Effet de la photo-isomérisation sur la cristallisation de
copolymères polyéthersulfone-azobenzène
Mémoire
Simon Provencher
Maîtrise en chimie
Maître ès Sciences (M.Sc.)
Québec, Canada
© Simon Provencher, 2014
iii
RÉSUMÉ
La cristallinité d’un matériau peut largement influencer les propriétés de ce dernier, notamment la résistance mécanique, chimique et thermique. Au cours des dernières années, le groupe de recherche de Josée Brisson a étudié les processus de cristallisation avec un système de copolymères rigides-flexibles. Cette approche a pour but d’induire à la fois une flexibilité dans la chaîne pour faciliter le repliement à des endroits précis et une rigidité pour favoriser les interactions favorables à la cristallisation.
Cette approche a permis notamment d’étudier les processus de cristallisation et de faire cristalliser un dérivé du poly(éthersulfone) (PES) qui nécessite généralement des conditions rigoureuses. Le présent projet portera sur l’utilisation d’un comonomère azobenzène pouvant se photo-isomériser sous irradiation lumineuse pour induire une flexion dans la chaîne et étudier si le changement de conformation peut influencer la cristallisation du polymère.
v
TABLE DES MATIÈRES
RÉSUMÉ ... iii
TABLE DES MATIÈRES ... v
LISTE DES FIGURES ... ix
LISTE DES TABLEAUX ... xi
ABBRÉVIATIONS ET SYMBOLES ... xiii
REMERCIEMENTS ... xv
Introduction ... 1
Les thermoplastiques dans l’industrie ... 1
L’étude de la cristallisation des polymères... 2
Application du système rigide-flexible aux PES ... 3
L’azobenzène, un colorant aux multiples facettes ... 4
Chapitre 2 : Synthèse et caractérisation ... 9
2.1 Synthèses des monomères éthersulfones ... 9
2.2 Synthèse du monomère d’azobenzène ... 11
2.3 Synthèses des copolymères éthersulfone-azobenzène ... 12
2.4 Caractérisation et instrumentation ... 14
2.4.1 Analyse thermogravimétrique (TGA) ... 14
2.4.2 Calorimétrie différentielle à balayage (DSC) ... 14
2.4.3 Diffraction des rayons X ... 16
2.4.4 Chromatographie d’exclusion stérique (CES) ... 16
2.4.5 Spectrométrie UV-visible ... 17
2.4.6 Irradiation des échantillons par une lampe ultraviolette ... 17
Chapitre 3 : Discussion sur les synthèses ... 19
3.1 Synthèse du monomère éthersulfone ... 19
3.2 Synthèse du monomère azobenzène ... 21
3.3 Synthèses des copolymères éthersulfone-azobenzène ... 22
Chapitre 4 : Discussion sur la photo-isomérisation ... 27
4.1 Notions générales sur la photo-isomérisation de l’azobenzène ... 27
vi
4.1.2 Le mécanisme d’inversion33 ... 28
4.2 Photo-isomérisation du copolymère éthersulfone-azobenzène ... 29
4.2.1 Photo-isomérisation des copolymères en solution ... 29
4.2.2 Photo-isomérisation des copolymères en phase solide, sous forme de films minces ... 33
4.3 Cinétique d’isomérisation dépendante de la puissance de la lampe d’irradiation ... 35
4.4 Stabilité dans le temps de l’isomère cis ... 38
4.4.1 Stabilité de l’isomère cis du polymère P4ArS-4Azo ... 39
4.4.2 Stabilité de l’isomère cis du polymère P8ArS-4Azo ... 40
4.5 Méthode utilisée pour comparer de manière quantitative la cinétique de reconversion des polymères dans le noir ... 40
4.5.1 Comparaison entre le P4ArS-4Azo et le P8ArS-4Azo ... 41
4.6 Conclusion du chapitre ... 45
Chapitre 5 : Étude des propriétés thermiques ... 47
5.1 Notions générales sur les propriétés thermiques ... 47
5.1.1 La température de transition vitreuse ... 47
5.1.2 La température de fusion ... 48
5.1.3 Température de décomposition ... 49
5.2 La stabilité thermique ... 49
5.3 La température de transition vitreuse ... 52
Chapitre 6 : Effet de la photo-isomérisation sur la cristallisation ... 57
6.1 Description des méthodes utilisées pour l’étude de la cristallisation ... 57
6.1.1 Cristallisation par évaporation de solvant ... 57
6.1.2 Cristallisation par évaporation de solvant sous irradiation continue ... 58
6.2 Techniques de cristallisation infructueuses ... 59
6.2.1 Cristallisation sous forme de film mince ... 59
6.2.2 Formation de films minces par évaporation de solvant sous irradiation continue ... 60
6.2.3 Irradiation d’un échantillon chauffé au-dessus de sa température de transition vitreuse ... 60
6.3 Explication concernant les résultats négatifs des techniques de la section 6.2 ... 61
6.4 Résultat de la cristallisation par évaporation de solvant sous irradiation ... 62
6.5 Effet de l’isomère sur la cristallisation ... 63
6.6 Pré-orientation des chaînes ... 63
6.7 Effet de l’orientation ortho/meta/para du polymère ... 65
vii
6.9 Conclusion de ce chapitre ... 67
Chapitre 7 : Conclusions et perspectives ... 69
7.1 Synthèses ... 69 7.2 Photo-isomérisation ... 70 7.3 Propriétés thermiques ... 70 7.4 Tentatives de cristallisation ... 71 ANNEXES ... 73 SPECTRES RMN ... 73 BIBLIOGRAPHIE ... 79
ix
LISTE DES FIGURES
Figure 1. Augmentation notable de la cristallinité d’un p(ES-b-butène), récemment publié par Faye,
Leduc et Brisson11. ... 4
Figure 2. La photo-isomérisation de l’azobenzène se produisant lors de l’absorption d’un photon. ... 5
Figure 3. Différence entre un polymère ayant un azobenzène en chaîne latérale (a) et en chaîne principale (b). Les azobenzènes sont représentés par les segments rouges. ... 6
Figure 4. Schéma général d’un polymère ayant des propriétés cristaux-liquides grâce aux azobenzènes. Les azos sont représentés par les segments rouges. ... 7
Figure 5. Schéma général de la synthèse de Williamson. ... 19
Figure 6. Synthèse du 4ArS-OMe, la forme protégée du 4ArS-OH. Le groupement méthoxy en fin de chaîne sert de groupement protecteur pour éviter une polymérisation non désirée... 20
Figure 7. Schéma général de la réaction de déprotection des monomères par le tribromure de bore. ... 20
Figure 8. Schéma de synthèse de l’azobenzène, d’après les travaux de Zhang et Jiao25. ... 21
Figure 9. Mécanisme réactionnel d’une substitution nucléophile aromatique impliquant la formation d’un complexe Meisenheimer. ... 21
Figure 10. Réaction générale d’une polycondensation de type A2B2. ... 22
Figure 11. Comparaison de la structure moléculaire d’un PES typique et du P4ArS-4Azo, un des polymères synthétisés dans le cadre de cette étude. ... 23
Figure 12. Schéma représentant la formation d’un complexe Meisenheimer réactif, puisqu’il est activé par un groupement R’ électroattracteur. ... 25
Figure 13. Schéma du mécanisme d’isomérisation par rotation. ... 28
Figure 14. Schéma du mécanisme d’isomérisation par inversion. ... 29
Figure 15. Positionnement des échantillons lors de l’irradiation. ... 30
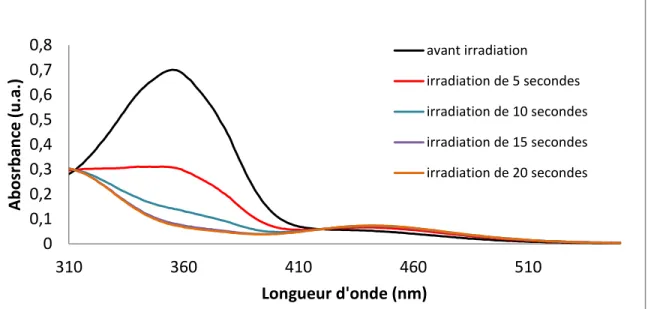
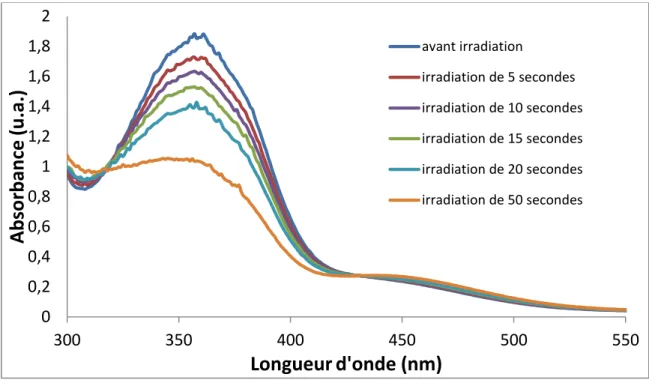
Figure 16. Spectre d’absorption UV-visible du P4ArS-4Azo en solution sous une lampe UV de 9W. 31 Figure 17. Spectre d’absorption UV-visible du P8ArS-4Azo en solution sous une lampe UV de 9W. 31 Figure 18. Graphique montrant la variation en unité d’absorbance du pic maximal d’absorption en fonction du temps d’irradiation. ... 33
Figure 19. Spectre d’absorption UV-visible du P4ArS-4Azo en film mince sous une lampe UV de 9W. ... 34
x
Figure 20. Vitesse d’isomérisation du P8ArS-4Azo avec une puissance de lampe de 9W (a) et 18W
(b). ... 35
Figure 21. Représentation graphique de la décroissance du maximum d’absorbance du P8ArS-4Azo en fonction du temps d’irradiation et de la puissance de la lampe (en Watt). ... 36
Figure 22. Décroissance du logarithme naturel de l’absorbance maximale en fonction du temps d’irradiation (en seconde) et de la puissance de la lampe d’irradiation (en Watt). ... 37
Figure 23. Évolution du spectre d’absorption du P4ArS-4Azo dans le noir en fonction du temps. ... 39
Figure 24. Évolution du spectre d’absorption du P8ArS-4Azo dans le noir en fonction du temps. ... 40
Figure 25. Structure moléculaire du P4ArS-4Azo (au-dessus) et du P8ArS-4Azo (en-dessous). ... 42
Figure 26. La croissance du pourcentage d’isomère trans en fonction du temps écoulé après l’irradiation. ... 44
Figure 27. Courbes TGA des trois principaux polymères synthétisés. ... 50
Figure 28. Schéma représentant, de haut en bas, le P2ArS-4Azo, le P4ArS-4Azo et le P8ArS-4Azo. ... 52
Figure 29. Courbes DSC des trois principaux polymères synthétisés. ... 53
Figure 30. Clichés de diffraction des rayons X du P4ArS-4Azo après cristallisation par évaporation de solvant sous irradiation continue. ... 62
Figure 31. Rétrécissement du pic amorphe lors de la cristallisation. ... 63
Figure 32. Schéma représentant le modèle de cristallisation par la phase mésomorphique selon Strobl39. ... 64
Figure 33. Différence entre l’orientation para et meta de l’azobenzène. ... 65
Figure 34. Reproduction de la figure 9 du chapitre 3 montrant le complexe de Meisenheimer. ... 65
Figure 35. Différence de l’effet résonant entre l’orientation para et meta. ... 66
Figure 36. Structure adoptée par l’isomère cis. Les groupements phényle ne sont pas dans le même plan. ... 67
xi
LISTE DES TABLEAUX
Tableau 1. Masses moléculaires moyennes en nombre (Mn) et en poids (Mw) et indice de
polydispersité Ip des différents polymères synthétisés. ... 24 Tableau 2. Ratios des deux polymères, calculés à partir des résultats des spectres des figures 23 et 24. ... 42 Tableau 3. Températures de dégradation des trois polymères synthétisés. ... 50 Tableau 4. Reproduction du tableau présenté au chapitre 3 montrant les différentes masses molaires des polymères synthétisés. ... 51 Tableau 5. Tableau compilant les différentes valeurs de température de transition vitreuse (Tg) des principaux polymères (températures de mi-transition). ... 53
xiii
ABBRÉVIATIONS ET SYMBOLES
° : Degré °C : Degré Celsius δ : Déplacement chimique % : Pourcentage Θ : Théta BBr3 : Tribromure de bore CDCl3 : Chloroforme deutéréCES : Chromatographie d’exclusion stérique CH2Cl2 : Dichlorométhane
CH3I : Iodure de méthane
d : doublet
DMAc : Diméthylacétamide
DSC : Calorimétrie différentielle à balayage éq. : Équivalent
g : gramme
g/mol : gramme par mole h : heure
H : Proton
HCl : Acide chlorhydrique K2CO3 : Carbonate de potassium
KOH : Hydroxyde de potassium M : molaire
m : multiplet mg : milligramme
MgSO4 : Sulfate de magnésium
mg/mL : milligramme par millilitre mL : Millilitre
mmol : Millimole nm : Nanomètre
xiv
PEK : Polyéthercétone PES : Polyéthersulfone ppm : Partie par million
RMN : Résonnance magnétique nucléaire RMN 1H : RMN proton
s : Singulet
Tf : Température de fusion
Tdécomp : Température de décomposition
Tg : Température de transition vitreuse
TGA : Analyse thermogravimétrique TP : Température pièce
UV : Ultraviolet
xv
REMERCIEMENTS
J’aimerais d’abord remercier ma directrice de maîtrise, la professeure Josée Brisson, de m’avoir accueilli dans son laboratoire de recherche pour la maîtrise mais aussi pour un stage d’été lors du baccalauréat. Son soutien, ses conseils, sa disponibilité et son écoute m’ont permis de mener à bien mon projet de maîtrise, et ainsi surmonter les obstacles rencontrés durant ces deux années.
Je tiens également à remercier les membres du groupe de recherche Brisson : Adrien, Mariane, François, Huan et David, pour l’esprit d’équipe, les échanges et les conversations qui ont embelli mon expérience des études supérieures.
Je tiens à remercier le CRSNG d’avoir fourni le financement nécessaire pour mener à terme ce projet.
Merci également aux différents membres du département de chimie pour m’avoir aidé avec plusieurs aspects techniques du projet. Je tiens à remercier Mme Rodica Plesu du CERMA pour son aide avec la chromatographie d’exclusion stérique, la thermogravimétrie et la calorimétrie différentielle à balayage. J’aimerais également remercier Serge Groleau et Marilyne Marois de m’avoir laissé utiliser leurs appareils de spectroscopie UV-Visible.
Finalement, je tiens à remercier ma famille et mes amis de m’avoir soutenu durant mes études universitaires. Merci à ma famille : Denis, Louise et Julien, d’avoir été présents pour moi. Merci à Stéphanie de faire partie de ma vie. Merci finalement à tous mes amis pour m’avoir permis de décrocher de temps à autres, et particulièrement à Marcelin pour ses conseils en chimie et son intelligence légendaire.
1
Introduction
Les thermoplastiques dans l’industrie
Depuis l’introduction des thermoplastiques, les matériaux traditionnels comme le métal, les oxydes de métaux et les alliages sont de plus en plus délaissés pour faire place à ces nouveaux matériaux ayant des propriétés mécaniques comparables, voire supérieures, tout en étant largement plus légers et plus faciles à usiner. Avec les récents avancements scientifiques dans le domaine des hautes technologies, il existe un besoin criant pour des matériaux résistants, robustes et qui sont à la fois légers et malléables.
Tout récemment, ce changement dans l’industrie s’est accéléré dans plusieurs secteurs, en particulier dans les pièces d’usinage. Ainsi, des géants comme BASF se lancent maintenant dans le marché des thermoplastiques hautes performances en misant sur les thermoplastiques bien établis tels que le polyétheréthercétone (PEEK), les polysulfones et les polyamides comme matériaux de choix pour la manufacture de pièces automobiles, entre autres1. Ce mouvement dans l’industrie s’est
également fait sentir dans la recherche académique, se manifestant par une progression constante du nombre d’articles concernant les thermoplastiques.
Parmi les polymères prometteurs pour une variété d’applications, se trouve le polyéthersulfone (PES), aussi parfois nommé polysulfone. Ce polymère d’ingénierie montre entre autres d’excellentes résistances thermique, mécanique et chimique. Il possède une température de transition vitreuse (Tg)
et une température de dégradation (Tdécomp) élevées, soit d’environ 200 °C et 500 °C,
respectivement2. Ces propriétés intéressantes en font un bon candidat pour des applications dans
des domaines très variés tels que l’aérospatiale3, les piles à combustibles4 et l’ultrafiltration5, 6, pour
en nommer quelques-uns.
L’utilisation du PES est cependant freinée par plusieurs contraintes, qui peuvent différer selon l’application visée. L’étude des propriétés de ce polymère est donc de première importance pour trouver des solutions possibles et permettre son utilisation dans des applications plus variées. En particulier, le présent projet se focalisera sur l’étude de l’introduction d’un groupement azo au sein de la chaîne de ce polymère, et de l’effet sur les propriétés thermiques et sur la cristallisation de ce
2
polymère. La cristallisation est un phénomène important qui a un impact majeur sur les propriétés mécaniques d’un matériau. Un polymère ayant une cristallinité élevée sera très résistant aux attaques chimiques et aux solvants, mais sera plus cassant et peu malléable. Il importe donc de bien comprendre et contrôler le processus de cristallisation d’un polymère pour obtenir un matériau ayant les propriétés voulues.
L’étude de la cristallisation des polymères
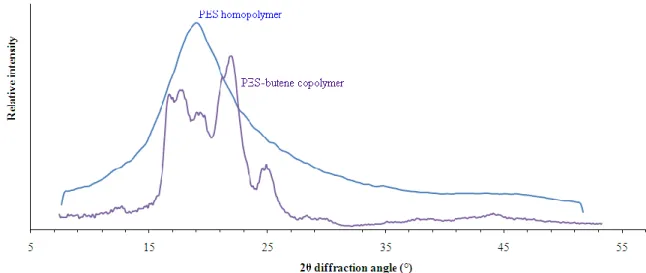
Le PES est un polymère connu pour avoir une structure amorphe, avec un pic large de diffraction des rayons X centré à l’angle 2θ = 20°7, Ce polymère est également résistant aux tentatives de
cristallisation par solvant ou par traitement thermique, en règle générale. Cependant, une étude a rapporté l’observation de croix de malte par microscopie optique, indiquant la présence de sphérolites cristallines8. Par contre, l’auteur fait part de la complexité du processus, indiquant que ces
observations ne sont possibles que lors d’une interaction avec du chloroforme. Le PES peut donc cristalliser, mais seulement dans des conditions très précises. Cela peut sembler surprenant, si on compare le PES avec son homologue structural, le poly(éthercétone) (PEK). En effet, les deux polymères sont semblables en terme de rigidité de la chaîne et également d’un point de vu chimique, avec comme différence notable la substitution du groupement sulfone par une cétone. En comparaison avec le PES, le PEK est un polymère semi-cristallin, avec un taux de cristallinité pouvant atteindre 30 % tout en conservant des propriétés mécaniques intéressantes pour certaines applications9.
D’un point de vue fondamental, il est impératif de comprendre les phénomènes de cristallisation des polymères afin de mieux contrôler ce processus, que ce soit pour l’inhiber ou pour le favoriser, dépendamment de l’utilisation visée. Pour cette raison, la cristallisation des polymères a été l’un des principaux sujets de recherche au sein du groupe de recherche de Josée Brisson ces dernières années. Les systèmes utilisés pour étudier la cristallinité sont des copolymères rigides-flexibles. Ces systèmes sont des polymères cristallisant par un mécanisme de repliement de chaîne, à des points précis dans la chaîne, dicté par la différence de flexibilité entre différentes unités. Plus précisément, il s’agit de copolymères alternés de type AB ayant un segment rigide, généralement composé de chaînes aromatiques, et d’un groupement flexible de faible taille et qui est souvent un simple alcane.
3
La présence d’un tel segment flexible crée des points de flexion naturels au sein de la chaîne, facilitant ainsi le repliement à des endroits précis que l’on peut contrôler en modifiant la structure et la longueur des segments rigides et des segments flexibles. Par exemple, une étude de notre groupe sur la relation entre la structure et la cristallisation a permis d’évaluer qu’une longueur de segment rigide correspondant à environ 8 répétitions du lien éther aromatique dans le cas du PEEK était suffisant pour obtenir un haut taux de cristallinité10. De plus, la longueur de l’alcane utilisé comme
comonomère avait également un impact sur la cristallisation, puisqu’il doit être suffisamment long pour permettre une flexion complète, mais pas trop long, afin d’éviter la formation d’une seconde phase cristalline. Dans le cas des chaînes alcanes, la longueur utilisée se trouvait donc entre 4 et 12 groupements CH210. Les premières études de ces copolymères ont été réalisées sur des monomères
de longueur précise dérivés du PEEK, polymère connu pour être semi-cristallin. L’une des suites logiques de ces travaux était de voir si ce système pouvait être utilisé pour induire une cristallisation dans un polymère tel que le PES qui, généralement, ne cristallise pas.
Application du système rigide-flexible aux PES
Le PES est en quelque sorte un homologue structural du PEEK, ayant comme différence la substitution du groupement cétone par un groupement sulfone. Cependant, contrairement au PEEK, le PES est généralement considéré comme un polymère amorphe qui cristallise peu, et seulement dans des conditions précises. L’idée était de déterminer si le système alternant les segments rigides et flexibles pourrait permettre d’induire une cristallisation du segment rigide dérivé du PES. Dans une étude récemment publiée dans notre groupe, ce système a été utilisé avec succès pour induire une cristallinité dans des copolymères de type PES et PEES. 11
4
Figure 1. Augmentation notable de la cristallinité d’un p(ES-b-butène), récemment publié par Faye,
Leduc et Brisson11.
En introduisant un groupement flexible à intervalles réguliers le long de la chaîne principale, le repliement de chaîne est facilité par rapport à l’homopolymère, permettant une augmentation significative de la cristallinité.
De ces résultats concluants, le projet de ce mémoire a émergé. Le questionnement initial était de se demander si l’introduction d’un groupement flexible était le seul moyen d’induire une cristallisation du polymère. Puisque l’un des rôles principaux de la flexibilité de l’alcane est de faciliter l’orientation les segments rigides selon une certaine conformation propice à la cristallisation, nous nous sommes demandé s’il existait d’autres moyens de forcer les segments rigides à adopter une conformation particulière. Pour répondre à cette question a été proposée l’idée d’utiliser l’azobenzène au lieu d’une chaîne d’alcane pour effectuer ce travail.
L’azobenzène, un colorant aux multiples facettes
Le bis(phényl)diazène, ou plus communément appelé azobenzène ou tout simplement « Azo », est une molécule qui a été étudiée et utilisée dans diverses applications depuis maintenant plusieurs décennies. D’un point de vue industriel, les azobenzènes sont des composés surtout utilisés comme pigments, dû à leur coloration très intense et la possibilité de moduler assez aisément leur coloration en substituant les protons des groupements phényle par différents groupements électro-donneurs ou électro-attracteurs12,13. Les azos sont également connus pour être des molécules rigides anisotropes,
5
faisant d’eux des candidats idéals pour des applications liquide-cristallines12, 13. Ce n’est que plus
tard que l’on observa les propriétés étonnantes de l’azobenzène qui est de se photo-isomériser efficacement et de manière réversible sous l’action de la lumière ultraviolette. Dans le cas d’un azobenzène non-substitué, le maximum d’absorption se situe autour de 360 nanomètres. Cette molécule absorbe donc bien les rayons de type UV-A.
L’azobenzène a deux isomères possibles. Un isomère trans thermodynamiquement stable et un isomère cis métastable. Cet isomère cis est obtenu lors de l’absorption d’un photon12. Cette
isomérisation se fait très rapidement, de sorte que les azobenzènes ne sont pas des molécules fluorescentes, sauf dans des cas bien précis. L’isomère cis est cependant dans un état métastable, et le système peut ensuite relaxer vers sa forme trans après un certain temps qui dépend largement du degré de substitution de l’azobenzène, de la viscosité du milieu et de la température.
Figure 2. La photo-isomérisation de l’azobenzène se produisant lors de l’absorption d’un photon.
De par cette dépendance à la température, tel qu’illustré à la figure 2, on peut accélérer le retour à l’isomère trans en augmentant la température. On peut également éclairer l’azobenzène avec de la lumière visible pour accélérer le processus13. Cette réaction réversible d’isomérisation est rapide,
quantitative et très stable dans le temps, dans le sens que plusieurs cycles peuvent être effectués avant d’observer une détérioration. Cette forme de photo-isomérisation réversible initiée par la lumière ouvre la possibilité à une variété d’applications dans divers domaines, en particulier dans le domaine des matériaux intelligents.
Initialement, certains chercheurs ont considéré l’azobenzène comme un comonomère potentiel dans les polymères conjugués, dû à son caractère conjugué, sa richesse en électrons due aux atomes d’azote et la rigidité structurale de la molécule. Un des premiers essais de polymérisation a été effectué sur le poly-paraphénylène par Izumi et al.14. Leurs travaux ont démontré que l’azobenzène
6
polymère pouvait s’oxyder et se réduire de manière réversible dans des solvants usuels15. Ils ont
également montré que la photo-isomérisation d’un azobenzène dans la chaîne principale n’était pas inhibée par une structure polymérique rigide.
Depuis quelques années, les publications concernant l’étude d’azobenzènes dans divers systèmes se sont multipliées et de nombreuses applications sont envisagées. Certaines applications sont particulièrement étudiées, tels que leur utilisation en tant que cristaux liquides16, 17, 18 et en tant que
commutateurs moléculaires19. D’autres applications plus exotiques sont proposées, par exemple
comme moyen rapide de séparer des nanotubes de carbone20, ou afin de modifier de manière
réversible l’hydrophobicité d’une surface21 ou encore de créer des micelles de manière réversible
avec la lumière pour le transport de médicament22.
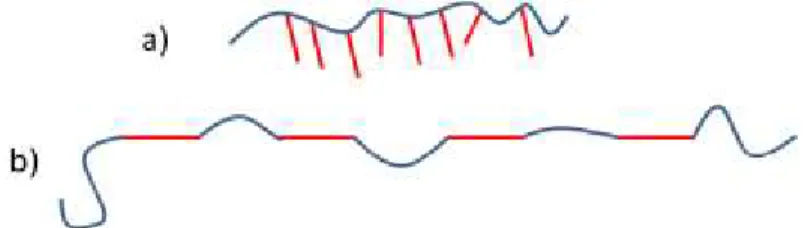
Dans la majorité des cas, les systèmes utilisés impliquent des azobenzènes en tant que chaîne latérale et non en tant que comonomère de la chaîne principale. La raison pour ceci est que la plupart des propriétés exploitables des azobenzènes implique que ces molécules se « voient » pour interagir entre elles.
Figure 3. Différence entre un polymère ayant un azobenzène en chaîne latérale (a) et en chaîne principale (b). Les azobenzènes sont représentés par les segments rouges.
En effet, très peu d’articles faisant l’étude des propriétés des azobenzènes en chaîne principale sont disponibles, en comparaison avec la quantité imposante d’articles traitant de chaînes latérales. Cependant, afin d’étudier l’effet de la photo-isomérisation des azobenzènes sur la cristallisation d’un polymère, le système à utiliser est le système en chaîne principale, tel que représenté à la figure 3-b. Si nous utilisons le système en chaîne latérale, comme à la figure 3-a, nous allons simplement observer une phase liquide-cristalline en faisant cristalliser les chaînes latérales lorsqu’elles sont bien organisées dans l’isomère trans.
7 Figure 4. Schéma général d’un polymère ayant des propriétés cristaux-liquides grâce aux
azobenzènes. Les azos sont représentés par les segments rouges.
Dans le cas de l’étude présente, nous étudierons l’effet de la photo-isomérisation des azobenzènes en tant que copolymère dans la chaîne principale contenant des segments rigides de poly(éthersulfone). En changeant l‘alignement des segments les uns par rapport aux autres, ainsi que la distance moyenne entre ces segments, nous désirons vérifier si la photo-isomérisation de l’azobenzène peut soit favoriser ou défavoriser la cristallisation de la chaîne. Nous voudrons donc étudier ce comportement face à la lumière et également vérifier si ce phénomène est réversible et rapide.
9
Chapitre 2 : Synthèse et caractérisation
Ce chapitre portera sur la description pratique des différentes synthèses ainsi qu’une brève description des différents appareils de caractérisation qui ont été utilisés dans le cadre de ce projet.
2.1 Synthèses des monomères éthersulfones
2.1.1 4-fluorophényl-4’-hydroxyphényl sulfone23
Dans un ballon rond de 100 mL équipé d’un barreau magnétique, on fait réagir 5,09 grammes (20,0 mmol) de bis(4-fluorophényl) sulfone avec 2,24 grammes (40,0 mmol) d’hydroxyde de potassium dans 25 mL de DMSO. Le milieu réactionnel est ensuite amené à 75 °C pour une durée de 20 heures sous une atmosphère inerte. Le mélange est ensuite dilué avec 100 mL d’eau légèrement basique et lavé avec trois petites portions de toluène. La couche aqueuse est ensuite acidifiée avec de l’acide chlorhydrique pour faire précipiter le produit. Ce produit est ensuite filtré, lavé à l’eau à quelques reprises et séché au four pendant la nuit à 60 °C pour récolter 2,674 grammes de produit pour un rendement de 53 %. RMN 1H (400 MHz, DMSO-d6, TMS,
température de la pièce (tp): δ 10,64 (singulet (s), 1H), 7,87-7,94 (multiplet (m), 2H), 7,72-7,75 (m, 2H), 7,35-7,41 (m, 2H), 6,87-6,91 (m, 2H) ppm.
2.1.2 4-fluorophényl-4’-methoxyphényl sulfone23
Dans un ballon rond de 100 mL équipé d’un barreau magnétique, on fait réagir 2,52 grammes (10,0 mmol) de 4-fluorophényl-4’-hydroxyphényl sulfone avec 2,07 grammes (15,0 mmol) d’hydroxyde de potassium et 0,62 millilitre (10,0 mmol) d’iodure de méthyle dans 20 mL de DMAc. Le milieu réactionnel est ensuite amené à 120 °C pour une durée de 18 heures sous une atmosphère d’argon. Le mélange est ensuite précipité dans l’hexane. Le produit résultant est par la suite filtré, puis lavé copieusement avec une solution diluée d’hydroxyde de potassium. Le produit est finalement séché au four à 60 °C durant une nuit pour récolter 2,423 grammes de produit, pour un rendement de 91%. RMN 1H (400 MHz, CDCl3, TMS, tp): δ 7,91-7,95 (m,
10
2.1.3 4, 4’-bis(p-méthoxyphénoxy)diphényl sulfone (4ArS-OMe)24
Dans un ballon rond de 100 mL équipé d’un barreau magnétique, on fait réagir 5,09 grammes (20,0 mmol) de 4,4’-difluorophényl sulfone avec 4,97 grammes (40,0 mmol) de 4-méthoxyphénol et 6,08 grammes (44,0 mmol) de carbonate de potassium. 30 mL de DMAc est ensuite ajouté et le milieu réactionnel est amené à 180 °C pour une durée de 20 heures. Le mélange est par la suite précipité en le versant dans 100 mL d’acide chlorhydrique 5%. Le produit résultant est ensuite filtré, lavé copieusement à l’eau et séché au four à 60 °C pour récolter 2,366 grammes de produit, pour un rendement de 52 %. RMN 1H (400 MHz, CDCl3, TMS, tp): δ
7,81-7,85 (m, 4H), 6,89-6,99 (m, 12H), 3,82 (s, 6H) ppm.
2.1.4 4,4’-bis(p-hydroxyphénoxy)diphényl sulfone (4ArS-OH)24
Dans un ballon sec de 100 mL équipé d’un barreau magnétique, on commence par dissoudre 7,23 grammes (15,6 mmol) de 4ArS-OMe dans 20 mL de dichlorométhane sec. On installe alors une ampoule à additionner sèche sur le ballon, on purge le montage à l’argon, puis on amène le montage à 0°C avec un bain de glace. Puis, 25,0 grammes (99,8 mmol) de tribromure de bore sont ajoutés goutte-à-goutte à la solution. Le mélange est gardé à 0°C pendant une heure, puis 5 heures additionnelles à température pièce. La réaction est ensuite arrêtée en versant le mélange goutte-à-goutte dans 300 mL d’eau froide. La poudre blanche ou beige résultante est filtrée, puis lavée copieusement avec de l’eau, jusqu’à ce que l’eau de lavage ne soit plus acide au papier pH. Le produit est finalement lavé au dichlorométhane puis séché au four à 60°C pour récolter 4,127 grammes de produit, pour un rendement de 98 %. RMN 1H (400MHz, DMSO-d6, TMS, tp): δ 9,52 (s, 2H),
7,84-7,88 (m, 4H), 6,94-7,03 (m, 8H), 6,79-6,83 (m, 4H) ppm.
11 Dans un ballon rond de 100 mL équipé d’un barreau magnétique, on fait réagir 4,35 grammes (10,0 mmol) de 4ArS-OH avec 5,33 grammes (20,0 mmol) de 4-fluorophényl-4-méthoxyphénylsulfone et 3,04 grammes (22,0 mmol) de carbonate de potassium. 40 mL de DMAc est ajouté puis le tout est amené à une température de 180°C pour une durée de 20 heures. Par la suite, le mélange est précipité en le versant dans 100 mL d’acide chlorhydrique 5%. La poudre obtenue est filtrée, lavée copieusement avec de l’eau et séchée au four à 60°C pour obtenir 7,062 grammes de produit, avec un rendement de 82 %. RMN 1H (400 MHz, CDCl3, TMS, tp): δ
7,89-7,93 (m, 12H), 6,99-7,09 (m, 20H), 3,87 (s, 6H) ppm.
2.1.6 4,4'-bis(4-(4-(4-hydroxyphénylsulfonyl)phénoxy)phénoxy)diphénylsulfone (8ArS-OH)24
Dans un ballon rond sec de 100 mL équipé d’un barreau magnétique, on commence par dissoudre 11,6 grammes (12,5 mmol) de 8ArS-OMe dans 30 mL de dichlorométhane sec. On installe alors une ampoule à additionner sèche sur le ballon, on purge le montage à l’argon, puis on amène le ballon à 0°C avec un bain de glace. Puis, 25,0 grammes (99,8 mmol) de tribromure de bore sont ajoutés goutte-à-goutte à la solution. Le mélange est gardé à 0°C pendant une heure, puis 5 heures additionnelles à température pièce. La réaction est ensuite arrêtée en versant le mélange goutte-à-goutte dans 500 mL d’eau froide. La poudre blanche ou beige résultante est filtrée, puis lavée copieusement avec de l’eau, jusqu’à ce que l’eau de lavage ne soit plus acide au papier pH. Le produit est finalement lavé au dichlorométhane puis séché au four à 60°C pendant 24 heures pour récolter 10,2 grammes de produit pour un rendement de 91 %. RMN 1H (400MHz, DMSO-d6,
TMS, tp): δ 9,52 (s, 2H), 7,84-7,88 (m, 4H), 6,94-7,03 (m, 8H), 6,79-6,83 (m, 4H) ppm.
2.2 Synthèse du monomère d’azobenzène
2.2.1 Bis(4-fluorophényl)diazène (4Azo)25
12
Dans un ballon rond de 100 mL équipé d’un barreau magnétique, 2,88 mL (30,0 mmol) de 4-fluoroaniline est dissous dans 10 mL de toluène. Puis, 0,218 mL (2,70 mmol) de pyridine et 0,129 gramme (0,90 mmol) de bromure de cuivre(I) sont ajoutés au milieu réactionnel. Le mélange est ensuite amené à 60 °C pour une durée de 24 heures en présence d’air ambiant (1 atm). Par la suite, le mélange est concentré brièvement sur évaporateur rotatif, puis purifié par chromatographie éclair sur une courte colonne de silice, avec comme éluent un mélange acétate d’éthyle/éther de pétrole en rapport 1 pour 4. La solution orange obtenue est alors évaporée avec l’évaporateur rotatif pour produire 2,58 grammes de produit, pour un rendement de 79%. RMN
1H (400 MHz, CDCl3, TMS, tp): δ 7,90-7,96 (m, 4H), 7,17-7,23 (m, 4H) ppm.
2.2.2 Bis(3-fluorophényl)diazène (3Azo)25
La synthèse est effectuée de la même façon que pour le 4Azo, seule la substitution du produit de départ étant différente (meta au lieu de para). La séparation par chromatographie éclair s’effectue de la même manière, à l’exception près que l’éluent a été modifié de sorte qu’un mélange hexanes/acétate d’éthyle 3:1 a été utilisé. Il faut noter toutefois que la différence est minime entre l’hexane et l’éther de pétrole, cette modification est survenue simplement parce que l’hexanes était disponible en plus grande quantité au laboratoire au moment de la synthèse.
2.3 Synthèses des copolymères éthersulfone-azobenzène
2.3.1 Synthèse du copolymère P4ArS-4Azo24
Dans un ballon à fond rond muni d’un barreau magnétique, on fait réagir 0,30 gramme (0,69 mmol) de 4ArS-OH avec 0,15 gramme (0,69 mmol) de 4Azo et 0,143 gramme (1.035 mmol) de carbonate de potassium dans 6 mL de DMAc. La réaction est amenée à 180 °C pour une durée de 24 heures sous atmosphère d’argon. La
13 réaction est ensuite arrêtée en laissant retourner à température pièce, puis le produit est précipité en versant le mélange dans une solution d’acide chlorhydrique 5%. Le produit jaune récolté est filtré et lavé abondamment avec de l’eau. Ensuite, le produit est lavé successivement avec du méthanol, puis de l’acétone pour éliminer les réactifs de départ et les plus petites chaînes de polymère. Le produit est finalement séché au four à 80 °C pendant 48 heures.
2.3.2 Synthèse du copolymère P2ArS-4Azo24
Dans un ballon à fond rond de 100 mL muni d’un agitateur magnétique, on fait réagir 0,574 gramme (2,29 mmol) du bis(4-hydroxyphényl)sulfone avec 0,50 gramme (2,29 mmol) et 0,633 gramme de carbonate de potassium dans 10 mL de DMAc. La réaction se poursuit durant 24 heures à 180 °C. Une fois la réaction terminée, on précipite le contenu du ballon dans 200 mL d’eau rendue légèrement acide par quelques gouttes d’acide chlorhydrique concentré. Le produit obtenu est une poudre jaunâtre. Ce produit est filtré sur Büchner, lavé abondamment avec de l’eau et du méthanol, puis séché au four à 80 °C pendant 48 heures ou plus.
2.3.3 Synthèse du copolymère P8ArS-4Azo24
Dans un ballon à fond rond de 100 mL muni d’un agitateur magnétique, on fait réagir 0,276 gramme (0,31 mmol) de 8ArS-OH avec 0,067 gramme (0,31 mmol) de 4Azo et avec 0,086 gramme (0,62 mmol) de carbonate de potassium dans environ 10 mL de DMAc. On laisse la réaction se produire à 180 °C pour 20 heures sous une atmosphère d’argon. Une fois la réaction terminée, on récolte une poudre jaunâtre lorsque le mélange est précipité dans l’eau rendue légèrement acide par ajout de HCl 5%. Le polymère est ensuite filtré sur Büchner et lavé abondamment avec de l’eau et du méthanol. Finalement, le produit est séché au four à 80 °C pendant 48 heures ou plus.
14
2.4 Caractérisation et instrumentation
2.4.1 Analyse thermogravimétrique (TGA)
Les analyses thermogravimétriques ont été effectuées sur l’appareil TGA/SDTA851e/SF/l100°C de Mettler Toledo. L’appareil est équipé d’une balance MT1 qui permet de peser et d’analyser des échantillons jusqu’à concurrence d’un gramme et ce, avec une précision au microgramme. Comme la balance possède une grande précision, il est possible d’utiliser une petite quantité afin de réduire la perte de produit, puisque cette méthode est destructrice. L’échantillon moyen pesait entre 1 et 2 milligrammes. Les creusets utilisés étaient faits d’une céramique de type alumine pouvant être utilisée pour des manipulations allant jusqu’à des températures de 800 °C sans problème. Puisque nos échantillons sont des polymères ou des oligomères organiques, ces creusets conviennent bien aux types d’analyses effectuées puisque la température de dégradation, qui est généralement associée à une perte de masse égale à 5%, ne dépassent généralement pas 500 °C. Les analyses ont été effectuées sous une atmosphère inerte (azote, 40 mL·min-1), pour des fins
de comparaison avec la littérature. Dans les faits, cette analyse ne représente pas la véritable température de dégradation du polymère si celui-ci est utilisé dans des conditions ambiantes, en présence d’oxygène. En effet, la présence d’oxygène va grandement faire diminuer la température de dégradation, dû à des réactions d’oxydation avec l’oxygène de l’air qui ne sont pas possibles sous une atmosphère inerte. Cette analyse n’est donc pas effectuée dans le but d’évaluer la gamme de température utilisable pour des fins pratiques, mais dans le but de comparer nos échantillons avec la littérature.
Les séquences de chauffage ont été définies et les analyses réalisées avec le programme STARe. Une expérience typique avait une température de départ de 50 °C. L’échantillon était ensuite chauffé jusqu’à 700 °C sous atmosphère inerte, avec un taux de chauffage de 10 °C par minute. Le programme STARe permet d’observer en temps réel la progression de l’analyse, la perte de masse étant affichée en fonction de la température de chauffage à l’aide d’une courbe. Le programme offre ensuite une fonction qui permet d’évaluer cette perte de masse en pourcentage par rapport à la masse initiale. La température de dégradation est ainsi déterminée comme étant la température à laquelle on observe une perte de masse de 5%. Certaines publications associent également la température de dégradation à une perte de 10%. Le pourcentage utilisé, en fin de compte, est un peu arbitraire et dépend surtout du type d’utilisation auquel l’échantillon est destiné.
2.4.2 Calorimétrie différentielle à balayage (DSC)
L’appareil utilisée est un DSC823e de Mettler Toledo et est contrôlé, comme dans le cas de la TGA, par le logiciel STARe. L’appareil fonctionne sous atmosphère inerte (azote, 80 mL·min-1) et est refroidi par un
15 système fonctionnant à l’azote liquide. Il est donc nécessaire de vérifier préalablement à l’utilisation de l’appareil si le contenant d’azote liquide Dewar de 50L est stable à la pression de travail, qui est d’environ 22 psi. Les capsules utilisées sont en aluminium et ont un volume de 40 μL. La nature de ces capsules fait en sorte que nous devons travailler à des températures inférieures à 600 °C, afin d’éviter de faire fondre la capsule et d’endommager les thermocouples du support à échantillon. Ces capsules conviennent parfaitement à notre projet, puisque les phénomènes que nous désirons étudier se déroulent à des températures bien inférieures à 600 °C.
Une méthode typique pour ce type d’analyse peut avoir une température initiale de 50 °C et une température finale de 200 °C, soit plusieurs dizaines de degrés en-dessous de la température de dégradation, avec un taux de chauffage et de refroidissement de 10 °C par minute. Une analyse typique comporte un premier chauffage, suivi d’un refroidissement afin d’éliminer « l’historique thermique » de l’échantillon, pour ensuite faire un second chauffage. Ce second chauffage est souvent utilisé pour analyser les différentes transitions de l’échantillon. Pour ce type d’analyse, il faut une quantité plus importante de produit que pour la TGA. Cependant, comme cette méthode n’est pas destructrice, il est possible de récupérer les produits une fois l’analyse complétée. Il est important de bien entasser l’échantillon sous forme de poudre dans le fond de la capsule, pour assurer un bon contact avec le thermocouple, afin d’éviter l’apparition d’artefacts lors de l’analyse.
L’appareil affiche l’analyse en temps réel à l’aide d’une courbe, qui correspond à la variation de la puissance nécessaire (en watt) pour conserver les deux capsules à une température égale en fonction de la température de chauffage. Ce changement dans la puissance nécessaire pour conserver la capsule vide et la capsule contenant l’échantillon à une température égale est ensuite associé, à l’aide du logiciel, à une énergie absorbée ou dégagée par l’échantillon. Il est alors possible de déterminer la présence de transitions thermiques et de calculer des enthalpies de fusion ou de cristallisation, selon le cas. La température de fusion d’un échantillon est déterminée comme étant la température correspondant au sommet du pic de fusion. La température de transition vitreuse (Tg) est déterminée comme étant la moyenne de la plage de température à
16
2.4.3 Diffraction des rayons X
La diffraction des rayons X a été effectuée sur un appareil de la compagnie Bruker. Le générateur Kristalloflex 760 est équipé d'un tube émettant une longueur d'onde de type Kα provenant du cuivre avec une longueur d'onde de 1,5418 Å, filtrée par du nickel. Les mesures ont été effectuées à grand angle (mode WAXS, «Wide
angle X-ray scattering») en mode transmission. Un détecteur bidimensionnel Hi-Star de la compagnie Bruker a
été utilisé, et le logiciel GADDS a servi à l'acquisition et l’analyse des données.
Les échantillons ont été analysés soit sous forme de poudre broyée et insérée dans un tube capillaire de verre spécial très mince, dédié aux mesures de diffraction des rayons X, et de 1 mm de diamètre, soit sous forme de film mince, maintenu en place par un support adéquat. Dans le cas de l’utilisation d’un capillaire, un diagramme de diffusion de ce tube capillaire vide était soustrait du diagramme de diffusion de l’échantillon pour éliminer la diffusion des rayons X des composantes du capillaire. Il est à noter que certains diagrammes de diffraction ont été obtenus sur les polymères et les oligomères tels que synthétisés, alors que certains ont été enregistrés suite à une cristallisation par solvant.
2.4.4 Chromatographie d’exclusion stérique (CES)
La chromatographie d’exclusion stérique a été effectuée sur un montage contenant une pompe HPLC 515, deux colonnes installées en série de type PL-Gel Mixed B-LS de la compagnie Agilent, un détecteur UV modèle 441 fonctionnant à 254 nm et une boucle d’injection de 100 μL. La technique est calibrée avec des standards de polystyrène monodisperses de masses molaires connues. Le logiciel traitant les informations du détecteur est le logiciel ASTRA version 4.70.07. Ce dernier affiche sous forme de graphique la variation de l’absorbance du détecteur UV en fonction du temps d’élution. L’acheminement des analytes au détecteur fait augmenter l’absorbance et se manifeste par des pics plus ou moins bien définis, selon l’indice de polymolécularité (Ip ou PDI en anglais) de l’échantillon.
Avant d’utiliser l’appareil, il faut préparer une quantité suffisante d’éluent (au moins un litre) et mettre en place les colonnes qui seront utilisées pour les purger avec l’éluent à un taux d’élution de 1,0 mL·min-1 pendant
quelques heures. Les analyses seront ensuite effectuées à un taux d’élution plus faible, soit 0,2 mL·min-1.
Ce types d’analyse requiert un solvant d’élution très propre (qualité HPLC, ne contenant pas d’impuretés qui donneraient un signal UV) qui sera préalablement filtré sur des filtres de 0,45 microns de PTFE (polytétrafluoroéthylène) pour retirer toute particule risquant de boucher les petites pores de la colonne et de faire monter la pression au sein des colonnes. Les échantillons sont également préparés avec un solvant de
17 qualité HPLC, à une concentration de 1 mg par 6 mL de solvant. Ces échantillons sont également filtrés pour éviter que des particules non solubles ne soient injectées dans les colonnes.
Lorsque l’élution est terminée, le logiciel ASTRA permet de transférer l’acquisition en un fichier ASCII qui peut être ensuite analysé par une fiche de calcul Excel ou d’un autre tableur. Une courbe de calibration est obtenue avec les standards de polystyrène. Cette courbe servira à associer un temps d’élution à une masse molaire spécifique pour un système d’éluent et de colonne donné.
2.4.5 Spectrométrie UV-visible
Les analyses spectrométriques UV-visible ont été effectuées sur un appareil Thermo Scientific Genesys 10S, un appareil portatif ayant un signal stable dans le temps et qui donne des résultats rapides. Les analyses ont été effectuées avec un support à échantillon pour les liquides, utilisant soit des cellules de verre ou de quartz, qui permet de travailler sans problème avec des longueurs d’onde de 300 nanomètres et plus. Les analyses ont été réalisées en solution diluée dans le dichlorométhane. Puisque l’objectif de ces analyses n’était pas de doser la quantité de groupements, mais plutôt d’observer les phénomènes, la concentration des solutions était ajustée de sorte que l’absorbance maximale de l’échantillon ne sature pas le détecteur, avec une valeur approximative d’environ une unité d’absorbance. L’appareil était utilisé en mode balayage rapide, avec l’acquisition d’une valeur d’absorbance par nanomètre. L’appareil est muni d’un port USB qui permet de transférer les données sous format texte, qui peut ensuite être analysé facilement dans un tableur (Excel a été utilisé dans le présent travail). Les fichiers textes exportés de l’appareil sont convertis en classeur Excel, pour créer un graphique et observer qualitativement l’allure du spectre d’absorption. Également, à certaines occasions, les valeurs d’absorption maximales ont été utilisées pour mesure de manière quantitative l’évolution de la structure de l’échantillon.
2.4.6 Irradiation des échantillons par une lampe ultraviolette
Bien que l’irradiation ne soit pas une technique de caractérisation en soi, une documentation détaillée du type de lampe utilisé pour étudier la photo-isomérisation est rarement disponible dans la littérature. Il semble cependant pertinent d’inclure une description détaillée de la lampe utilisée au cours de ce projet, à des fins de reproductibilité. Pour que la photo-isomérisation soit efficace, il faut deux éléments clé : la bonne longueur d’onde et une puissance de lampe suffisamment élevée. La longueur d’onde nécessaire pour isomériser un azobenzène est d’environ 365 nanomètres. Dans les faits, la longueur d’onde la plus efficace peut varier en fonction du type de substitution des groupements phényle de l’azobenzène, de sorte qu’il peut être utile
18
d’effectuer une analyse par spectrométrie UV-visible avant de choisir la longueur d’onde à utiliser pour isomériser le groupement azo. La bande la plus intense sur le spectre d’absorption UV-visible de la molécule, qui est généralement centrée autour de 360 nanomètres, est la transition qui doit être effectuée lors de l’irradiation pour isomériser l’azobenzène. Si l’on désire donc optimiser la photo-isomérisation, il faut ajuster la longueur d’onde d’irradiation en fonction du maximum du spectre d’absorption de l’échantillon.
À des fins pratiques, la très grande majorité des études utilise des ampoules de longueur d’onde de 365 nanomètres. Ce type d’ampoule est facilement achetable et peu dispendieux, puisqu’il a des applications dans divers domaines en science autant que dans la vie de tous les jours. Le modèle de lampe utilisé dans le cadre de cette étude est une lampe préalablement destinée à la pose d’ongles en résine. Ce type de lampe est robuste et peu dispendieux et est disponible pour moins de 50 dollars. De plus, cette lampe possède quatre supports à ampoule de 9 Watts chacune, permettant une puissance maximale de 36 Watts, ce qui est largement suffisant aux études avec les polymères synthétisés. Certaines études rapportent la puissance en fonction du volume irradié (W·cm-3), ce qui peut être utile pour comparer plusieurs systèmes entre eux.
Cependant, tel qu’observé dans la littérature, la puissance par volume utilisée pour les études varie énormément, de sorte qu’un calcul rigoureux de la puissance par volume ne semble pas nécessaire14, 26, 27.
Dans le cadre de cette étude, la puissance utilisée a été variée de 9 Watts à 36 Watts, et chacune de ces puissances est suffisante pour permettre une photo-isomérisation appréciable en quelques secondes. De plus amples détails sur le positionnement des échantillons, ainsi que quelques images pour un support visuel, sont disponibles à la section 4.2.1.
19
Chapitre 3 : Discussion sur les synthèses
Ce chapitre discutera des différentes synthèses de monomères et de polymères. Ces synthèses seront décrites d’un point de vue réactionnel et certaines considérations mécanistiques quant à la réactivité des monomères seront discutées. En effet, la réactivité des monomères est un facteur important quant à l’obtention de masses moléculaires importantes en polycondensation.
3.1 Synthèse du monomère éthersulfone
Les méthodes de synthèse utilisées pour la préparation des monomères éthersulfone sont des méthodes adaptées d’un précédent projet dans notre laboratoire, développées en majeure partie par Jean-Michel Benoit et inspiré d’un article paru dans Organic Letters en 200624. La stratégie était une
approche par étapes, en utilisant des réactions de protection-déprotection successives pour atteindre une longueur de segment très précise. La majorité des réactions avaient comme objectif de créer un lien éther aromatique par substitution nucléophile aromatique (SNAr), en utilisant une variante de la
synthèse d’éther de Williamson.
Figure 5. Schéma général de la synthèse de Williamson.
Le principe général de cette réaction est la formation d’un alcoolate par l’action d’une base sur un groupement alcool. Ce groupement devient donc plus nucléophile et peut ensuite attaquer un site réactionnel électrophile comportant un groupement partant, tel qu’un halogénure. Au final, un groupement éther est créé à partir d’un alcool et il y a dégagement d’une petite molécule HX, où X est le groupement partant.
Les premiers tests de la réaction ont été effectuées avec le bis(4-hydroxyphényl)sulfone (BPS) qui est commercialement disponible. Ce monomère a été utilisé pour synthétiser le copolymère P2ArS-4Azo dans le présent projet. La réactivité du BPS est quelque peu différente de celle du macromonomère éthersulfone terminé par des groupements hydroxyl, le 4ArS-OH, le groupement sulfone étant en position para à l’alcool. Le BPS a été utilisé pour optimiser les conditions de
20
réaction de polymérisation et a également servi à quelques occasions comme réactif de départ pour synthétiser des monomères éthersulfones plus longs, comme le 4ArS-OH ou le 8ArS-OH, comprenant respectivement quatre et huit cycles aromatiques.
Le monomère qui a été le plus utilisé est le 4,4’-bis(p-hydroxyphénoxy)diphénylsulfone (4ArS-OH), principalement dû au fait que sa synthèse nécessitait quelques étapes en moins et est également un monomère intermédiaire lors de la synthèse du 8ArS-OH, le plus long monomère utilisé dans le cadre de ce projet. Le 4ArS-OH s’obtient rapidement en synthétisant d’abord le 4ArS-OMe avec des réactifs de départ commerciaux, soient le 4,4’-difluorodiphényl sulfone et le 4-methoxyphénol, en utilisant le carbonate de potassium comme base dans un solvant aprotique polaire ayant un haut point d’ébullition pour permettre de hautes température réactionnelles, tel qu’illustré à la figure 6.
Figure 6. Synthèse du 4ArS-OMe, la forme protégée du 4ArS-OH. Le groupement méthoxy en fin de chaîne sert de groupement protecteur pour éviter une polymérisation non désirée.
Les synthèses de Williamson ont été effectuées avec des groupements méthoxy-terminés, comme dans le cas du 4ArS-OMe illustré dans le Schéma de la figure 6, afin d’éviter une polymérisation non désirée qui serait possible vu les températures réactionnelles utilisées. Ces groupements étaient ensuite déprotégés en groupements alcools à l’aide du tribromure de bore, un puissant agent déméthylant pour groupements méthoxy aromatiques, tel qu’illustré dans le Schéma de la figure 7.
Figure 7. Schéma général de la réaction de déprotection des monomères par le tribromure de bore.
Quoi qu’il existe certaines méthodes alternatives plus douces28, 29 pour déméthyler un groupement
méthoxy aromatique, aucune méthode n’est aussi simple et aussi efficace que celle utilisant le tribromure de bore. Elle permet une déprotection quantitative, réduisant grandement le besoin de purification. De plus, la réaction est rapide et simple à effectuer. Cette étape de déprotection précédait soit une polymérisation ou une synthèse de Williamson supplémentaire selon la même
21
stratégie pour allonger davantage le monomère, tout dépendant de la structure finale désirée du polymère.
3.2 Synthèse du monomère azobenzène
Ensuite, il fallait synthétiser l’azobenzène qui servirait de comonomère lors de la polymérisation A2B2.
Il existe certaines méthodes plus traditionnelles30 pour synthétiser efficacement une vaste gamme
d’azobenzènes. Ces synthèses sont efficaces mais nécessitent plusieurs étapes et plusieurs réactifs. Un groupe de recherche a développé une méthode plus directe pour synthétiser une vaste gamme d’azobenzènes avec d’excellents rendements25. Dû à sa simplicité, sa versatilité et ses excellents
rendements, cette méthode a été principalement utilisée lors de la synthèse des monomères azobenzène dans le présent travail.
Figure 8. Schéma de synthèse de l’azobenzène, d’après les travaux de Zhang et Jiao25.
L’utilisation du fluor en tant que groupement partant lors d’une substitution nucléophile peut sembler contre-intuitif puisqu’en général, parmi les halogènes, l’atome de fluor est généralement le moins bon groupe partant dû à la stabilité du lien C-F et la dureté de l’atome de fluor en général. Cependant, il a fréquemment été mis en évidence31 que, dans le cas des substitutions nucléophiles aromatiques
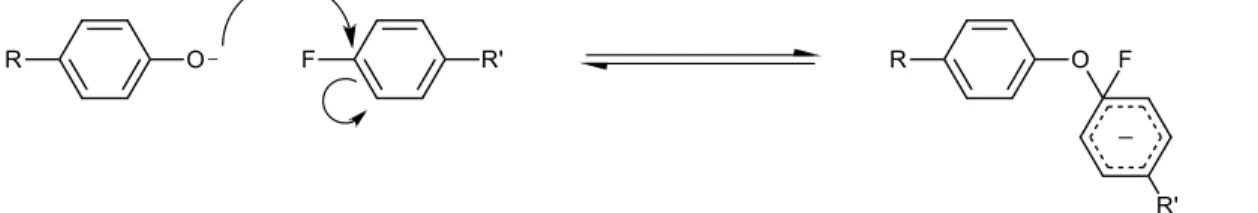
utilisées couramment lors des synthèses industrielles des PES, des PEK et PEEK, le mécanisme de réaction le plus probable est la création d’un complexe Meisenheimer, tel qu’illustré à la figure 9, suivie d’une élimination et que les atomes de fluor et de chlore semblent être les meilleurs atomes d’halogène pour favoriser la formation de ce complexe.
Figure 9. Mécanisme réactionnel d’une substitution nucléophile aromatique impliquant la formation d’un complexe Meisenheimer.
22
Tel qu’illustré à la figure 9, le complexe intermédiaire de la réaction de Williamson vient créer une charge négative partielle sur le groupement phényle attaché à l’halogénure. Cet intermédiaire vient en quelque sorte briser l’aromaticité de la molécule. De ce fait, cette réaction nécessite une grande énergie d’activation, ce qui explique le besoin, dans la plupart des cas, d’utiliser des températures réactionnelles élevées.
Plus la charge négative partielle générée par l’étape intermédiaire est stabilisée, plus l’énergie d’activation de ce complexe diminue. C’est pourquoi l’atome de fluor est le meilleur choix comme groupement partant, puisque dans ce cas précis, il est le meilleur agent stabilisateur pour l’état intermédiaire, grâce à son grand pouvoir polarisant. La littérature montre que la réactivité de cette réaction diminue grandement si l’on utilise des groupements chlorés et devient quasiment nulle lors de l’utilisation de groupements bromés ou iodés31. Cependant, certains articles ont obtenu des
masses molaires supérieures à celles obtenues dans le cadre de ce projet en utilisant des groupements chlorés32.
Cette différence peut s’expliquer par plusieurs facteurs autres que la réactivité des monomères, telle la pureté des monomères et du solvant utilisés. Plusieurs chercheurs ajoutent une fraction de toluène au solvant pour permettre une distillation azéotropique des traces d’eau avec un montage Dean-Stark32 ce qui n’a pas été fait dans le présent travail. Pour conclure ce point, le choix du groupement
partant est donc crucial pour l’obtention de bonnes masses molaires, pour favoriser au maximum la réactivité des monomères tout en minimisant les possibilités de réactions secondaires non désirées.
3.3 Synthèses des copolymères éthersulfone-azobenzène
Le type de synthèse pour les copolymères éthersulfone-azobenzène est une polycondensation de type A2B2.
Figure 10. Réaction générale d’une polycondensation de type A2B2.
Une réaction de type A2B2 implique une réaction de condensation dans laquelle chacun des
23
ne peut y avoir la perte d’une petite molécule XX ou YY à la figure 10 lors de la réaction. Ce type de synthèse permet donc l’obtention d’une structure régulière, alternée et linéaire.
La synthèse avait pour objectif de polymériser un éthersulfone et un azobenzène en créant un lien éther aromatique entre les monomères. De plus, il fallait obtenir des polymères de masses molaires suffisamment élevées pour créer des films minces ayant une bonne résistance mécanique, ce qui permettrait de les utiliser pour effectuer certains tests de cristallisation et d’irradiation en phase solide. Il fallait aussi que les polymères résultants soient solubles dans les solvants usuels pour faciliter la formation de films et la caractérisation des phénomènes d’isomérisation.
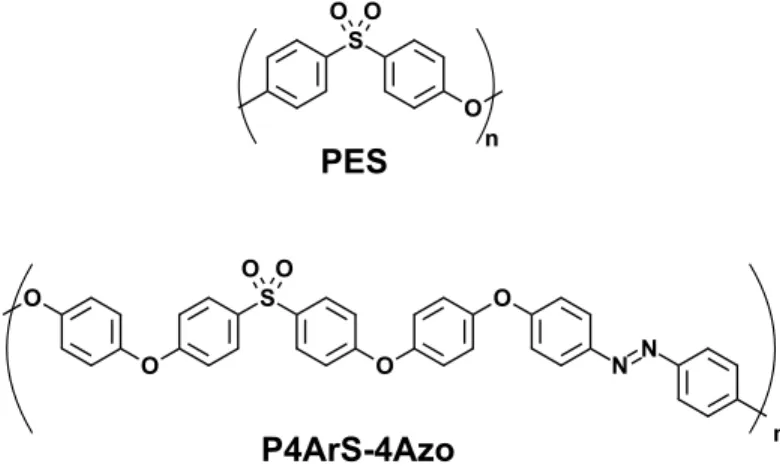
De plus, la création d’un lien éther aromatique ne dénature pas la structure primaire d’un PES, qui est une succession de liens entre des groupements éther aromatiques et des groupements sulfone aromatiques, tel qu’on peut le voir à la figure 11, qui présente une comparaison de ces unités de répétition.
Figure 11. Comparaison de la structure moléculaire d’un PES typique et du P4ArS-4Azo, un des polymères synthétisés dans le cadre de cette étude.
La stratégie de synthèse reste la même pour la polymérisation que lors de la construction du monomère éthersulfone. En effet, la polymérisation était, à la base, une synthèse de Williamson. Pour les mêmes considérations mécanistiques qu’à la section 3.2, le choix de l’atome halogène agissant comme groupe partant s’est arrêté sur le fluor. En effet, quoi que contre-intuitif, la sélection de cet halogène permet la formation des polymères les plus réguliers et offre la meilleure réactivité possible selon la littérature31. Malheureusement, malgré ces considérations, la réactivité de cette
24
élevées. Le Tableau 1 permet de comparer les masses molaires de trois différents polymères synthétisés, soient le P2ArS-4Azo, le P4ArS-4Azo et le P8ArS-4Azo.
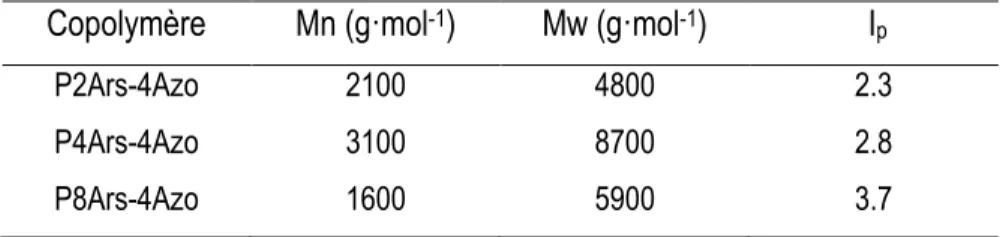
Tableau 1. Masses moléculaires moyennes en nombre (Mn) et en poids (Mw) et indice de polydispersité Ip des différents polymères synthétisés.
Copolymère Mn (g·mol-1) Mw (g·mol-1) Ip
P2Ars-4Azo 2100 4800 2.3
P4Ars-4Azo 3100 8700 2.8
P8Ars-4Azo 1600 5900 3.7
Tel que présenté dans le Tableau 1, les masses molaires varient entre 1600 g·mol-1 et 5900 g·mol-1.
Ces masses molaires signifient que, si l’on tient compte des pertes de masse dues à l’éjection de la petite molécule HX lors de la polymérisation, ces échantillons ont un degré de polymérisation (DP) qui varie de 2 à 5, approximativement. Ces masses molaires sont relativement faibles par rapport à celles généralement obtenus avec ce type de réaction de polymérisation. Plusieurs tentatives ont été effectuées pour augmenter ces masses molaires. Parmi ces tentatives, les réactions ont été menées à des températures supérieures à 180 °C, le temps de réaction a été prolongé, le solvant réactionnel a été modifié et l’ajout progressif des monomères a été tenté. Les temps de réaction plus longs ont résulté en l’obtention de polymères insolubles dans les solvants usuels, probablement en raison d’une haute masse moléculaire, ce qui n’a cependant pas été vérifié en raison précisément de cette insolubilité. Afin d’écarter la possibilité d’un changement de solubilité dû à une cristallisation lors de la synthèse, ces échantillons insolubles ont été analysés en diffraction des rayons X. Ces échantillons n’étaient cependant pas cristallins, écartant ainsi cette hypothèse.
Les masses molaires rapportés au Tableau 1 ne sont donc probablement pas les plus élevées obtenues dans le cadre de ce projet, mais ce sont les masses molaires des échantillons utilisés pour les caractérisations et les expériences subséquentes, puisque ces polymères nous permettaient de créer des films qui se tenaient bien et étaient solubles dans le chloroforme, caractéristiques très utiles pour la plupart des tests qui seront détaillés au chapitre 4. De plus, comme il sera discuté au chapitre 5 sur les propriétés thermiques, il semble que malgré des masses molaires relativement faibles, les propriétés thermiques sont assez bonnes, particulièrement pour le P8ArS-4Azo.
25
Certains facteurs peuvent expliquer la faible réactivité des monomères en utilisant cette réaction, conduisant à des masses molaires relativement faibles. Dans le cas de la polycondensation aromatique de type éther de Williamson, la littérature explicite le besoin d’avoir un groupement électro-attracteur en position ortho ou para de l’atome d’halogène pour favoriser la réaction31. Tel
qu’illustré à la figure 9 et expliqué précédemment, il se crée une charge négative sur le cycle aromatique de l’halogène lors de l’attaque de l’alcoolate. Cette charge est délocalisée à travers le cycle aromatique et stabilisée par l’effet électro-attracteur de l’atome d’halogène. Par conséquent, malgré l’effet électro-attracteur sur la réactivité, la réaction est tout de même peu favorisée. La littérature montre que la présence d’un groupement électro-attracteur supplémentaire en position
ortho ou para permet de favoriser davantage l’état intermédiaire, ainsi que le départ de l’atome de
fluor31.
Figure 12. Schéma représentant la formation d’un complexe Meisenheimer réactif, puisqu’il est activé par un groupement R’ électroattracteur.
La figure 12 montre un groupement électro-attracteur en position -para, mais il pourrait également se trouver en ortho de l’atome d’halogène, puisque cette réaction est orientée ortho/para31. Dans notre
cas, la stratégie de synthèse fait en sorte que l’atome d’halogène se trouve sur l’azobenzène et que le groupement diazène est un groupement électro-donneur au lieu d’électro-accepteur, diminuant ainsi davantage la réactivité de la polymérisation. L’ajout d’un groupement sulfonate en position ortho aurait pu grandement augmenter la réactivité de la réaction. Cependant, ce genre de groupements est susceptible d’inhiber la cristallisation d’un polymère en raison de l’encombrement qu’il induirait, favorisant une structure amorphe. Étant donné notre intention d’étudier des phénomènes de cristallisation par la lumière dans ce projet, cette option n’a donc pas été utilisée.
26
Une seconde alternative est d’utiliser une haute température de polymérisation (180 °C) sur une période de 24 heures, ce qui a été effectué dans le cadre du présent travail. Malgré les masses molaires relativement faibles obtenues, les études que nous voulions faire étaient tout de même possibles et nous étions malgré tout capable de créer des films polymériques minces par évaporation de solvant ayant une résistance mécanique acceptable, permettant leur manipulation avec des pinces sans les briser.
27
Chapitre 4 : Discussion sur la photo-isomérisation
4.1 Notions générales sur la photo-isomérisation de
l’azobenzène
Les azobenzènes et leurs dérivés sont connus pour la possibilité d’isomériser leur lien double de conformation trans du groupement diazène vers la forme cis, à température pièce sous l’action d’une irradiation ultra-violette. Ce processus est réversible dans le noir, dû à la grande stabilité thermodynamique de l’isomère trans33. Le retour vers l’isomère trans peut également être accéléré
par l’application de chaleur ou avec une irradiation de lumière visible.
Typiquement, la photo-isomérisation de l’isomère trans à l’isomère cis se fait avec une lampe suffisamment puissante, en utilisant une longueur d’onde de 365 nm. Celle-ci induit une transition π-π* permise par la symétrie de l’isomère trans33. La transition n- π*, non-permise par la symétrie,est
également visible à environ 450 nm, mais est de plus faible intensité. Lors de l’isomérisation, on observe une diminution très marquée du maximum d’absorbance à 360 nm. Cette diminution est le principal indicateur de l’isomérisation efficace de l’échantillon. En plus, on observe une légère augmentation de l’absorbance de la bande centrée vers 450 nm. Ceci correspond à la transition n- π*
de l’isomère cis qui absorbe plus fortement, à cette longueur d’onde, que l’isomère trans35, 36. Ces
deux observations combinées permettent donc de conclure qu’il y a bien eu isomérisation de l’échantillon. La différence notable entre les deux bandes associées respectivement aux isomères
trans et cis à 360 et 450 nm correspond à la différence du coefficient d’absorptivité molaire entre les
deux espèces. L’isomère trans absorbe bien la lumière, tandis que l’isomère cis est un moins bon fluorophore.
Il existe quatre mécanismes pour expliquer comment la photo-isomérisation se produit. Les quatre mécanismes sont la rotation, l’inversion, l’inversion concertée et la rotation assistée par une inversion. Une explication détaillée de ces quatre mécanismes est disponible dans l’excellente revue de la littérature sur l’azobenzène écrite par H. M. Dhammika Bandara et Shawn C. Burdette33. Une