d'une structure secon daire dans les cellules HeLa intervient dans le mécanisme d'exclusion de l'exon 6B.. Quelles sont
Although we do not disregard visual documents depicting past life at this trading port in general, we simulated the crowd mainly based on our rudimentary knowledge about the groups
