Université de Sherbrooke
Mécanismes de signalisation d’AT1R médiés par des analogues cycliques de
l’angiotensine II
Par David St-Pierre
Programme de pharmacologie
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de maitre ès sciences (M. Sc.)
en pharmacologie
Sherbrooke, Québec, Canada Août, 2017
Membres du jury d’évaluation
Pr Richard Leduc, programme de pharmacologie Pr Pierre Lavigne, programme de biochimie Pr Jean-Luc Parent, programme de pharmacologie
Pr Jana Stankova, programme d’immunologie
R
ÉSUMÉMécanismes de signalisation d’AT1R médiés par des analogues cycliques de
l’angiotensine II Par
David St-Pierre
Programme de pharmacologie
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de maitre ès sciences (M.Sc.) en pharmacologie, Faculté de médecine et des
sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 L'angiotensine II (Ang II) joue un rôle important dans la régulation du système cardiovasculaire par l’activation de plusieurs voies de signalisation. L’activation de ces voies passe par le récepteur de l'angiotensine II de type 1 (AT1R). Ce récepteur fait partie de la famille des récepteurs couplés aux protéines G (GPCRs). De plus, il est maintenant connu que certains ligands peuvent lier le récepteur et induire une conformation qui permet d'activer certaines voies de signalisation tout en n’étant pas favorable à l'activation d'autres voies. Il est alors question de sélectivité fonctionnelle, aussi appelée signalisation biaisée. Ainsi, avec cette approche, il est possible de cibler les voies qui produiront les effets thérapeutiques désirés sans toutefois activer les voies qui seraient responsables des effets indésirables. Nous avons émis l’hypothèse que de cycliser des ligands va restreindre les conformations possibles lors du couplage avec AT1R et induire un agonisme biaisé. Ainsi, des analogues cycliques de l’AngII substitués aux positions 3 et 5 par des cystéines et des homocystéines ont été synthétisés : [Sar1Hcy3,5]AngII, [Sar1Cys3Hcy5]AngII et [Sar1Cys3,5]AngII. D’abord, la capacité de ces analogues cycliques à activer la voie Gq a été évaluée par la mesure de la production des inositol phosphates. Puis, la capacité à activer les voies G12, le recrutement des β-arrestines (1 et 2) ainsi que l’activation de ERK1/2 a également été évaluée. Nos travaux ont montré que l’analogue cyclique [Sar1Hcy3,5]AngII a une puissance et une efficacité maximales sur toutes les voies testées à l'exception de la voie Gq. Des simulations de dynamique moléculaire ont été effectuées pour nous permettre de comprendre comment la conformation du ligand influence la structure d’AT1R et donc l’activation des différentes voies de signalisation. Les simulations en dynamique moléculaire ont montré que la barrière énergétique associée à l'insertion du résidu Phe8 de l’AngII dans le coeur hydrophobe d'AT1R est augmentée avec [Sar1Hcy3,5]AngII, pouvant expliquer que cet analogue active moins bien la voie Gq. D’autres analogues cyclisés aux positions 3 et 5 de l’AngII ont été synthétisés; [Sar1Hcy3Ile4Hcy5]AngII, [Sar1Hcy3,5Ile8]AngII et [Sar1Hcy3Cys5]AngII. Leur capacité à activer les voies Gq, ERK1/2 et le recrutement des β-arrestines (1 et 2) a été évaluée. L’analogue [Sar1Hcy3Cys5]AngII semblait bien activer la voie ERK1/2, mais pas les voies G12 et β-arrestines. Ces résultats suggèrent que le fait de contraindre les mouvements des déterminants moléculaires d’un ligand en introduisant des structures cycliques peut entraîner un biais dans la signalisation en stabilisant différentes structures du récepteur. Mots clés : AT1R, angiotensine II, signalisation biaisée, analogues cycliques.
S
UMMARYAT1R signaling mechanisms mediated by angiotensin II cyclic analogs
By David St-Pierre Pharmacology Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Master degree diploma maitre ès sciences (M.Sc.) in pharmacology, Faculty of medicine and
health sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 Angiotensin II (Ang II) has an important role in the regulation of the cardiovascular system by its ability to activate several signaling pathways. The activation of these pathways occurs via the angiotensin II receptor type 1 (AT1R). This receptor belongs to the family of G protein-coupled receptors (GPCRs). Moreover, it is now known that certain ligands can bind to the receptor and induce a conformation that allow the activation of certain signaling pathways while not promoting the activation of other pathways. This concept is known as functional selectivity or biased signaling. With this approach, it is possible to target the signaling pathways that produce the desired therapeutic effects rather than activating the pathways responsible for adverse effects. We hypothesized that cyclizing ligands would restrict possible conformations when coupled with AT1R and induce biased agonism. Thus, cyclic AngII analogs substituted at positions 3 and 5 by cysteines and homocysteines were synthesized: [Sar1Hcy3,5]AngII, [Sar1Cys3Hcy5]AngII and [Sar1Cys3,5]AngII. First, the ability of these cyclic analogs to activate the Gq pathway was measured by the inositol phosphates production. Then, the G12 pathway activation, β-arrestin (1 and 2) recruitment and the ability of these analogs to activate the ERK1/2 pathway was evaluated. Our work has shown that [Sar1Hcy3,5]AngII has maximum potency and efficacy on all of the evaluated pathways, except for the Gq pathway. Molecular dynamic simulations were used to understand how a distinct ligand conformation influences the AT1R structure and the activation of signaling pathways. These studies have shown that the energy barrier associated with the insertion of the Phe8residue of AngII within the hydrophobic core of AT1R is increased with [Sar1Hcy3,5]AngII, possibly explaining why this analog is less potent in activating the Gq pathway. Other analogues cyclized at positions 3 and 5 of AngII were synthesized; [Sar1Hcy3Ile4Hcy5]AngII, [Sar1Hcy3,5Ile8]AngII and [Sar1Hcy3Cys5]AngII. Their ability to activate Gq, ERK1/2 and recruitment of β-arrestins (1 and 2) was evaluated. The analog [Sar1Hcy3Cys5]AngII appeared to activate the ERK1/2 pathway but not the G12 and β-arrestin pathways. These results suggest that constraining the movements of molecular determinants of a ligand by introducing cyclic structures can lead to a signaling bias by stabilizing different structures of the receptor.
T
ABLE DES MATIÈRESRésumé ... ii
Summary ... iii
Table des matières ... iv
Liste des figures ... vi
Liste des tableaux ... viii
Liste des abréviations ... ix
1. Introduction ... 1
1.1 Les récepteurs couplés aux protéines G (GPCRs) ... 1
1.1.1 Activation des GPCRs ... 2
1.1.2 Transduction du signal des GPCRs ... 3
1.1.3 Les GPCRs de classe A ... 4
1.1.4 Mécanismes d’activation des GPCRs de classe A ... 5
1.2 Le système rénine-angiotensine ... 6
1.2.1 Angiotensine II ... 8
1.2.2 Effets physiologiques de l’angiotensine II ... 9
1.2.3 Récepteurs de l’angiotensine II ... 10
1.2.4 Structure et mécanismes d’activation d’AT1R ... 11
1.3 Signalisation d’AT1R ... 14
1.3.1 Voies dépendantes des protéines G ... 15
1.3.2 Voies indépendantes des protéines G et voies des MAPK/ERK1/2 ... 15
1.4 Sélectivité fonctionnelle ... 17
1.4.1 Intérêt de la sélectivité fonctionnelle ... 17
1.4.2 Bases structurales de la sélectivité fonctionnelle d’AT1R ... 19
2. Hypothèse/problématique ... 20
Objectifs ... 21
3. Article ... 22
3.1 Summary ... 24
3.2 Introduction ... 25
3.3 Materials and methods ... 27
3.3.1 Materials ... 27
3.3.2 Constructs ... 27
3.3.4 Binding Experiments ... 28
3.3.5 Measuring inositol-1 phosphate production ... 28
3.3.6 BRET-based biosensor assays ... 28
3.3.7 ERK1/2 activation assay ... 29
3.3.8 Data analysis ... 30
3.3.9 Molecular dynamic simulations ... 30
3.3.10 Trajectory analysis ... 31
3.4 Results ... 32
3.4.1 Binding properties of cyclic AngII analogs ... 32
3.4.2 IP production via Gq signaling ... 32
3.4.3 β-arrestin recruitment ... 33
3.4.4 G12 activation ... 35
3.4.5 PKC-dependent and EGFR-dependent ERK1/2 activation ... 36
3.4.6 Quantification of ligand bias ... 39
3.4.7 Molecular dynamics simulations of the AngII-AT1R and [Sar1Cys3Hcy5]AngII-AT1R complexes ... 42
3.4.8 Environments explored by the C-terminal Phe8 of AngII and [Sar1Hcy3,5]AngII in the AT1R... 46
3.4.9 2D probability distribution functions reveals a link between the conformation of the backbone of residues Val3 to Ile5 and the position of side-chain of Phe8 in the AT 1R ... 50
3.5 Discussion ... 54
4. Résultats (hors article) ... 63
4.1 Propriétés de liaison des analogues cycliques de l’AngII ... 63
4.2 Production des IP via la signalisation Gq ... 64
4.3 Recrutement des β-arrestines ... 65
4.4 Activation PKC-dépendante et EGFR-dépendante de ERK1/2 ... 67
5. Discussion ... 70
5.1 Sélectivité fonctionnelle des analogues cycliques de l’AngII ... 70
5.2 Effet de la structure des analogues sur la sélectivité fonctionnelle ... 74
5.3 Comparaison du complexe AngII-AT1R avec le complexe [Sar1Hcy3,5]AngII-AT1R ... 75
5.4 Insertion du résidu Phe8 de l’analogue cyclique [Sar1Hcy3,5]AngII dans le cœur hydrophobe ... 75
6. Conclusions et perspectives ... 78
L
ISTE DES FIGURESIntroduction
Figure 1. Le système rénine-angiotensine ... 7
Figure 2: Angiotensine II ... 8
Figure 3. Effets physiologiques de l'angiotensine II ... 10
Figure 4: Signalisation du récepteur AT1. ... 14
Figure 5: Principales voies de signalisation du récepteur AT1 ... 17
Figure 6. Signalisation biaisée ... 18
Article
Figure 7 : Inositol 1-phosphate production induced by AngII analogs ... 33Figure 8 : βarr1 recruitment to the AT1R by AngII analogs ... 34
Figure 9 : βarr2 recrutment to the AT1R by AngII analogs ... 35
Figure 10 : G12 activation by AngII analogs. ... 36
Figure 11 : PKC-dependent ERK activation by AngII analogs ... 37
Figure 12 : EGFR-dependent ERK activation by AngII analogs ... 38
Figure 13 : Effects of AngII analogs on AT1R signaling pathways ... 40
Figure 14 : 3D representation of the three cyclic analogs and probability distribution of the distance between the Cβ atoms of residues in positions 3 and 5 of the cyclic analogs. ... 43
Figure 15 : Probability distribution of the distance between the Cβ atoms of residues in positions 3 and 5 for ligands in complex with AT1R ... 45
Figure 16 : Examples of the strand, γ-turn and extended conformations of AngII. The β-strand and γ-turn conformations ... 46
Figure 17 : Positioning of Phe8 of AngII outside or inside of the AT1R hydrophobic core. 47 Figure 18 : Probability distribution of the distance between the side chain of residue Phe8 of the ligand and the side chain of residue F772.53 of the AT1R ... 49
Figure 19 : 2D probability of the distance between the side chain of residue Phe8 of the ligand and the side chain of residue F772.53 of the AT1R.and the distance between the Cβ atoms of residues in positions 3 and 5 of AngII or [Sar1, Hcy3,5]AngII ... 53
Résultats (hors article)
Figure 20: Production d'inositol 1-phosphate induite par les analogues de l'AngII ... 65
Figure 21: Le recrutement de βarr1 à l'AT1R par des analogues de l’AngII ... 66
Figure 22 : Le recrutement de βarr2 à l'AT1R par des analogues de l’AngII ... 67
Figure 23 : Activation PKC-dépendante de ERK par des analogues de l’AngII. ... 68
L
ISTE DES TABLEAUXArticle
Table 1: Binding proprieties of AT1R ligands ... 32 Table 2 : Activation of Gq, βarr1, βarr2, G12, PKC-dependent ERK1/2 (PKC-ERK) and EGFR-dependent ERK1/2 (EGFR-ERK) by AT1R ligands ... 39 Table 3 : Transduction ratios of AT1R ligands ... 41 Table 4 : Bias factors of AT1R ligands ... 42
Résultats (hors article)
Tableau 5 : Propriétés de liaison des ligands AT1R ... 64 Tableau 6 : Activation de Gq, βarr1, βarr2, PKC-ERK et EGFR-ERK par des ligands d’AT1R ... 69
L
ISTE DES ABRÉVIATIONS AMPc AngII AngIII AngIV AT1R AT2R Cys DAG DM ECA EGF EGFR ERK1/2 GDP GEF GPCR GRK GTP Hcy IP1 IP3 IP3R Jak MAPK MEK MJH PIP2 PKC RafAdénosine monophosphate 3’, 5’-cyclique Angiotensine II
Angiotensine III Angiotensine IV
Récepteur à l’angiotensine II de type 1 Récepteur à l’angiotensine II de type 2 Cystéine
Diacylglycérol
Dynamique moléculaire
Enzyme de conversion de l’angiotensine Facteur de croissance épidermique
Récepteur du facteur de croissance épidermique Extracellular signal-regulated kinases
Guanosine diphosphate Facteur d’échange de guanine Récepteur couplé aux protéines G
Kinase de récepteurs couplés aux protéines G Guanosine triphosphate
Homocystéine Inositol phosphate
Inositol 1, 4, 5-trisphosphate
Récepteur inositol 1, 4, 5-trisphosphate Janus kinase
Mitogen-acitvated protein kinase MAP kinase kinase
Major H-bond network
Phosphatidylinositol 4,5-bisphosphate Protéine kinase C
Ras RhoA RhoGEF ROCK TM VFD
Petite GTPase, isolée de sarcome de Rat Ras homolog family member A
Guanine exchange factors for Rho proteins Rho-associated protein kinase
Domaine transmembranaire
1. I
NTRODUCTION1.1 Les récepteurs couplés aux protéines G (GPCRs)
Les récepteurs couplés aux protéines G (GPCRs) sont des récepteurs transmembraires souvent appelés récepteurs à 7 domaines transmembranaires. Ces récepteurs peuvent donc capter un stimulus à l’extérieur d’une cellule pour en faire la transduction et produire une réponse à l’intérieur de la cellule. Ces stimuli peuvent entre autres être des hormones, des stimulations sensorielles ou des neurotransmetteurs. Les GPCRs sont responsables de différents processus physiologiques contrôlant la réponse immunitaire et inflammatoire, la neurotransmission, la prolifération cellulaire, la contraction cellulaire, la différenciation cellulaire, la migration cellulaire, la sécrétion d’hormones et le métabolisme cellulaire (Wettschureck, 2005).
Souvent appelée la superfamille des GPCRs, elle représente la plus grande famille de protéines membranaires dans le génome humain. Les GPCRs sont la cible de 30 à 40 % des médicaments disponibles sur le marché en faisant donc un sujet très étudié (Kobilka, 2007; Stevens et al., 2013).
Cette superfamille est divisée en 7 familles : A, B, C, D, E, F et les GPCRs d’adhésion, basée sur l’homologie de séquence en acides aminés (Flower, 1999; Jacoby et al., 2006). Les sept domaines transmembranaires qui composent les GPCRs ont une structure secondaire hélice-α reliées par trois boucles intracellulaires et trois boucles extracellulaires. Leur structure est composée d’une portion extracellulaire avec une terminaison amine (N-terminale) ainsi qu’une une portion intracellulaire carboxy-terminale (C-terminale). Les 7 familles se distinguent principalement par leurs extrémités N-terminales et C-terminales (Kobilka, 2007). Les GPCRs de classe A, aussi nommés « Rhodopsin-like » à cause de leur homologie avec la structure de la Rhodopsine, sont la plus grande famille de GPCRs. La classe A des GPCRs est responsable de plusieurs fonctions telles que la neurotransmission, la vision, l’odorat, la réponse immunitaire et la production d’hormones. La liaison du ligand se fait à l’intérieur des domaines transmembranaires plus ou moins profondément dans la pochette de liaison en fonction de la nature du ligand (Zhang et al., 2015). Les GPCRs de classe B ou famille des récepteurs à la
sécrétine sont des récepteurs activés majoritairement par des ligands de type peptidiques et la liaison du ligand se fait sur la partie N-terminale du récepteur (Hoare, 2005). La famille de GPCRs de classe C ou récepteurs au glutamate sont des récepteurs ayant un domaine extracellulaire exceptionnellement grand qui contient un module nommé «Vénus
attrape-mouche» (Venus flytrap, VFT) puisque ce module ressemble à une plante carnivore qui porte ce
même nom. Le module VFT de la partie N-terminale contient les sites orthostériques de liaison des ligands, mais les GPCRs de classe C ont également des sites de liaison allostériques parmi les 7 domaines transmembranaires. La partie N-terminale contient également des sites riches en cystéine (cystein rich domain) situés en aval du module VFT (Chun et al., 2012). Les GPCRs de classes D et E sont des récepteurs de phéromones et ne se retrouvent pas chez les humains (Flower, 1999). Les GPCRs de la classe F contiennent les récepteurs Frizzled et Smoothened qui sont des récepteurs impliqués principalement dans le développement de l’organisme au moment de l’embryogenèse. Les récepteurs Frizzled sont activés par des ligands de la famille des Wnts qui se lient en N-terminal sur les récepteurs Frizzled (Nichols et al., 2013). Le récepteur
Smoothened n’a quant à lui aucun ligand connu, il est activé par la protéine membranaire Patched qui est activée par le ligand Sonic Hedgehog (Wilson and Chuang, 2010; Shen et al.,
2013). Finalement, la famille des GPCRs d’adhésion comporte une structure formée d’une longue queue N-terminale avec de multiples domaines fonctionnels. Cette famille de GPCRs joue un rôle dans le système immunitaire et certaines évidences montrent que les GPCRs d’adhésion auraient un rôle important dans le cortex nerveux central (Bjarnadóttir et al., 2007; Paavola and Hall, 2012).
1.1.1 Activation des GPCRs
L’unité fonctionnelle des GPCRs est formée d’un récepteur, d’une protéine G hétérotrimérique (Gα, Gβ et Gγ) et d’un effecteur. À l’état basal, la sous-unité Gα liée à la molécule de guanosine diphosphate (GDP) est liée au complexe formé des sous-unités Gβ et Gγ. Une fois activé par un agoniste, le GPCR agit comme facteur d’échange de guanine (GEF) et permet l’échange d’une molécule de GDP pour une molécule de guanosine triphosphate (GTP) qui se lie à la sous-unité Gα. Suite à la liaison du GTP, la sous-unité Gα change de conformation et permet la dissociation de la sous-unité Gα du complexe Gβγ, puis Gα et Gβγ peuvent activer
divers effecteurs. La terminaison du signal se fait par l’hydrolyse du GTP en GDP qui se lie à la sous-unité Gα et qui s’associe à nouveau avec le complexe Gβγ (N. Wettschureck, 2005).
1.1.2 Transduction du signal des GPCRs
Il existe quatre principales familles de protéines Gα hétérotrimériques qui vont permettre l’activation de différentes voies de signalisation : Gαs, Gαi/ο, Gα12/13 et Gαq/11. Ainsi, les protéines de la famille Gαq/11 activent la phospholipase C (PLCβ) qui hydrolyse le phosphatidylinositol-4,5-bisphosphate (PIP2) en diacylglycérol (DAG) et en inositol-1,4,5-trisphosphate (IP3). L’IP3 entraîne une augmentation du calcium intracellulaire et DAG active la protéine kinase C (PKC). La protéine Gαs active l’adénylate cyclase qui cause une augmentation de l’AMP cyclique, ce qui active la protéine kinase A (PKA). Inversement, la protéine Gαi/o inhibe l’adénylate cyclase ce qui entraîne une diminution de l’AMP cyclique. Finalement, les protéines de la famille Gα12/13 activent la kinase RhoA impliquée dans le remodelage du cytosquelette (Vilardaga et al., 2010).
La transduction du signal des GPCRs ne se fait pas exclusivement via l’activation de la protéine G hétérotrimérique et peut également se faire par des voies de signalisation qui sont indépendantes des protéines G. La signalisation indépendante des protéines G peut se faire via le recrutement des β-arrestines (isoformes 1 et 2). Les β-arrestines sont connues pour permettre la terminaison du signal des GPCRs en impliquant les kinases de GPCR (GRKs) qui phosphorylent le GPCR et empêchent ainsi la liaison du récepteur avec la protéine G. En effet, lors de son activation, un changement de conformation dans le GPCR expose le site de liaison des GRKs, permettant la phosphorylation du récepteur (Huang et al., 2011). Ce mécanisme joue un rôle essentiel dans le processus de désensibilisation des GPCRs en permettant le découplage des GPCRs liés à un agoniste. Ensuite, ce découplage promeut l’internalisation des GPCRs par endocytose, dans des vésicules de clathrine, vers la dégradation ou le recyclage (Luttrell and Lefkowitz, 2002; Vilardaga et al., 2010). Il est également possible pour les β-arrestines d’activer la voie ERK1/2, indépendamment des protéines G. La signalisation indépendante des protéines G peut également se faire via la transactivation du récepteur à l’EGF (EGFR) ou l’activation de kinases telles que Src, Jak et ERK1/2. Il est cependant connu que la transactivation de l’EGFR
peut également se faire par des mécanismes dépendants de la protéine G, suite à l’activation de la PKC (Hunyady and Catt, 2006; Kenakin, 2011; Overland and Insel, 2015).
De plus, les protéines Gα ne seraient pas les seules impliquées dans la signalisation des GPCRs. En effet, le complexe Gβγ peut également jouer un rôle dans la signalisation des GPCRs en interagissant avec des effecteurs, les récepteurs et également avec les GRKs. Le complexe Gβγ agit essentiellement comme modulateur dans la signalisation de protéines G passant par des interactions protéine-protéine (Smrcka, 2008). Le premier effecteur identifié pour le complexe Gβγ est le canal potassique Kir3. Le complexe Gβγ a également été identifié pour son implication dans la régulation des canaux calciques voltages-dépendants. Le complexe Gβγ joue aussi un rôle dans l’activation de la phospholipase C (PLC) et permet le recrutement de GRKs. Ces GRKs phosphorylent le GPCR et promeut le recrutement des β-arrestines (isoformes 1 et 2) qui peuvent servir de protéines adaptatrices et d’échafaudage pour l’activation de plusieurs voies de signalisation comme Src, ERK, JNK, p38 et Akt (DeWire et al., 2007). La signalisation Gβγ est également connue pour son implication dans l’activation de la cascade des mitogen-activated
protein Kinases (MAPK) par les récepteurs de facteurs de croissance, localisés à la surface
cellulaire (Khan et al., 2013).
1.1.3 Les GPCRs de classe A
Les GPCRs de classe A sont les plus connus jusqu’à maintenant. Cette classe compte environ 700 GPCRs humains et inclut des ligands qui peuvent être des peptides, des lipides, des neurotransmetteurs aminergiques ou autre. La classe A des GPCRs est également nommée «rhodopsin-like» à cause de son homologie avec la protéine rhodopsine, un GPCR responsable de la sensibilité de l’œil à la lumière (McBee et al., 2001). La rhodopsine est le premier GPCR à avoir été cloné en 1983 (Nathans and Hogness, 1983) et cristallisé en 2000 (Palczewski et al., 2000). C’est seulement lors du clonage et séquençage du récepteur β2-adrénergique que l’homologie de séquence avec la rhodopsine bovine fût prédite (Dixon et al., 1986). Cette hypothèse a été confirmée lorsque la structure cristalline par diffraction des rayons X de la rhodopsine fût comparée avec celle du récepteur β2-adrénergique (Cherezov et al., 2007). Depuis, une trentaine d’autres GPCRs de classe A ont vu leur structure cristalline être résolue par diffraction des rayons X en complexe avec des ligands. La structure des différents récepteurs
de cette classe a ainsi montré des différences mineures au niveau conformationnel, ce qui confirme également la forte homologie dans les sept domaines transmembranaires (Katritch et
al., 2013). De plus, le mécanisme d’activation des GPCRs semble impliquer des résidus
hautement conservés au sein de la classe A (Venkatakrishnan et al., 2016). Parmi les GPCRs de classe A, l’homologie de séquence entre les régions transmembranaires des structures élucidées expérimentalement est de 35 à 100%. Par contre, les GPCRs de classe A ont une homologie de séquence faible avec les autres GPCRs, soit de 20 à 30 % (Cvicek et al., 2016).
1.1.4 Mécanismes d’activation des GPCRs de classe A
Les GPCRs ont plusieurs motifs conservés qui sont impliqués dans leur activation. Les structures cristallines des GPCRs de classe A montrent que certains résidus peuvent former des ponts hydrogènes entre eux ou à l’aide de molécules d’eau. Cela forme un réseau appelé le
Major H-bond network (MHN). Le MHN semble jouer un rôle important dans l’activation des
GPCRs de classe A (Rosenbaum et al., 2009; Cabana et al., 2015). De plus la structure des GPCRs de classe A est composée de plusieurs motifs jouant un rôle majeur dans leur activation. Les principaux motifs impliqués dans l’activation des GPCRs de classe A sont le motif E/DRY dans la partie cytoplasmique du TM3, le motif CWxP dans le TM6 et le motif NPxxY dans le TM7. Le motif E/DRY (Glu/Asp-Arg-Tyr) se trouve dans l’extrémité de la région cytoplasmique du TM3 et joue un rôle important dans l’activation et la désensibilisation des GPCRs en formant un pont salin avec le résidu conservé E (acide glutamique) en position 6.30 dans le TM6. Cette interaction ionique entre l’aspartate en position 3.49 (nomenclature de Ballesteros) (Ballesteros and Weinstein, 1995), l’arginine en position 3.50 du TM3 et l’acide glutamique en position 6.30 sur le TM6 se nomme le « ionic lock » et a pour effet de stabiliser l’état inactif du GPCR pour empêcher la liaison de la protéine G dans la région cytoplasmique (Audet and Bouvier, 2012; Latek et al., 2012; Zhang et al., 2015). En effet, des études ont montré avec la structure cristalline de la rhodopsine (Palczewski et al., 2000) et celle du récepteur β2-adrénergique que lorsque cette interaction est rompue, le TM6 s’éloigne du TM3 et favorise la liaison de la protéine G hétérotrimérique (Yao et al., 2006).
Le motif NPxxY (Asn-Pro-Xaa-Xaa-Tyr) situé dans le bas du TM7 est également impliqué dans l’activation des GPCRs de classe A. Les résidus de ce motif et particulièrement le
résidu tyrosine serviraient d’interrupteur appelé le « tyrosine toggle switch » en entraînant un changement de conformation qui cause l’inclinaison de l’hélice du TM7 (Palczewski, 2006; Trzaskowski et al., 2012). Ce mouvement vers l’intérieur du récepteur permet le repositionnement de la tyrosine en position 7.53 du motif NPxxY, ce qui prévient le mouvement inverse du TM6 et promeut ainsi la stabilisation de la conformation active du GPCR en étendant le réseau de ponts hydrogène (Rosenbaum et al., 2009; Audet and Bouvier, 2012; Trzaskowski et
al., 2012).
Le motif CWxP (Cys-Trp-Xaa-Pro) se trouve sur le TM6 et son résidu tryptophane en position 6.48 est impliqué dans un réarrangement des TM dans la pochette de liaison du ligand. Ainsi, le résidu Trp 6.48 est impliqué dans l’interrupteur moléculaire « transmission switch »qui, lors de la liaison du ligand, réorganise les résidus Trp 6.48 et et Phe 6.44 vers les résidus Leu 5.51 et Pro 5.50, puis éloigne le résidu Leu/Ile 3.40 de Pro 5.50. Le TM6 se rapproche donc du TM5 et le TM5 est éloigné du TM3. Le réarrangement des TM3, 5 et 6 causé par cet interrupteur semble être un mécanisme commun de l’activation des GPCRs de classe A (Deupi and Standfuss, 2011; Trzaskowski et al., 2012; Rasmussen et al., 2015).
En résumé, lors de l’activation des GPCRs de classe A, il y a réarrangement des TM3, 5, 6 et 7 par le changement des différentes interactions causées principalement par les interrupteurs localisés dans les motifs conservés des GPCRs. Ainsi, la rotation du TM6 par l’interrupteur «
transmission switch » dans le motif CWxP perturbe une barrière hydrophobe et permet la
propagation de ponts hydrogènes de la pochette de liaison à l’interrupteur moléculaire « ionic
lock » du motif E/DRY sur le TM3. Puis, le « tyrosine toggle switch » du motif NPxxY vient
remplir l’espace laissé par le réarrangement de la barrière hydrophobe et étend le réseau de ponts hydrogènes (Rasmussen et al., 2015).
1.2 Le système rénine-angiotensine
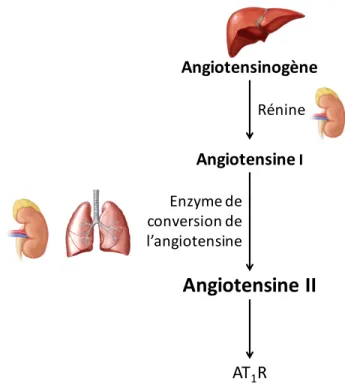
Le système rénine-angiotensine est une cascade enzymatique qui a pour principale fonction de réguler la pression artérielle. La rénine est une enzyme sécrétée par les reins qui initie la cascade lorsqu’il y une baisse du volume sanguin, de la pression artérielle, la rétention d’eau ou de la concentration de sodium. La rénine vient cliver son substrat, l’angiotensinogène, une
protéine sécrétée par le foie. Le clivage produit l’angiotensine I (AngI) qui est un décapeptide (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu). L’AngI n’a pas de fonction biologique connue, mais est par la suite convertie en AngII par l’enzyme de conversion de l’angiotensine (ECA), exprimée principalement dans l’endothélium pulmonaire et rénal. L’AngII, un octapeptide (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe), est l’hormone active du système rénine-angiotensine permettant l’activation du récepteur à l’angiotensine II de type I (AT1R), un GPCR (figure 1). L’AngII peut être métabolisée en angiotensine III (AngIII) (Arg-Val-Tyr-Ile-His-Pro-Phe) par l’aminopeptidase A, puis en angiotensine IV (AngIV) (Val-Tyr-Ile-His-Pro-Phe) par l’aminopeptidase N (de Gasparo et al., 2000; Hunyady and Catt, 2006). Avec l’aide d’endopeptidases, une autre forme de l’ECA, l’ECA-2 peut cliver l’AngI en angiotensine 1-7 (Ang (1-7)). L’Ang (1-7) est essentiellement l’angiotensine II sans l’acide aminé en position 8 (Asp-Arg-Val-Tyr-Ile-His-Pro) et qui active plutôt le récepteur Mas, un GPCR impliqué dans l’inflammation (Simões E Silva et al., 2013).
Figure 1. Le système rénine-angiotensine. Représentation schématique de la production d’angiotensine II. Angiotensinogène AngiotensineI
Angiotensine II
Rénine Enzyme de conversion de l’angiotensine AT1R1.2.1 Angiotensine II
L’angiotensine II (AngII), hormone active du système rénine-angiotensine, est un octapeptide composé de huit acides aminés : Asp-Arg-Val-Tyr-Ile-His-Pro-Phe (figure 2). Les principaux effets physiologiques de l’AngII proviennent de l’activation du récepteur AT1 (AT1R) (Miura et al., 2003).
Figure 2: Angiotensine II. Représentation tridimensionnelle de l'octapeptide angiotensine II. Les acides aminés qui composent l’AngII lui confèrent son activité biologique en interagissant avec les différents résidus d’AT1R. Plusieurs études ont permis d’évaluer l’importance de chacun des acides aminés qui composent l’AngII sur son activité biologique. Ainsi, l’acide aspartique en position 1 ne semble pas impliqué dans l’activité biologique de l’AngII puisque l’analogue Sar1AngII qui comporte une sarcosine en position 1, est aussi puissant et efficace que l’AngII (Domazet et al., 2015). De plus, l’AngIII (AngII 2-8) qui ne comporte pas d’acide aspartique en position 1 a une puissance semblable à l’AngII (de Gasparo
et al., 2000). La position 2 semble être plus importante pour l’activité biologique de l’AngII
puisque l’AngIV (AngII 3-8) sans acide aminé en position 2 devient un agoniste partiel avec une faible affinité pour AT1 (Wright et al., 2008). Des études réalisées en substituant les résidus Tyr4 et His6 de l’AngII ont montré que ces résidus sont également importants pour l’activité biologique de l’AngII. En effet, ces modifications ont essentiellement causé une diminution importante de l’affinité pour AT1R (Miura and Karnik, 1999; Oliveira et al., 2007). Le résidu aromatique Phe8 en C-terminal du peptide semble le plus important pour l’activité biologique de l’AngII (de Gasparo et al., 2000). Ainsi, la substitution du résidu Phe8 par un résidu aliphatique
comme Ile8 antagonise l’effet vasoconstricteur de l’AngII (Hata et al., 1982; Miura and Karnik, 1999).
1.2.2 Effets physiologiques de l’angiotensine II
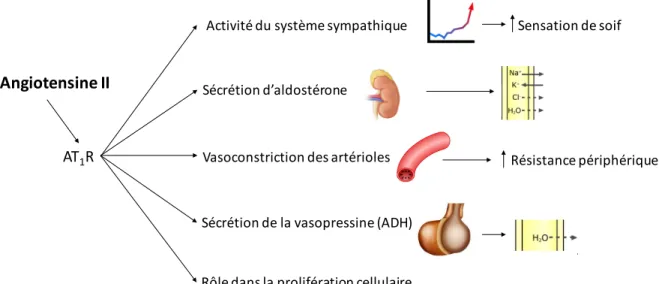
L’AngII active AT1R et entraîne une élévation de la pression artérielle par plusieurs mécanismes physiologiques. Parmi ces différents mécanismes, l’AngII est impliquée dans l’activation neuronale par l’augmentation du système nerveux sympathique en stimulant la soif, ce qui favorise l’absorption d’eau et augmente le volume sanguin. L’AngII stimule la sécrétion d’aldostérone par le cortex de la glande surrénale et favorise la réabsorption tubulaire de sodium (Na+) et de chlore (Cl-), puis l’excrétion de potassium (K+). L’AngII est aussi connu pour son rôle de vasoconstriction des artérioles qui cause une augmentation de la résistance périphérique. L’activation d’AT1R par l’AngII stimule également la sécrétion de vasopressine par le lobe postérieur de l’hypophyse et favorise la réabsorption d’eau. Tous ces mécanismes contribuent ainsi à l’élévation de la pression artérielle. Aussi, l’AngII joue un rôle qui n’est pas lié à l’augmentation de la pression artérielle par son implication dans la prolifération cellulaire (figure 3) (Mehta and Griendling, 2007; Bader, 2010).
Il est possible de réduire les effets physiologiques de l’AngII, pour traiter l’hypertension par exemple, avec certains traitements disponibles sur le marché. L’hypertension peut mener à des dommages cardiaques comme l’hypertrophie du muscle cardiaque. Ainsi, ces traitements agissent principalement sur le système rénine-angiotensine. Il existe deux classes principales de traitements: les inhibiteurs de l’ECA et les antagonistes (aussi nommés bloqueurs) d’AT1R. L’inhibition de l’ECA empêche la formation de l’AngII et prévient donc les principaux effets du système rénine-angiotensine pour permettre de diminuer la pression artérielle. Ces traitements sont indiqués pour l’hypertension, l’insuffisance cardiaque et pour le traitement de maladies rénales chroniques. Puisque l’ECA est impliquée dans différents processus physiologiques, son inhibition peut par contre entraîner des effets indésirables comme l’angio-œdème, causé par l’augmentation plasmatique de bradykinine qui ne peut plus être clivé par l’ECA (Burnier and Brunner, 2000). Pour bloquer les effets hypertenseurs de l’AngII, des antagonistes d’AT1R, comme le Losartan, peuvent être utilisés. Ces antagonistes empêchent l’activation d’AT1R et
donc l’effet vasoconstricteur ainsi que la relâche d’aldostérone ce qui permet de diminuer également la pression artérielle, mais ces antagonistes n’empêchent pas la formation de l’AngII (Barreras and Gurk-Turner, 2003).
Figure 3. Effets physiologiques de l'angiotensine II. Représentation schématique des effets physiologiques suite à la production d'angiotensine II.
1.2.3 Récepteurs de l’angiotensine II
Chez l’humain, on retrouve deux récepteurs à l’AngII : le récepteur à l’angiotensine II de type 1 (AT1R) et également le récepteur à l’angiotensine II de type 2 (AT2R). AT1R est principalement impliqué dans la régulation de la pression artérielle et est exprimé dans le cerveau, les reins, le foie, les poumons, les vaisseaux sanguins et dans la glande surrénale. AT2R est quant à lui exprimé dans plusieurs tissus également, mais principalement lors du développement fœtal. AT2R possède une homologie de séquence d’environ 30% avec AT1R (de Gasparo et al., 2000). La signalisation du récepteur AT2 n’est toujours pas résolue, mais la littérature suggère qu’AT2R aurait plutôt des effets opposés aux effets de la signalisation d’AT1R. Dans le cas d’AT1R, la phosphorylation des résidus tyrosines ou des résidus sérines/thréonines permet l’activation des MAPK, impliquées dans la prolifération cellulaire. Contrairement à AT1R, AT2R serait donc impliqué dans l’inhibition de la prolifération cellulaire en engageant des tyrosines ou des sérines/thréonines phosphatases. AT2R aurait également des effets pro-apoptotiques et, à l’opposé d’AT1R, AT2R aurait un rôle dans la vasodilatation causant
Résistance périphérique Sensation de soif Angiotensine II Activité du système sympathique Sécrétion d’aldostérone Vasoconstriction des artérioles Sécrétion de la vasopressine (ADH) Rôle dans la prolifération cellulaire AT1R
plutôt une diminution de la pression artérielle (Bedecs et al., 1997; Yamada et al., 1998; D’Amore et al., 2005). AT2R est également connu pour diminuer l’expression de cytokines pro-inflammatoires et augmenter l’expression des cytokine anti-pro-inflammatoires au niveau du cerveau et du système cardiovasculaire (Gallo-Payet et al., 2011; Dhande et al., 2015). Certaines évidences montrent également une diminution de l’expression d’AT2R dans les maladies comme l’Alzheimer et le Parkinson (Gallo-Payet et al., 2011; Guimond and Gallo-Payet, 2012). De plus, l’implication potentielle d’AT2R dans le cancer du pancréas à récemment été découverte lors de l’utilisation de souris déficientes en AT2R (Ishiguro et al., 2015).
1.2.4 Structure et mécanismes d’activation d’AT1R
AT1R est un GPCR de classe A, de par sa forte homologie avec le récepteur de la rhodopsine, il est donc constitué de 7 domaines transmembranaires de type hélice α et comporte un domaine N-terminal extracellulaire et un domaine C-terminal intracellulaire. AT1R est composé de 359 acides aminés et son poids est de 41 kDa. Le domaine extracellulaire d’AT1R contient 3 sites de glycosylation (Asn4, Asn176 et Asn188) et 4 résidus cystéine qui forment 2 ponts disulfures essentiels à la liaison de l’AngII sur le récepteur (Miura et al., 2003; Fillion et
al., 2013). Le domaine intracellulaire est constitué de la queue C-terminale et de 3 boucles
intracellulaires. La boucle C-terminale contient des résidus riches en sérine/thréonine qui peuvent être phosphorylés par les GRKs. La phosphorylation par les GRKs permet le recrutement de l’arrestine et donne lieu à la désensibilisation et l’internalisation du récepteur (Oliveira et al., 2007; Tobin, 2008).
Pour accommoder un ligand, les sept domaines transmembranaires forment une cavité appelée pochette de liaison. Ainsi, les petits ligands non peptidiques vont se lier plus profondément dans la pochette de liaison sur les domaines transmembranaires. Pour les ligands peptidiques plus volumineux, comme l’AngII, la liaison aux domaines transmembranaires formant la pochette de liaison joue un rôle majeur dans l’activation d’AT1R, mais les régions extracellulaires contribuent également de façon importante à la liaison de l’AngII (de Gasparo et
al., 2000). Comme mentionné dans la section 1.1.4, l’activation des GPCRs de classe A dépend
de plusieurs interrupteurs moléculaires sur différents motifs hautement conservés pour leur activation (E/DRY, NPxxY et CWxP). La structure cristalline par diffraction des rayons X
d’AT1R obtenue en 2015 révèle que le « ionic lock » du motif E/DRY qui forme un pont salin entre l’arginine 3.50 et l’acide glutamique en position 6.30 n’est pas possible, puisque dans le cas d’AT1R il n’y a pas de résidu acide en position 6.30 (Zhang et al., 2015). Le motif E/DRY semble tout de même jouer un rôle important dans la signalisation des protéines G et dans le recrutement des β-arrestines. En effet, lorsque l’aspartate 3.49 est substitué par une alanine, cela cause une désensibilisation constitutive d’AT1R et affecte de façon importante la signalisation
via les protéines G, mais n’affecte pas la signalisation indépendante des protéines G comme le
recrutement des β-arrestines (Wilbanks et al., 2002; Gáborik et al., 2003). De plus, le mouvement des TM3 et TM6 est connu pour être une caractéristique importante de l’activation d’AT1R. Lors de la liaison de l’AngII à AT1R, une des étapes importantes est l’éloignement entre le TM3 et le TM6 qui favorise une conformation active et permet le couplage à la protéine G (Balakumar and Jagadeesh, 2014). Ainsi, lors de l’activation d’AT1R, le TM6 pivote et s’éloigne du TM3 qui fait un léger mouvement dans le sens antihoraire exposant ainsi des résidus qui permettent maintenant au récepteur de passer de l’état inactif à l’état actif (Martin et al., 2004, 2007).
Les différents résidus qui composent l’AngII sont également importants dans la signalisation d’AT1R. Comme discuté à la section 1.2.1, les résidus 2, 4, 6 et 8 semblent avoir une influence majeure sur l’activité biologique de l’AngII. Des évidences montrent que l’interaction entre le résidu Asn111 dans le TM3 d’AT1R et le résidu Tyr4 de l’AngII joue un rôle majeur dans l’activation de la voie Gq et la production des inositols phosphates (Petrel and Clauser, 2009). La mutation de l’Asn111 pour Gly111 ou Ala111 (N111A ou N111G) entraîne une activation constitutive d’AT1R puisque l’alanine et la glycine sont des résidus moins encombrants favorisant l’état actif du récepteur (Groblewski et al., 1997; Feng et al., 1998). Le résidu His256 dans le TM6 semble également avoir une importance majeure dans l’activation de la voie Gq puisqu’il interagit avec le résidu Phe8, largement connu pour son rôle dans l’activation de la voie Gq médiant les principaux effets hypertenseurs d’AT1R (Miura et al., 2011). Des études sur AT1R montrent que l’interaction entre le résidu Tyr113 sur le TM3 et le résidu His256 sur le TM6 est importante pour stabiliser AT1R dans un état inactif. L’activation de la voie Gq, qui peut être mesurée par la production des inositols phosphates, est empêchée par ces interactions lors de la liaison de l’agoniste inverse Olmesartan (Miura et al., 2006).
Aussi, le MHN joue également un rôle important dans l’activation d’AT1R. Le résidu Asp74 du TM2 agit comme interrupteur moléculaire et forme un pont hydrogène avec le résidu Asn111 du TM3 et serait impliqué dans la stabilisation de l’état inactif d’AT1R. En effet, le réseau de ponts hydrogènes formé de Asn111(TM3), Asp74(TM2) et Asn295(TM7) serait associé avec l’état inactif d’AT1R. Par contre le réseau de ponts hydrogènes impliquant Asp74(TM2), Asn46(TM1) et Asn295(TM7) serait quant à lui associé avec l’état actif d’AT1R. La formation d’un pont hydrogène entre Asp74 dans le TM2 et Asn46 dans le TM1 semble donc important pour l’activation d’AT1R (Cabana et al., 2013, 2015; Fillion et al., 2013). Une mutation au niveau du résidu Asp74 (TM2) pour Asn74 (D74N) réduit le couplage aux protéines G, mais maintient la signalisation des β-arrestines (1 et 2) après stimulation avec l’AngII. Par contre, la mutation du résidu Asn111 (TM3) dans le MHN pour Trp111 (N111W) rend AT1R inactivable et empêche la signalisation via les protéines G et les β-arrestines (1 et 2) (Auger-Messier et al., 2003; Fillion et
al., 2013; Cabana et al., 2015).
Finalement, la structure du complexe récepteur-ligand (AT1R-AngII) est très dynamique et des études de photomarquage ont montré que lorsque l’AngII se lie à AT1R, le résidu Phe8 de l’AngII s’insère profondément dans la pochette de liaison du récepteur. Le complexe AngII-AT1R adoptant ainsi un mode de liaison vertical, il peut y avoir des contacts concomitants à travers la surface extracellulaire et en profondeur dans le noyau de domaines transmembranaires du récepteur. Les études de photomarquage révèlent également l’importance du contact entre l’Arg2 de l’AngII et le résidu Asp281(TM7) d’AT1R et confirment l’importance de la position Tyr4 de l’AngII et de son interaction avec le résidu Arg167(TM4)du récepteur. Le résidu Phe8 de l’AngII est bien connu pour son importance dans la signalisation dépendante des protéines G d’AT1R (de Gasparo et al., 2000). Les études de photomarquage montrent que Phe8 formerait un pont salin avec le résidu Lys199 et cette interaction place le résidu Phe8 à proximité du résidu Trp253(TM6) d’AT1R qui est le « transmission switch » du motif CWxP, un interrupteur moléculaire important dans l’activation des GPCRs de classe A (Arsenault et al., 2010; Fillion et
1.3 Signalisation d’AT1R
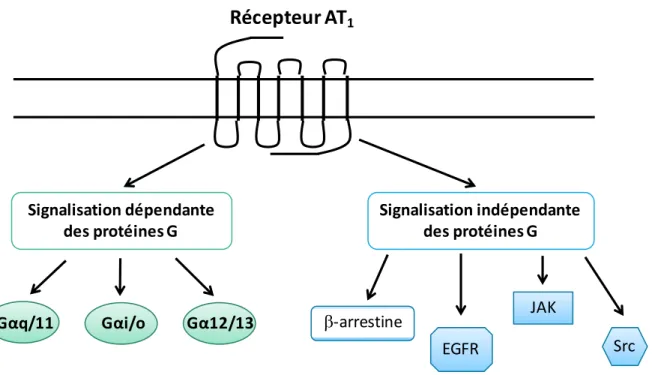
Suite à la formation du complexe AT1R-AngII, AT1R interagit avec la protéine Gα et permet l’activation de voies de signalisation dites dépendantes des protéines G. Ces voies passent par l’activation des protéines hétérotrimériques Gq/11, Gi/o et G12/13 qui activent à leur tour divers effecteurs. Il est aussi connu qu’AT1R ne signale pas exclusivement via les protéines G permettant également une signalisation indépendante des protéines G. Ainsi, AT1R peut permettre le recrutement des β-arrestines (β-arrestine-1 et β-arrestine-2) et l’activation du récepteur à l’EGF menant à l’activation de MAP kinases (EGFR) (figure 3). L’activation d’AT1R par l’AngII est également connue pour être impliquée dans l’activation des kinases de la famille Jak et de la famille Src. Les kinases Jak phosphorylent et activent les facteurs de transcription STAT tandis que les kinases Src peuvent également mener à l’activation de MAP kinases (Guo et al., 2001; Hunyady and Catt, 2006).
Figure 4: Portrait global de la signalisation d’AT1R. AT1R peut activer différentes voies de signalisation de façon dépendante des protéines G ou de façon indépendante des protéines G.
Récepteur AT
1 Gαq/11 Gα12/13 b-arrestine Signalisation dépendante des protéines G Gαi/o Signalisation indépendante des protéines G EGFR JAK Src1.3.1 Voies dépendantes des protéines G
Suite à la liaison de l’AngII, AT1R interagit avec la protéine Gαq et active la phospholipase Cβ (PLCβ). La PLCβ hydrolyse le phosphatidylinositol-4,5-bisphosphate (PIP2) en diacylglycérol (DAG) et en inositol-1,4,5-trisphosphate (IP3). Ainsi, suite à la production du second messager IP3, il y a relâche de calcium intracellulaire (Ca2+) via l’activation du récepteur/canal IP3R (Mikoshiba, 2007). Cela stimule alors la contraction musculaire lisse (Wynne et al., 2009). Le second messager DAG permet l’activation de la protéine kinase C (PKC). La PKC est une famille de protéines kinase qui contrôle différentes fonctions de protéines par la phosphorylation des résidus sérine/thréonine sur ces protéines (Nishizuka, 1984; Freeley et al., 2011). De cette façon, la voie Gq peut entrainer plusieurs effets au niveau cardiaque tel que l’hypertrophie du muscle cardiaque (Keys et al., 2002). Le AT1R peut interagir avec la protéine G12/13 qui va activer RhoGEF qui active à son tour RhoA et ROCK qui jouent un rôle dans la réorganisation du cytosquelette (Siehler, 2009). Le récepteur peut également interagir avec la protéine hétérotrimérique Gi qui cause une diminution de l’adénosine monophosphate cyclique (AMPc) via l’inhibition de l’adénylate cyclase (de Gasparo et al., 2000).
1.3.2 Voies indépendantes des protéines G et voies des MAPK/ERK1/2
AT1R peut permettre l’activation de voies de signalisation indépendamment des protéines G, comme le recrutement des β-arrestines (β-arrestine-1 et β-arrestine-2). Ainsi, chaque agoniste promeut des changements de conformation distincts dans les GPCRs et certains agonistes vont permettre l’activation des GRKs qui peuvent phosphoryler AT1R, ce qui aura pour effet de faciliter le recrutement des β-arrestines et de stopper la signalisation via les protéines G (Turu et
al., 2006). Les β-arrestines sont également impliquées dans l’internalisation et la translocation
des GPCRs (van Koppen, 2004).
AT1R peut activer des voies de signalisation qui impliquent les MAP kinases (MAPK), jouant un rôle dans la prolifération cellulaire (Liu et al., 2014). L’activation des voies impliquant les MAPK se fait par des cascades de signalisation. Ainsi, les récepteurs tyrosine kinases sont activés par des facteurs de croissance qui activent la protéine MAP-kinase-kinase-kinase (Raf) pour ensuite phosphoryler sur les résidus sérine/thréonine et finalement activer
MAP-kinase-kinase (MEK) qui phosphoryle sur résidus tyrosine et thréonine et active MAPK (extracellular
signal regulated kinase) ERK (Bokemeyer et al., 1996). L’activation de ERK1/2 par AT1R peut
se faire de façon dépendante de la protéine Gq, via la PKC. La voie ERK1/2 activée par la PKC suite à l’activation de la voie Gq, entraîne l’activation de la cascade de kinases Raf, MEK et ERK1/2 (Chiu et al., 2003). L’activation de ERK1/2 peut être indépendante des protéines G suite au recrutement des arrestines. L’activation de la voie ERK1/2 passant par les arrestines se fait par la formation de complexes stables entre AT1R et les β-arrestines (1 et 2) suite à la stimulation et l’internalisation d’un ligand. La formation de ces complexes entraînent ainsi l’augmentation de l’activation des MAP kinases (Tilley, 2011). Finalement, l’activation de la voie ERK1/2 peut se faire par la transactivation de l’EGFR causé par la relâche de Ca2+, par la PKC et par la transactivation dépendante de la kinase Src. La kinase Src peut entraîner l’activation de métalloprotéinases (MMP) transmembranaires. Ces MMP sont responsables de la relâche du ligand EGF lié à l’héparine (HB-EGF) qui va permettre l’activation du récepteur à l’EGF (EGFR). L’activation de l’EGFR entraîne l’activation de la petite protéine G Ras qui active la cascade de MAP kinases Raf, MEK et ERK1/2 (Hunyady and Catt, 2006; Overland and Insel, 2015). Récepteur AT1 Récepteur AT1 AngII
ERK1/2
EGFR Voies dépendantes des protéines G Raf MEK Raf Ras MEK β γ Gα Voies indépendantes des protéines G PIP2 PLCβ IP3 Ca2+ PKC DAG Gαq Gα12/13 RhoA Gαi cAMP b-arrestineFigure 5: Signalisation d’AT1R. Schématisation de l'activation d’AT1R par l'AngII qui permet l'interaction avec différents effecteurs pour produire des réponses physiologiques. AT1R peut activer des voies de signalisation dépendantes ou indépendantes des protéines G.
1.4 Sélectivité fonctionnelle
Historiquement, les ligands étaient connus pour interagir avec un récepteur afin d’activer la protéine G et entraîner la signalisation d’un récepteur. Maintenant, il est connu que les GPCRs peuvent adopter un grand nombre de conformations suite à leur stimulation par divers ligands pour permettre le couplage à différentes protéines G. Ce couplage à différentes protéines G permet ainsi l’activation de différentes voies de signalisation. Il est donc logique de penser que des ligands qui lient un récepteur peuvent donner lieu à des conformations différentes, affectant alors la signalisation du GPCR. Un ligand peut donc lier un récepteur et induire des conformations qui activeront certaines voies de signalisation en interassigant avec leurs effecteurs respectifs, mais sans activer nécessairement toutes les voies connues de ce récepteur. C’est le concept de la sélectivité fonctionnelle où il y a une activation sélective des voies de signalisation (Galandrin et al., 2007). AT1R peut activer plusieurs voies de signalisation suite à la liaison de l’AngII. L’AngII arrive donc à stabiliser AT1R dans une conformation qui permet l’activation des différentes voies de signalisation. Le développement de nouveaux ligands d’AT1R a montré que ces ligands peuvent stabiliser AT1R dans différentes conformations. La sélectivité fonctionnelle ou signalisation biaisée d’AT1R a montré que certains ligands permettent de stabiliser le récepteur dans certaines conformations qui promeuvent une activation préférentielle de certaines voies tout en ne permettant pas l’activation d’autres voies (Urban et
al., 2007; Kenakin, 2011).
1.4.1 Intérêt de la sélectivité fonctionnelle
La sélectivité fonctionnelle est un concept de plus en plus exploité pour la conception de nouveaux médicaments en raison du potentiel thérapeutique de l’activation de voies de signalisation produisant les effets plutôt bénéfiques sur l’organisme tout en éteignant les voies pouvant avoir des effets délétères. Par exemple, le recrutement des β-arrestines peut avoir un effet cardioprotecteur en augmentant la résistance au stress mécanique subit par le cœur (Noor et
al., 2011). De plus, la compagnie Trevena Inc., qui travaille sur le développement de ligands
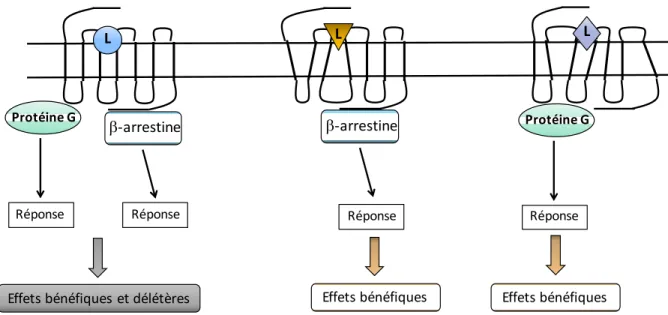
biaisés a développé un ligand biaisé pour les β-arrestines, le TRV027 ([Sar1D-Ala8]AngII). Le TRV027 montre des effets cardioprotecteurs par le recrutement des β-arrestines. Ce recrutement pourrait donc réduire la pression sanguine et augmenter les performances du cœur, ce qui n’est pas observé avec le Losartan (Violin et al., 2010). Par contre, ce ligand a une courte demi-vie allant de 2,4 à 13,2 minutes (Soergel et al., 2013). À l’opposé, l’activation de la voie Gq amène des effets plutôt délétères lors de l’activation d’AT1R puisque cette voie est responsable de l’augmentation de la pression sanguine et de l’hypertrophie cardiaque (N Wettschureck, 2005). Il est donc d’intérêt de bloquer sélectivement certaines voies ayant des effets négatifs dans un contexte pathologique sans toutefois bloquer les voies qui ont un rôle bénéfique pour le système cardiovasculaire. Certains ligands pourraient donc atteindre plus sélectivement la cible désirée en étant des agonistes sur des voies particulières, mais ayant un effet antagoniste sur les voies produisant des effets adverses (van der Westhuizen et al., 2014) (figure 6).
Figure 6. Signalisation biaisée. Représentation schématique de différents ligands (L) qui peuvent induire différentes conformations d’un récepteur. Chacune des conformations du récepteur permet donc d’activer différentes voies de signalisation produisant diverses réponses.
L L L b-arrestine Protéine G Réponse Réponse Effets bénéfiques et délétères b-arrestine Réponse Effets bénéfiques Effets bénéfiques Protéine G Réponse
1.4.2 Bases structurales de la sélectivité fonctionnelle d’AT1R
Depuis plusieurs années, beaucoup de travaux sont consacrés au concept de la signalisation biaisée. Ces travaux ont permis de montrer le potentiel de certaines modifications aux acides aminés qui forment l’AngII. En effet, des travaux ont montré que les modifications aux positions 4 et 8, par exemple ([Sar1Ile4]AngII et [Sar1Ile8]AngII) entraînent une signalisation biaisée pour certaines voies de signalisation (Domazet et al., 2015). La modification à la position 4 de l’AngII semble affecter davantage l’affinité du ligand et permet l’activation de la voie des ERK1/2 (Domazet et al., 2015). La modification de la position 8 de l’AngII empêche quant à elle l’activation de la voie Gq puisque l’insertion du résidu Phe8 dans le cœur hydrophobe d’AT1R est un déterminant important pour l’activation de cette voie (Cabana et al., 2015). Le composé TRV027 a atteint la phase clinique 2b, mais s’est montré inefficace dans la cardioprotection en ne montrant pas de différence d’efficacité en comparaison avec un placebo (Pang et al., 2017). Son développement pour l’indication d’insuffisance cardiaque aiguë a donc été arrêté.
Il reste donc beaucoup de travail à faire sur la sélectivité fonctionnelle pour développer des ligands qui vont s’avérer efficaces dans l’activation des voies qui ont un effet bénéfique sur l’organisme. Mieux comprendre l’effet des ligands sur la signalisation, de même que les mécanismes moléculaires impliqués dans la signalisation d’AT1R, permettront certainement de développer des ligands plus efficaces dans l’optique du concept de la sélectivité fonctionnelle, qui présente des avantages indéniables sur l’organisme.
2. H
YPOTHÈSE/
PROBLÉMATIQUELa sélectivité fonctionnelle est un concept de plus en plus exploité afin d’obtenir des effets beaucoup plus sélectifs sur la cible voulue. Comme mentionné en introduction, il est maintenant connu que des ligands peuvent antagoniser certaines voies de signalisation tout en permettant l’activation d’autres voies pour un même GPCR. AT1R est un GPCR largement étudié dans ce contexte et plusieurs ligands d’AT1R sont connus pour discriminer certaines voies de signalisation. Les acides aminés aux positions 2, 4, 6 et 8 de l’AngII sont importants pour l’activité de l’octapeptide. Plusieurs travaux dans le laboratoire ont montré que des changements dans les acides aminés des positions 4 et 8 de l’AngII, importants pour la signalisation dépendante des protéines G, peuvent amener une sélectivité fonctionnelle (Domazet et al., 2015). De plus, des changements aux positions 3 et 5 de l’AngII semblent bien tolérés dans la signalisation de ce ligand et ne semble pas affecter l’activité du ligand (Fillion et al., 2010). Ainsi, sachant que la cyclisation d’une molécule peptidique va augmenter la rigidité, la cyclisation de l’AngII devrait favoriser certaines interactions moléculaires avec AT1R et influencer l'activation des voies de signalisation. Afin de comprendre l’interaction de différents ligands avec AT1R et pour tenter d’expliquer leur profil signalétique, des simulations de dynamique moléculaire (DM) permettent l’exploration d’une partie de l’espace conformationnel des différents complexes ligand-récepteur. Cette approche rend possible la comparaison des conformations adoptées par le complexe AT1R-AngII avec le complexe formé par des ligands avec AT1R (Cabana et al., 2013, 2015). L’hypothèse principale du projet est donc que la substitution des acides aminés aux positions 3 et 5 de l’AngII, dans le but de cycliser l’AngII entre ces positions, va augmenter la rigidité structurale de l’AngII et induire un biais dans la signalisation d’AT1R.
Objectifs
Objectif #1
Le premier objectif du projet consiste à évaluer les effets de la restriction conformationnelle imposée par la cyclisation d’analogues de l’AngII sur la signalisation d’AT1R. Ainsi, la puissance (EC50) et l’efficacité (Emax) des ligands cycliques dans l’activation des voies Gq, G12, ERK1/2 et pour le recrutement de β-arrestine-1 et de β-arrestine-2 ont été mesurées.
Objectif #2
Le second objectif consistait à expliquer comment la restriction conformationnelle imposée par la cyclisation de ligands affecte la signalisation. Des simulations en DM ont donc été réalisées en explorant une partie de l’espace conformationnel de chaque ligand cyclique pour le comparer avec l’espace conformationnel du complexe AT1R-AngII. Ces simulations en DM ont donc servi à expliquer les différences dans la signalisation des ligands cycliques en comparaison avec la signalisation de l’AngII.
3. A
RTICLEAngiotensin II cyclic analogs as tools to investigate AT1R biased signaling mechanisms
Auteurs de l’article: David St-Pierre, Jérôme Cabana, Brian J. Holleran, Emanuel Escher, Gaétan Guillemette, Pierre Lavigne et Richard Leduc
Statut de l’article: Soumis à Mol Pharmacol le 05 mai 2017.
Avant-propos: J’ai une participation majeure dans la rédaction du 1er manuscrit. J’ai réalisé tous les essais fonctionnels in vitro et le calcul du biais de signalisation, mais pas la partie in silico de dynamique moléculaire. J’ai contribué de façon importante au montage des figures.
Résumé : Les récepteurs couplés aux protéines G (GPCR) produisent des effets pléiotropes par leur capacité à engager de nombreuses voies de signalisation une fois activés. La sélectivité fonctionnelle (également appelée signalisation biaisée), où des composés spécifiques peuvent amener les GPCR à adopter des conformations qui permettent le couplage à des molécules de signalisation sélectives, continue d'être étudiée de manière significative. Cependant, un aspect important, mais souvent négligé de la sélectivité fonctionnelle est la capacité des ligands tels que l'angiotensine II (AngII) à adopter des conformations spécifiques qui peuvent préférentiellement se lier à des structures sélectives de GPCR. La compréhension de la conformation des récepteurs et des ligands est de la plus haute importance pour la conception de nouveaux médicaments ciblant les GPCR. Dans cette étude, nous avons examiné les propriétés des analogues AngII pour transmettre un agonisme biaisé sur AT1R. Nous avons évalué la capacité des analogues cycliques de l’AngII auxquels les positions 3 et 5 ont été substituées par des résidus de cystéine et d'homocystéine ([Sar1Hcy3,5]AngII, [Sar1Cys3Hcy5]AngII et [Sar1Cys3,5]AngII) à activer Gαq, Gα12, ERK1/2 et β-arrestin (βarr) via AT1R. Fait intéressant, [Sar1Hcy3,5]AngII a montré une puissance et une efficacité sur toutes les voies testées à l'exception de la voie Gq. Les simulations dynamiques moléculaires ont montré que la barrière énergétique associée à l'insertion du résidu Phe8 d'AngII dans le noyau hydrophobe d'AT1R, associée à l'activation Gq, est augmentée avec [Sar1Hcy3,5]AngII. Ces résultats suggèrent que de contraindre les mouvements des déterminants moléculaires d’un ligand donné en introduisant des structures cycliques peut conduire à la génération de nouveaux ligands fournissant un agonisme biaisé plus efficace.
Angiotensin II cyclic analogs as tools to investigate AT1R biased signaling mechanisms
David St-Pierre, Jérôme Cabana, Brian J. Holleran, Emanuel Escher, Gaétan Guillemette, Pierre Lavigne and Richard Leduc
Department of Pharmacology-Physiology, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Quebec, Canada, J1H 5N4
Running title: AT1R biased signaling by cyclic Angiotensin II analogs
Corresponding author: Richard Leduc, Ph.D. Department of Pharmacology-Physiology, Faculty of Medicine and Health Sciences, Université de Sherbrooke, 3001 12th Avenue North, Sherbrooke, Quebec, Canada, J1H 5N4. Tel.: (819) 564-5413 Fax: (819) 564-5400, E-mail: Richard.Leduc@USherbrooke.ca
Abbreviations: AngII, angiotensin II; AT1R, angiotensin II receptor type 1; BRET, bioluminescence resonance energy transfer; DAG, diacylglycerol ; DMEM, Dulbecco’s modified Eagle’s medium; EGFR, epidermal growth factor receptor; ERK1/2, extracellular signal– regulated kinases 1/2; FBS, fetal bovine serum; GPCR, G protein-coupled receptor; GRK, G protein–coupled receptor kinases; Hcy, homocysteine; HEK, human embryonic kidney; IP1, inositol 1-phosphate; PEI, polyethyleneimine; PKC, Protein kinase C; RLucII, Renilla luciferase II.
3.1 Summary
G protein coupled receptors (GPCRs) produce pleiotropic effects by their capacity to engage numerous signaling pathways once activated. Functional selectivity (also called biased signaling), where specific compounds can bring GPCRs to adopt conformations that enable coupling to selective signaling molecules, continues to be widely investigated. However, an important but often overlooked aspect of functional selectivity is the capability of ligands such as angiotensin II (AngII) to adopt specific conformations that may preferentially bind to selective GPCRs structures. Understanding both receptor and ligand conformation is of the utmost importance for the design of new drugs targeting GPCRs. In this study, we examined the properties of AngII analogs to impart biased agonism to the angiotensin type 1 receptor (AT1R). We evaluated the ability of cyclic AngII analogs whereby positions 3 and 5 were substituted for cysteine and homocysteine residues ([Sar1Hcy3,5]AngII, [Sar1Cys3Hcy5]AngII and [Sar1Cys3,5]AngII) to activate the Gαq, Gα12, ERK1/2 and β-arrestin (βarr) signaling pathways
via AT1R. Interestingly, [Sar1Hcy3,5]AngII exhibited potency and efficacy on all pathways
tested with the exception of the Gq pathway. Molecular dynamic simulations showed that the energy barrier associated with the insertion of the residue Phe8 of AngII within the hydrophobic core of AT1R, associated with Gq activation, is increased with [Sar1Hcy3,5]AngII. These results suggest that constraining the movements of molecular determinants within a given ligand by introducing cyclic structures may lead to the generation of novel ligands providing more efficient biased agonism.
3.2 Introduction
The octapeptide hormone angiotensin II (AngII) is the active component of the renin-angiotensin system, responsible for controlling blood pressure and water retention via smooth muscle contraction and ion transport. It exerts a wide variety of physiological effects, including vascular contraction, aldosterone secretion, neuronal activation, and cardiovascular cell growth and proliferation. Virtually all the known physiological effects of AngII are produced through the activation of the AT1 receptor (AT1R), which belongs to the G protein-coupled receptor (GPCR) superfamily (de Gasparo et al., 2000) and whose structure in complex with a selective AT1R antagonist has recently been elucidated (Zhang et al., 2015).
The AT1R interacts with Gq/11 leading to the activation of phospholipase C (PLC), in turn leading to the formation of diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3). IP3 binds to the IP3 receptor on the endoplasmic reticulum, whereupon Ca2+ is released into the cytosol. Together, Ca2+ and DAG allow the activation of protein kinase C (PKC) (Hunyady and Catt, 2006). Also, AT1R interacts with G12, thereby activating RhoA and ROCK, via RhoGEF regulation, leading to the reorganization of the cytoskeleton (Siehler, 2009). The AT1R can also activate the ERK1/2 kinase pathway mediated by PKC (G protein-dependent) or by EGFR transactivation, which is G protein-independent (Luttrell, 2002; Chiu et al., 2003). Following receptor activation, G protein-coupled receptor kinases (GRKs) phosphorylate the AT1R, facilitating β-arrestin (βarr) recruitment and terminating G protein signaling (Turu et al., 2006). βarrs are involved in the internalization and translocation of GPCRs (van Koppen, 2004) but also serve as scaffolds for further GPCR signaling to ERK1/2 for example (Lefkowitz and Whalen, 2004).
Biased signaling is the ability of a ligand to stabilize a receptor under a particular conformation that promotes activation of specific signaling pathways over others (Urban et al., 2007; Kenakin, 2011). The therapeutic potential of functional selectivity is increasingly exploited for the design of new drugs since some signaling pathways produce beneficial effects on the body while others can have harmful consequences. For example, activation of the Gq/11 pathway by AngII may cause adverse effects to the failing heart by increasing blood pressure,


![Figure 7 : Inositol 1-phosphate production induced by AngII analogs. HEK293 cells expressing the AT 1 R were stimulated with increasing concentrations of AngII (A), [Sar 1 Hcy 3,5 ]AngII (B), [Sar 1 Cys 3 Hcy 5 ]AngII (C) and [Sar 1 Cys 3,5 ]AngII (D) fo](https://thumb-eu.123doks.com/thumbv2/123doknet/3570405.104627/44.918.192.714.90.447/figure-inositol-phosphate-production-expressing-stimulated-increasing-concentrations.webp)
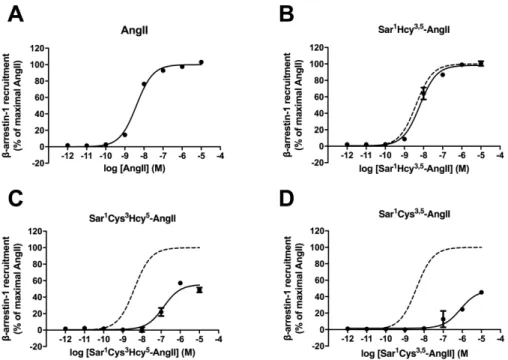
![Figure 9 : βarr2 recrutment to the AT 1 R by AngII analogs. HEK293 cells co-transfected with fusion the protein RLucII-βarr2 and the reporter AT 1 R-GFP10 were stimulated with increasing concentrations of AngII (A), [Sar 1 Hcy 3,5 ]AngII](https://thumb-eu.123doks.com/thumbv2/123doknet/3570405.104627/46.918.185.700.122.492/figure-recrutment-analogs-transfected-reporter-stimulated-increasing-concentrations.webp)
![Figure 11 : PKC-dependent ERK1/2 activation by AngII analogs. HEK293 cells expressing the AT 1 R were pretreated with 250 nM PD168393 for 30 min and then were stimulated with increasing concentrations of AngII (A), [Sar 1 Hcy 3,5 ]Ang](https://thumb-eu.123doks.com/thumbv2/123doknet/3570405.104627/48.918.219.719.274.645/figure-dependent-activation-expressing-pretreated-stimulated-increasing-concentrations.webp)
