Régulation du transcriptome codant et non-codant chez Schizosaccharomyces pombe: facteurs et mécanismes impliqués dans la maturation 3’ des ARNs
et la terminaison de la transcription
Par
Jean-François Lemay Programme de Biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de Philosophiae Doctor (Ph.D.) en Biochimie
Sherbrooke, Québec, Canada Décembre, 2016
Membres du jury d’évaluation
Professeur François Bachand, Ph.D. – Département de Biochimie Professeur Simon Labbé, Ph.D. – Département de Biochimie
Professeur Sherif Abou Elela, Ph.D. – Département de Microbiologie et Infectiologie
Professeur Jacques Côté, Ph.D. – Centre de Recherche sur le Cancer, Département de Biologie Moléculaire, Biochimie Médicale et Pathologie, Université Laval
Le succès n’est pas la clé du bonheur. Le bonheur est la clé du succès. Si vous aimez ce que vous faites, vous réussirez. - Albert Schweitzer
R
ÉSUMÉ
Régulation du transcriptome codant et non-codant chez Schizosaccharomyces pombe: facteurs et mécanismes impliqués dans la maturation 3’ des ARNs
et la terminaison de la transcription
Par
Jean-François Lemay Programme de Biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de Philosophiae Doctor (Ph.D.) en Biochimie,
Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
La synthèse d’un ARNm eucaryotique dépend d’une suite d’étapes qui inclut notamment l’ajout d’une queue poly(A) à son extrémité 3’. Au noyau, la queue poly(A) des ARNms est liée par PABPN1 (poly(A)-binding protein nuclear 1). PABPN1 fut notamment caractérisée, d’après des études in vitro, pour stimuler la réaction de polyadénylation en plus de contrôler la taille ultime des queues poly(A). Cela dit, la ou les fonction(s) biologique(s) de PABPN1 est/sont cependant largement méconnue(s). Chez Schizosaccharomyces pombe (S. pombe), Pab2 est l’orthologue présumé de PABPN1. Or, mes travaux indiquent que Pab2 est fonctionnellement différente de PABPN1 à l’égard de son rôle sur le processus général de polyadénylation. Ainsi, in vivo, l’absence de Pab2 entraîne l’expression et l’accumulation d’un groupe limité d’ARNs hyperadénylés parmi lesquels se trouvent de nombreux petits ARNs nucléolaires non-codants (snoRNAs) lesquels constituent normalement un groupe abondant d’ARN poly(A)-. Mes résultats supportent ainsi un mécanisme par lequel des snoRNAs immatures poly(A)+, sont convertis en une forme mature poly(A)- par le biais de Pab2 et de l’activité 3’5’ exoribonucléase de l’exosome à ARN. Ces observations sont inusitées dans la mesure où elles associent une fonction pour une PABP dans la maturation d'ARNs non-codants, contrairement à la notion que les PABPs travaillent exclusivement au niveau des ARNms, en plus de procurer une nouvelle perspective face au mécanisme de recrutement de l'exosome à ARN à des substrats poly(A)+.
La formation de l’extrémité 3’ d’un ARN est un processus étroitement lié à la terminaison de sa transcription. Pour les gènes codants, la terminaison transcriptionnelle est initiée par le clivage endonucléolytique du pré-ARNm. Ce clivage génère une extrémité d’ARN 5’ libre laquelle sera ciblée par une exoribonucléase 5’3’ afin de mener à bien l’éviction de l’ARNPII de la matrice d’ADN (terminaison transcriptionnelle de type torpedo). Au contraire, chez Saccharomyces cerevisiae (S. cerevisiae), la majorité des gènes non-codants, incluant les snoRNAs, dépendent plutôt du complexe NNS (Nrd1/Nab3/Sen1)
pour la terminaison de leur transcription. Cela dit, il est incertain si le complexe NNS est conservé chez d’autres espèces. À cet égard, mes travaux indiquent que S. pombe est dépourvu d’un mécanisme de terminaison de la transcription de type NNS. Seb1, l’orthologue présumé de Nrd1 chez S. pombe, s’associe plutôt à la machinerie de clivage et de polyadénylation et influence la sélection de site de polyadénylation à l’échelle du génome. Mes résultats supportent ainsi l’utilisation de la machinerie de maturation 3’ des ARNms comme principal vecteur de terminaison transcriptionnelle chez S. pombe et identifient Seb1 comme un facteur clé de ce processus.
L’évènement transcriptionnel étant hautement complexe, des erreurs peuvent arriver de manière stochastique menant à l’accumulation d’ARNs aberrants potentiellement néfastes pour la cellule. Or, mes travaux ont mis en lumière un mécanisme de surveillance co-transcriptionnel des ARNs impliquant l’exosome à ARN et lié à la terminaison de la transcription. Pour ce faire, l’exosome à ARN promeut la terminaison transcriptionnelle via la dégradation d’une extrémité 3’ libre d’ARN devenue émergente suite au recul de l’ARNPII le long de la matrice d’ADN (phénomène de backtracking). Mes résultats supportent ainsi une terminaison de la transcription de type torpedo inversé (3’5’) réévaluant par la même occasion le concept voulant que la terminaison de la transcription s’effectue uniquement selon une orientation 5’3’.
Somme toute, mes travaux de doctorat auront permis d’identifier et de caractériser plus en détail les facteurs et mécanismes impliqués dans la maturation 3’ et la terminaison de la transcription des gènes codants et non-codants chez l’organisme modèle S. pombe.
Mots-clés: Schizosaccharomyces pombe, PABPN1, Pab2, exosome à ARN, Seb1, maturation 3’, polyadénylation alternative, terminaison transcriptionnelle.
T
ABLE DES MATIÈRES1 Résumé ... iii
2 Table des matières ... v
4 Liste des figures ... ix
5 Liste des tableaux ... xi
6 Liste des abréviations ... xii
7 Introduction ... 6
1. La levure Schizosaccharomyces pombe en tant qu'organisme modèle ... 6
2. L'expression génique ... 8
3. L'évènement transcriptionnel ... 9
3.1 L'ARN polymérase ADN-dépendante (ARNP) ... 9
3.1 Le domaine CTD de l'ARNPII ... 10
3.1 Les modifications post-traductionnelles du domaine CTD durant le cycle transcriptionnel et leur influence sur les évènements de maturation co-transcriptionnels de l'ARN ... 10
4. La maturation 3' de l'ARNm ... 14
4.1 Les éléments en cis nécessaires à la maturation 3’ de l'ARNm ... 14
4.2 La reconnaissance des PAS par des facteurs protéiques chez les mammifères 16 4.2.1 Le complexe CPSF ... 16
4.2.2 Le complexe CstF ... 17
4.2.3 Le complexe CFI ... 18
4.2.3 Le complexe CFII ... 18
4.3 La reconnaissance des PAS par des facteurs protéiques chez la levure ... 19
5. Les protéines liant les queues poly(A) (PABP) ... 20
5.1 Les domaines protéiques de PABPN1 ... 21
5.2 Fonction nucléaire canonique de PABPN1 ... 22
5.3 Pab2: l’orthologue putatif de PABPN1 chez S. pombe ... 25
6. La terminaison de la transcription ... 25
6.1.1 La terminaison de la transcription de type torpedo ... 26
6.1.2 La terminaison de la transcription de type allostérique ... 28
6.1.3 Unification des modèles de terminaison transcriptionnelle ... 29
6.1.4 L’association entre la pause transcriptionnelle et la terminaison de la transcription ... 32
6.2 La terminaison transcriptionnelle d’ARN non-codants chez la levure ... 33
6.2.1 La terminaison transcriptionnelle NNS-dépendante ... 33
7. Les petits ARN nucléolaires (snoRNAs) ... 36
8. La biogénèse des snoRNAs ... 39
9. L'exosome à ARN ... 41
9.1 La structure de l'exosome à ARN ... 42
9.2 Les activités enzymatique de l'exosome à ARN ... 43
9.2.1 Dis3 ... 44
9.2.2 Rrp6 ... 45
9.3 Les isoformes de l'exosome à ARN chez la levure ... 45
9.3 Les voies de dégradation/maturation 3’5’ de l’exosome à ARN nucléaire .. 45
10. Les cofacteurs de l'exosome à ARN ... 48
11. Les déterminants dictant le choix entre Dis3 et Rrp6 ... 50
12. Hypothèses et objectifs des travaux de recherche ... 50
8 Résultats ... 53 Article 1 ... 53 Contribution ... 54 Résumé ... 55 Abstract ... 56 Introduction ... 57 Results ... 60 Discussion ... 78 Experimental procedures ... 84 Acknowledgments ... 86 References ... 86
Supplementary references ... 97 Supplementary tables ... 98 Article 2 ... 102 Contribution ... 103 Résumé ... 104 Abstract ... 105 Introduction ... 106 Results ... 108 Discussion ... 133 Experimental procedures ... 137 Acknowledgments ... 139 References ... 139
Supplementary experimental procedures ... 145
Supplementary references ... 153 Supplementary tables ... 155 Article 3 ... 173 Contribution ... 174 Résumé ... 175 Abstract ... 176 Introduction ... 177 Results ... 179 Discussion ... 201 Acknowledgments ... 205 Author contributions ... 205 References ... 205 Experimental procedures ... 209 Supplementary references ... 217 Supplementary tables ... 220 Discussion ... 222 Article 1 ... 222
La polyadénylation lors de la biogénèse des snoRNAs ... 223
Pab2: recruteur ou activateur de l’exosome à ARN? ... 224
Une fonction gène-spécfique pour Pab2 ... 226
Pab2 et ses partenaires dans la biogénèse des snoRNAs ... 227
Les mécanismes entourant la biogénèse des snoRNAs chez S. pombe ... 229
Article 2 ... 231
Divergence fonctionnelle entre Nrd1 et Seb1 ... 231
Simplification du mécanisme de maturation 3’/terminaison transcriptionnelle chez S. pombe ... 233
Les déterminants dictant le recrutement de Seb1 au complexe d’élongation ... 234
La relation entre Seb1 et la viabilité cellulaire ... 237
Le mécanisme d’action de Seb1 lors de l’évènement de maturation 3’ de l’ARN ... 239
Une conservation de fonction chez les organismes supérieurs? ... 242
Article 3 ... 242
L’exosome à ARN nucléaire : un complexe aux multiples fonctions ... 242
L’importance du backtracking de l’ARNPII lors de la terminaison de la transcription médiée par l’exosome à ARN ... 243
Les causes potentielles du backtracking ... 245
La conservation du mécanisme de terminaison de la transcription de type torpedo inversé ... 246
La présence d’un mécanisme de terminaison de la transcription de type «double torpedo» ... 247
L'intégration des travaux de thèse sur la conception actuelle de la terminaison transcriptionnelle chez les eucaryotes ... 249
Conclusion ... 251
Remerciements ... 253
L
ISTE DES FIGURESIntroduction
Figure 1. Schéma de la modulation des modifications post-traductionnelles du domaine CTD et des évènements de maturation co-transcriptionnels de l’ARN naissant au cours du cycle
transcriptionnel chez la levure ... 11
Figure 2. Schéma des domaines protéiques présents chez PABPN1 et Pab2 ... 22
Figure 3. Schéma du mécanisme de polyadénylation PABPN1-dépendant ... 24
Figure 4. Schéma de la terminaison transcriptionnelle de type torpedo ... 27
Figure 5. Schéma de la terminaison transcriptionnelle de type allostérique ... 30
Figure 6. Schéma de l’unification des mécanismes de terminaison transcriptionnelle torpedo et allostérique ... 31
Figure 7. Schéma de la terminaison transcriptionnelle NNS-dépendante ... 36
Figure 8. Schéma des différents types de snoRNA ... 39
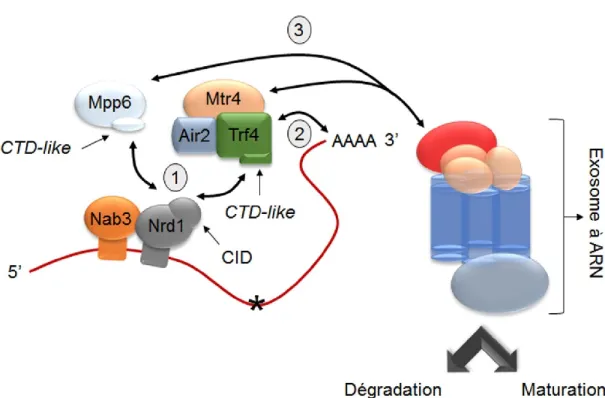
Figure 9. Schéma décrivant la connexion entre la terminaison transcriptionnelle NNS-dépendante et la maturation/dégradation d’ARNnc ... 41
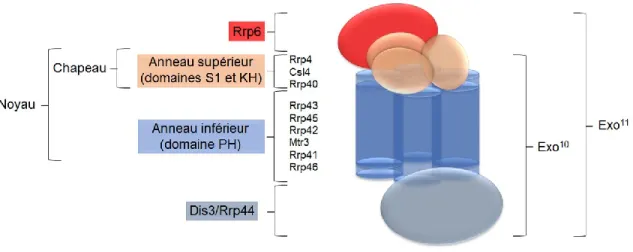
Figure 10. Schéma de l’architecture de l’exosome à ARN…... ... 43
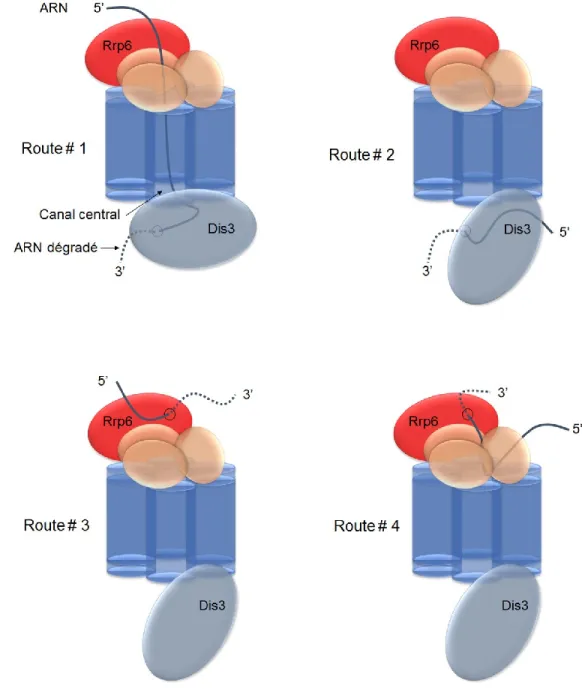
Figure 11. Schéma des voies de dégradation/maturation 3’5’ de l’exosome à ARN nucléaire ... 47
Figure 12. Résumé général des travaux de thèse ... 252
Article 1 Figure 1. Gene-specific changes in expression level in the pab2∆ strain ... 61
Figure 2. Accumulation of 3’-extended polyadenylated snoRNAs in pab2 cells ... 62
Figure 3. 3’-extended snoRNAs accumulate in discrete foci in pab2 cells ... 68
Figure 4. Functional and physical associations between Pab2 and the nuclear exosome ... 69-70 Figure 5. Pab2 is required for Rrp6-dependent processing, but functions in a pathway distinct from the core exosome ... 73
Figure 6. Recruitment of Pab2 to the 3’-end of snoRNA genes ... 77
Figure 7. Model for the role of Pab2 in snoRNA synthesis ... 83
Figure S1-S3. Density of RNA Pol II along snoRNA genes in WT and pab2 cells ... 66
Figure S4. Overlap of upregulated genes from pab2, rrp6, and dis3-54 cells ... 69
Figure S5. Rrp6 directly interacts with Pab2 ... 71
Figure S6. Cid14-independent accumulation of nuclear poly(A) bodies in pab2 cells .... 74
Figure S7. Absence of 3’-extended snoRNA accumulation in ski7 cells ... 74
Figure S8. The H/ACA snoRNP dyskerin associates with polyadenylated H/ACA snoRNAs ... 81
Article 2 Figure 1. Transcription termination defects in Seb1-depleted cells ... 110
Figure 2. Seb1 interacts with the 3’ end processing machinery and is enriched at the 3’ end of genes ... 115
Figure 3. Seb1 levels affect poly(A) site selection ... 118
Figure 4. Seb1 requires functional CID and RRM domains for accurate 3’ end processing and transcription termination ... 122
Figure 5. Seb1 binds to GUA-containing motifs downstream of p(A) sites ... 126
Figure 6. Seb1 levels affect the co-transcriptional assembly of the cleavage/polyadenylation machinery ... 129
Figure 7. Transcription kinetics contributes to Seb1-dependent polyadenylation site selection ... 132
Figure S1. Read-through transcription by RNAPII in Seb1-deficients cells ... 111
Figure S2. Seb1 is not the functional ortholog of Nrd1p ... 112
Figure S3. Genome-wide distribution of Seb1 at the 3’ end of RNAPII genes ... 116
Figure S4. Alternative p(A) sites (APA) utilization in Seb1-deficient cells ... 119
Figure S5. Transcription termination defects upon mutations of the CID and RRM domains of Seb1 ... 124
Figure S6. Mapping Seb1-HTP-bound RNAs by the CRAC technic ... 127
Figure S7. 3’ end processing factors are aberrantly recruited in Seb1-depleted condition 130 Article 3 Figure 1. Cells deficient in core exosome subunits, but not the TRAMP complex, accumulate read-through RNAs ... 182
Figure 2. Widespread production of 3′-extended transcripts in exosome-depleted cells .. 184
Figure 3. The core exosome promotes termination of RNAPII transcription ... 187
Figure 4. Transcription-dependent recruitment of core exosome subunits to genes ... 190
Figure 5. The 3′5′ exonucleolytic activity of Dis3 and the exosome central channel are required for transcription termination ... 193
Figure 6. Transcription termination by the exosome is mechanistically linked to RNAPII backtracking ... 198
Figure 7. Model of co-transcriptional RNA surveillance by the RNA exosome by competition with 3′ end processing ... 204
Figure S1. Exosome depletion results in the production of read-through RNAs ... 180
Figure S2. Read-through RNAs detected in exosome-depleted cells are cleaved and polyadenylated downstream of motifs associated with 3 end processing and can result in the production of chimeric transcripts ... 181
Figure S3. The Rrp6 exoribonuclease does not extensively contribute to the suppression of read-through transcript ... 183
Figure S4. Read-through transcription by RNAPII in exosome-deficient cells ... 188
Figure S5. Expression and growth phenotypes of D166N (endo-), D516N (exo-), and D166N D516N (endo- exo-) versions of S. pombe Dis3 ... 192
Figure S6. The central channel is essential for viability and snoRNA processing, but does not impair exosome assembly, in S. pombe ... 195
Figure S7. Transcription termination by the RNA exosome is mechanistically linked to RNAPII backtracking and pausing ... 200
L
ISTE DES TABLEAUXArticle 1
Supplementary Table 1. List of up- and down-regulated genes in pab2 cells ... 98
Supplementary Table 2. S. pombe strains used in this study ... 101
Article 2 Supplementary Table 1. List of proteins identified in the Seb1-HTP purification ... 155
Supplementary Table 2. List of Seb1-HTP -associated proteins with putative functions in 3' end processing and transcription termination of mRNAs ... 165
Supplementary Table 3. List of the top 100 genes displaying 3' UTR lengthening upon Seb1-depletion ... 166
Supplementary Table 4. List of the top 100 k-mer and their associated Z-score derived from pyMOTIF analysis of Seb1-HTP CRAC experiments ... 168
Supplementary Table 5. List of S. pombe strains used in this study ... 171
Supplementary Table 5. List of S. cerevisiae strains used in this study ... 172
Supplementary Table 5. List of plasmids used in this study ... 172
Article 3 Supplementary Table 1. S. pombe strains used in this study ... 220
L
ISTE DES ABRÉVIATIONS 3’READS 3’ UTR(s) 6-AU ADN APA ARN(s) ARNm(s) ARNnc(s) ARNPI/II/III ARNr ARNsb ARNdb ARNt CFI/II CFIA/B ChIP CID CPF CPSF CRAC CstF CTD CUT(s) DSE(s) E. coli EE FISH3' region extraction and deep sequencing 3’ untranslated region(s) 6-azauracil Acide désoxyribonucléique Alternative polyadenylation Acide(s) ribonucléique(s) ARN(s) messager(s) ARN non-codant(s)
ARN polymérase ADN-dépendante I/II/III ARN ribosomal
ARN simple brin ARN double brins ARN de transfert Cleavage factor I/II Cleavage factor I A/B
Chromatin immunoprecipitation CTD-interacting domain
Cleavage and polyadenylation factor
Cleavage and polyadenylation specificity factor Cross-linking and cDNA analysis
Cleavage stimulation factor Carboxy-terminal domain Cryptic unstable transcript(s) Downstream sequence element(s) Escherichia coli
Efficiency element
LCD MTREC NEXT NLS NNS PAP Poly(A) PABP PABPN/C PAS PE PFI Pré-ARNm(s) qPCR RIP RNB RRM SDE snoRNA(s) snRNA(s) S. pombe S. cerevisiae TRAMP TSS WT
Low complexity domain Mtl1-Red1 core
Nuclear exosome targeting Nuclear localization signal Nrd1-Nab3-Sen1
Poly(A) polymerase
Succession de ribonucléotides de type adénosine (A) Poly(A)-binding protein
Nuclear/cytoplasmic poly(A)-binding protein Poly(A) site
Positioning element Polyadenylation factor I
Précurseur(s) d’ARN(s) messager(s) Quantitative polymerase chain reaction RNA immunoprecipitation
RNAse II catalytic domain RNA recognition motif Site-determining element small nucleolar RNA(s) small nuclear RNA(s)
Schizosaccharomyces pombe Saccharomyces cerevisiae TRf-Air-Mtr4 Polyadenylation
Site d’initiation de la transcription / transcription start site Wild-type
I
NTRODUCTION1. La levure Schizosaccharomyces pombe en tant qu’organisme modèle
L’utilisation des levures comme vecteur de recherche pour l’identification et la caractérisation des processus fondamentaux de la cellule a été propulsée par les nombreux avantages qu’offrent ces micro-organismes. Au contraire des bactéries, tel qu’Escherichia coli (E. coli) qui fut l’un des premiers organismes modèles utilisés, les levures se distinguent par leur statut d’organisme unicellulaire eucaryotique. En plus d’accorder un avantage indéniable sur le plan technique en offrant la possibilité d’obtenir un nombre imposant d’individus dans un court laps de temps, au contraire d’organismes pluricellulaires, les levures, en tant qu’organisme eucaryotique, ont également l’avantage de permettre l’étude de processus conservés chez l’humain, mais absents chez les bactéries, tels la biogenèse d’organelles et l’organisation du cytosquelette, ainsi que l’étude de mécanismes divergents significativement de leur équivalent bactérien, tels que ceux liés à la transcription, la traduction ou encore la réplication d’ADN. Ces études ont particulièrement bénéficié de la capacité qu’ont les levures à subir des réarrangements génétiques lors de leur croissance végétative, notamment par le biais de la recombinaison homologue. L’habilité à recombiner de l’ADN exogène à des endroits spécifiques du génome a ainsi permis la génération d’un nombre considérable de souche mutante (délétion/insertion ou modification de gène ainsi que la création de fusion traductionnelle). De plus, la transition aisée entre les états d’haploïdie et de diploïdie, de même que l’aptitude à maintenir la présence de plasmide réplicatif autonome sont d’autres avantages qui ont favorisé l’essor des levures, principalement S. cerevisiae et S. pombe, en tant qu’organismes modèles.
S. pombe, une levure à fission d’après son mode de division cellulaire (scissiparité ou division binaire), fut initialement isolée de la bière de millet, un breuvage traditionnel communément retrouvé en Afrique. De nos jours, S. pombe est isolée d’une variété de sources naturelles et est présente mondialement (Jeffares et al., 2015). L’étendue et l’importance limitée des applications associées à S. pombe, comparativement à S. cerevisiae, ont toutefois
conféré à cette dernière une certaine préférence au sein de la communauté scientifique. L’utilisation de S. pombe en recherche a néanmoins permis d’importantes avancées notamment dans les domaines du cycle cellulaire et de la dynamique des chromosomes ainsi que sur l’influence de l’épigénétique sur le contrôle de l’expression génique (Hoffman et al., 2015). Son importance en recherche est aussi soulignée par un partage de caractéristiques biologiques plus important avec les cellules mammifères qu’avec S. cerevisiae. S. pombe possède notamment de larges centromères modulaires composés majoritairement d’ADN répété qui s’étendent sur plusieurs milliers de paires de bases, rappelant ceux d’organismes supérieurs. En comparaison, les centromères chez S. cerevisiae ne s’étendent que sur quelques paires de bases et sont dépourvus d’ADN répété (Wood et al., 2002). S. pombe a de plus des origines de réplication et un contenu intronique s’approchant davantage de ceux retrouvés chez les eucaryotes supérieurs que de ceux de chez S. cerevisiae (Mojardin et al., 2013, Wood et al., 2002). S. pombe possède également quelques 338 protéines conservées chez les vertébrés, mais absentes chez S. cerevisiae, parmi lesquelles se trouvent notamment les composantes de la machinerie d’interférence à l’ARN (Aravind et al., 2000, Wood et al., 2012)1. Ces gènes font partie des 66% (3399/5138) possédant une équivalence chez les
vertébrés (Wood et al., 2012)2. De manière intéressante, parmi ces gènes, 501 sont présumés
associés à des maladies humaines (Wood et al., 2012)3 et de ce nombre 50 gènes sont
uniquement présents chez S. pombe, n’étant pas conservés chez S. cerevisiae (Wood et al., 2012)4. Avec en prime la complétion du séquençage et de l’annotation du génome (Dutrow
et al., 2008, Rhind et al., 2011, Wilhelm et al., 2008) ainsi qu’un accès grandissant à des études transcriptomiques, protéomiques, génétiques et phénotypiques à large échelle (caractérisation de la localisation cellulaire de l’ORFeome (Matsuyama et al., 2006), détermination des gènes essentiels pour la viabilité cellulaire (Kim et al., 2010), quantification absolue du protéome, du phosphoprotéome et du transcriptome (Carpy et al.,
1 http://www.pombase.org/spombe/query/builder. Éléments de recherche: PBO:0011070 (conserved in
vertebrates) et PBO:0000055 (no apparent S. cerevisiae ortholog). En date d’avril 2016.
2 http://www.pombase.org/spombe/query/builder. Éléments de recherche: Feature Type (protein coding) et
PBO:0011070 (conserved in vertebrates). En date d’avril 2016.
3 http://www.pombase.org/spombe/query/builder. Éléments de recherche: Feature Type (protein coding),
PBO:5000000 (disease associated) et PBO:0011070 (conserved in vertebrates). En date d’avril 2016.
4 http://www.pombase.org/spombe/query/builder. Éléments de recherche: Feature Type (protein coding),
PBO:5000000 (disease associated), PBO:0011070 (conserved in vertebrates) et PBO:0000055 (no apparent S.
2014, Marguerat et al., 2012), identification des sites de clivage et de polyadénylation (Mata, 2013, Schlackow et al., 2013), détermination du translatome lors de la différenciation sexuelle (Duncan et Mata, 2014), caractérisation du réseau d’interaction protéique (Vo et al., 2016) et caractérisation des paramètres cinétiques du métabolisme d’ARN (Eser et al., 2016)) S. pombe peut donc être considéré comme un système de prédilection pour l’étude des principes de bases de la cellule et un vecteur de connaissances important pour une meilleure compréhension d’organismes plus complexes tels que les humains.
2. L’expression génique
L’ensemble des instructions nécessaires au maintien des activités cellulaires se trouve inscrit à l’intérieur du code génétique. L’expression génique renvoie donc au processus global par lequel l’information contenu à l’intérieur d’une région définie d’ADN, appelée gène, est utilisée pour la synthèse de matériel biochimique. Ce produit peut être soit une protéine ou encore un ARN selon le type de gène étudié (codant versus non-codant respectivement). Les processus qui dictent l’expression génique sont particulièrement complexes et, à bien des égards, l’activité génique, définie par la quantité du produit d’un gène donné, est le reflet des fluctuations temporelles des taux relatifs d’évènements de synthèse et de dégradation. Virtuellement n’importe quelle étape du processus d’expression génique peut être modulée. Cette régulation peut avoir lieu lors du processus transcriptionnel ou traductionnel, mais également lors des évènements dictant la maturation co- et post-transcriptionnelle de l’ARN, le transit intracellulaire de l’ARN ou encore l’ajout de modifications post-traductionnelles aux protéines (Komili et Silver, 2008). Ces points de régulation sont d’autant plus importants qu’ils servent, en autre, à assurer l’intégrité du contenu ribonucléique de la cellule, mais aussi à conférer une capacité d’adaptation face aux changements environnementaux.
3. L’évènement transcriptionnel
Les gènes eucaryotiques sont transcrits par l’entremise d’un cycle composé de trois phases séquentielles, mais interconnectées, débutant avec la phase d’initiation et se poursuivant respectivement avec les phases d’élongation et de terminaison de la transcription. Chacune de ces phases dépend d’un nombre imposant de facteurs, soulignant ainsi la nature complexe et l’important potentiel de régulation qu’offre l’évènement transcriptionnel dans son ensemble. Cela dit, la protéine centrale du processus transcriptionnel est l’ARN polymérase ADN-dépendante.
3.1. L’ARN polymérase ADN-dépendante (ARNP)
La polymérisation de ribonucléotides libres en une chaîne d’acide ribonucléique chez les eucaryotes est sous le contrôle de trois ARNs polymérases ADN-dépendantes nucléaires, l’ARNPI, II et III, chacune ayant un rôle primordial au bon fonctionnement de la cellule. L’ARNPI transcrit la plupart des ARNs ribosomaux (ARNr) (5.8S, 18S et 25S) (Russell et Zomerdijk, 2005) tandis que l’ARNPIII transcrits les ARNs de transfert (ARNt), l’ARNr 5S, le petit ARN nucléaire U6 ainsi qu’une variété d’autres ARNs de petites tailles (Dieci et al., 2013). Combiné, la transcription médiée par l’ARNPI et III équivaut à plus de la moitié du débit transcriptionnel de la cellule (Warner, 1999). Malgré une contribution absolue moindre, l’activité transcriptionnelle de l’ARNPII reste la plus variée, allant de la transcription des gènes codant pour des protéines à la transcription d’un nombre important d’ARNs non-codants (ARNncs) tels que les petits ARNs nucléaires et nucléolaires (snRNAs et snoRNAs respectivement) de même que les transcrits d’origine hétérochromatique ou cryptique (intergénique, antisense).
Les ARNPs sont des enzymes aux sous-unités multiples. Celles-ci sont structurellement liées par la présence d’un noyau multiprotéique conservé auquel vient s’associer des sous-unités additionnelles (Cramer et al., 2008). L’ARNPII est composée de 12 polypeptides (Rpb1 à Rpb12) dont 10 s’assemblent en un noyau auquel vient se greffer en périphérie un sous-complexe formé de l’hétérodimère Rpb4/7 (Armache et al., 2005). Des 12 sous-unités
formant l’ARNPII, 5 sont communes aux 3 ARNPs alors que les 7 autres possèdent des similarités de séquence et de structure à des sous-unités présentes chez l’ARNPI et III. L’ARNPII se distingue de l’ARNPI et III par la présence, au niveau de sa sous-unité Rpb1, d’un domaine carboxy-terminal (CTD) flexible situé à proximité de l’emplacement où l’ARN naissant émerge de l’ARNPII (Allison et al., 1985, Chen et al., 2009, Corden et al., 1985, Cramer et al., 2001, Spahr et al., 2009).
3.2. Le domaine CTD de l’ARNPII
Le domaine CTD de l’ARNPII est présent chez un large éventail d’organismes (Liu et al., 2010). La grande majorité de ce domaine est composé de répétitions de l’heptapeptide Y1S2P3T4S5P6S7 dont le nombre varie parmi les taxa allant de 26 chez S. cerevisiae (Allison
et al., 1985) à 29 chez S. pombe (Azuma et al., 1991) et jusqu’à 52 chez les mammifères (Corden et al., 1985).
Le domaine CTD est essentiel à la viabilité cellulaire. Toutefois, chez les levures et les cellules mammifères un nombre limité d’heptades peut être enlevé sans conséquences phénotypiques notables. Ainsi, chez S. cerevisiae, 10 des 26 répétitions sont nécessaires pour l’obtention d’une croissance similaire à une souche sauvage (WT) (West et Corden, 1995) alors que ce nombre est respectivement de 16 et 29 chez S. pombe et la souris (Bartolomei et al., 1988, Schneider et al., 2010). Cette capacité à tronquer le domaine CTD illustre la redondance fonctionnelle des répétitions de ce domaine.
3.3. Les modifications post-traductionnelles du domaine CTD durant le cycle transcriptionnel et leur influence sur les évènements de maturation co-transcriptionnels de l’ARN
Durant l’évènement transcriptionnel, le domaine CTD de l’ARNPII sert au recrutement d’une gamme diversifiée de facteurs (Hirose et Manley, 1998, Hsin et Manley, 2012, Jeronimo et al., 2013, McCracken et al., 1997). Cette capacité à interagir avec des facteurs donnés nécessite des modifications post-traductionnelles spécifiques du domaine CTD
lesquelles sont dynamiquement modulées en fonction de la progression du cycle transcriptionnel, un concept connu comme le «code» du domaine CTD (Buratowski, 2003). Des 7 acides aminés composant un heptade, 5 peuvent être phosphorylés (Y1, S2, T4, S5 et S7)
tandis que les prolines (P3 et P6) peuvent isomériser entre une conformation cis et trans
(Heidemann et al., 2013). De nombreuses études ont répertorié les transitions des marques de phosphorylation durant l’évènement transcriptionnel chez l’humain et S. cerevisiae (Bataille et al., 2012, Kim et al., 2010, Komarnitsky et al., 2000, Mayer et al., 2012, Mayer et al., 2010, Tietjen et al., 2010) (Figure 1).
Figure 1. Schéma de la modulation des modifications post-traductionnelles du domaine CTD et des évènements de maturation co-transcriptionnels de l’ARN naissant au cours du cycle transcriptionnel chez S. cerevisiae.
Au promoteur des gènes, l’ARNPII est sous forme hypo-phosphorylée (pré-initiation), mais est rapidement phosphorylée au niveau de la S5 et S7 au cours de la phase d’initiation de la
transcription. La phosphorylation de la S5 est primordiale pour l’ajout de la structure coiffe
à l’extrémité 5’ du pré-ARNm. Durant la phase d’élongation, la phosphorylation de la S5
diminue graduellement au profit de la phosphorylation de la S2 et de la Y1. Cette transition
est associée au recrutement de facteurs stimulant l’élongation de la transcription ainsi que des facteurs nécessaires à l’épissage du pré-ARNm. Au contraire de la phosphorylation de la Y1 qui chute drastiquement en amont du site de polyadénylation (PAS), la phosphorylation
de la S2 culmine au PAS ce qui coïncide avec le recrutement de facteurs impliqués au niveau
du clivage et de la polyadénylation du pré-ARNm et la terminaison de la transcription. Le profil de la phosphorylation de la T4 est pour sa part largement confiné au cadre de lecture
alors que celui de la S7 est prévalent sur l’ensemble de l’unité transcriptionnelle. Durant la
afin de débuter un nouveau cycle transcriptionnel. Les patrons de distribution des modifications du domaine CTD sont dérivés d’expériences de ChIP lesquelles utilisent des anticorps modification-spécifique. Ces patrons représentent l’enrichissement relatif pour chacune des modifications et ne peuvent donc pas servir de base de comparaison entre celles-ci. À cet égard, le niveau de phosphorylation de la Y1, de la T4 et de la S7 est largement
sous-représenté par rapport à celui de la S2 et S5 lorsque quantifié par spectrométrie de masse.
TSS : site d’initiation de la transcription. Le schéma n’est pas à l’échelle.
Durant la phase d’initiation de la transcription, une version hypo-phosphorylée de l’ARNPII est recrutée au promoteur des gènes et s’assemble avec des facteurs généraux de transcription afin de former un complexe de pré-initiation (Grunberg et Hahn, 2013). Les premières marques de phosphorylation à apparaître se produisent au niveau de la S5 et de la
S7. Celles-ci résultent de l’action de la kinase cycline-dépendante Cdk7 (Kin28 et Mcs6 chez
S. cerevisiae et S. pombe respectivement), une sous-unité du facteur générale de transcription TFIIH (Akhtar et al., 2009). La principale fonction de la phosphorylation de la S5 consiste à
recruter la machinerie enzymatique nécessaire à l’ajout de la structure coiffe à l’extrémité 5’ du pré-ARNm naissant (Cho et al., 1997, Ho et Shuman, 1999, McCracken et al., 1997, Yue et al., 1997). À cet égard, chez S. pombe, la substitution de l’ensemble des S5 en alanine
(S5A5) entraine un phénotype d’inviabilité. Or, ce phénotype peut être contourné par le
truchement d’une liaison covalente entre le domaine CTD et le complexe enzymatique nécessaire à l’ajout de la structure coiffe (Schwer et Shuman, 2011). La phosphorylation de la S5 participe également au recrutement des facteurs nécessaires pour la terminaison des
gènes non-codants chez S. cerevisiae (Kubicek et al., 2012, Vasiljeva et al., 2008) (voir la section 6.2.1). La présence de cette modification est intimement liée à la distance parcourue par l’ARNPII en cours d’élongation comme l’est d’ailleurs l’ensemble des modifications du domaine CTD. La présence de S7 phosphorylé est, quant à elle, relativement stable sur
l’ensemble de l’unité transcriptionnelle (Bataille et al., 2012, Kim et al., 2010, Mayer et al., 2010, Tietjen et al., 2010). Au contraire de la S5, l’implication de la phosphorylation de la S7
sur les évènements de maturation co-transcriptionnels de l’ARN chez S. pombe et S. cerevisiae sont inexistantes. Par contre, chez l’humain, cette modification est liée à la maturation des snRNAs par le complexe Integrator (Egloff et al., 2007, Egloff et al., 2010). Pour sa part, la phosphorylation de la Y1 s’accroit graduellement le long de l’unité
de restreindre le recrutement des facteurs liés aux évènements de maturation 3’ et de terminaison de la transcription à la région immédiate du PAS (Mayer et al., 2012). Le domaine CTD est également phosphorylé au niveau de la T4. Chez S. cerevisiae, cette
modification est limitée aux cadres de lecture alors qu’elle est principalement localisée juste en amont du PAS chez l’humain (Hintermair et al., 2012, Mayer et al., 2012). La phosphorylation de la T4 est notamment nécessaire à la maturation 3’ des ARNms d’histones
chez des cellules de poulet (Hsin et al., 2011), à l’élongation de la transcription chez l’humain (Hintermair et al., 2012) ainsi qu’au remodelage de la chromatine chez certains gènes chez S. cerevisiae (Rosonina et al., 2014). Suite à la phosphorylation de la S5 apparaît celle de la
S2. La phosphorylation de la S2 s’accroît graduellement à partir du site d’initiation de la
transcription (TSS) pour culminer dans l’environnement du PAS (Bataille et al., 2012, Kim et al., 2010, Mayer et al., 2010, Tietjen et al., 2010). L’émergence de cette modification concorde avec l’entrée de l’ARNPII en un mode d’élongation (Bowman et Kelly, 2014). Chez les levures, la présence de S2 phosphorylée dépend de l’action de deux kinases: une
majeure, Ctk1/Lsk1 (S. cerevisiae/S. pombe), et une mineure, Bur1/Cdk9 (Qiu et al., 2009). Celles-ci coordonnent le recrutement des facteurs favorisant l’élongation de la transcription, l’épissage, l’export des ARNms au cytoplasme ainsi que la maturation 3’ du pré-ARNm et la terminaison de la transcription (Jeronimo et al., 2013).
Le «code» du domaine CTD apparait cependant passablement moins complexe qu’envisagé. Ainsi, chez la levure et l’humain, des analyses protéomiques effectuées sur une version modifiée du CTD permettant son analyse par spectrométrie de masse ont révélé que le domaine CTD est principalement phosphorylé en S2 et S5 avec peu ou pas de
phosphorylation au niveau de la Y1, T4 et S7 (Schuller et al., 2016, Suh et al., 2016).
L’utilisation de cette approche a notamment l’avantage de ne pas dépendre d’anticorps modification-spécifique dont l’affinité pourrait être influencée positivement ou négativement par des modifications environnantes. Ces données n’excluent toutefois pas un rôle pour la phosphorylation de la Y1, T4 et S7 sur le processus transcriptionnel et/ou les évènements de
maturation co-transcriptionnels de l’ARN, mais suggèrent que la modification post-traductionnelle de ces acides aminés est plus dynamique/transitoire ou encore limitée à des classes particulières de gènes.
4. La maturation 3’ de l’ARNm
La formation de l’extrémité 3’ des précurseurs d’ARNms est une étape essentielle dans le processus d’expression génique. La maturation 3’ des pré-ARNms eucaryotiques, à l’exception des ARNms d’histones, se caractérise par une réaction en deux étapes comprenant le clivage du pré-ARNm naissant par une endonucléase et la synthèse d’une queue poly(A) par une poly(A) polymérase (PAP) au niveau de l’extrémité 3’ du produit de clivage en amont. En apparence simple, l’évènement de maturation 3’ implique une combinaison d’éléments en cis au niveau du transcrit naissant ainsi qu’une machinerie protéique formée de plusieurs dizaines de sous-unités.
4.1. Les éléments en cis nécessaires à la maturation 3’ de l’ARNm
Pour que la maturation 3’ s’exécute correctement, le pré-ARNm doit être pourvu d’éléments en cis afin de guider le positionnement adéquatement de protéines effectrices. L’ensemble de ces éléments, présents à la fois en amont et en aval du site de clivage, défini le PAS (Tian et Graber, 2012). Chez les mammifères, les PAS sont composés de plusieurs éléments en cis distincts. De ces éléments, 3 se trouvent en amont du site de coupure. Le premier élément est formé d’un motif hexamérique, principalement AATAAA ou ATTAAA, positionné environ -10 à -35 nucléotides du site de coupure (Beaudoing et al., 2000). Le second élément est situé entre les nucléotides -40 à -100 et se compose de plusieurs répétitions du motif TGTA. Finalement, le troisième élément est caractérisé par une composition nucléotidique riche en T et est positionné entre l’élément hexamérique et le site de clivage. Des éléments en cis sont également trouvés en aval du site de coupure. Parmi ceux-ci, se trouvent les éléments DSEs (pour downstream elements) composés de séquences riches en T et en GT. Ceux-ci sont préférentiellement enrichis dans une fenêtre d’environ 40 nucléotides suivant le site de coupure avec une tendance pour les séquences riches en GT à y être positionnés plus près. Les PAS des mammifères possèdent également un élément en cis riche en G à la suite des éléments DSEs (Hu et al., 2005).
La grande majorité des connaissances entourant la composition des PAS chez les levures provient d’études effectuées chez S. cerevisiae. Le modèle actuel inclue 4 éléments en cis: un élément EE, PE ainsi que deux éléments riches en T. L’élément EE (pour efficiency element) se situe en amont du site de clivage et est caractérisé par un contexte nucléotidique riche en TA (TATATA étant la séquence optimale) (Russo et al., 1991). Ce motif sert à accroitre l’efficacité des éléments situés en aval. Parmi ceux-ci se trouvent, l’élément PE (pour positioning element) (Russo et al., 1993). Le motif PE est équivalent au motif hexamérique des PAS chez les mammifères et sert donc à définir l’emplacement du site de clivage. AATAAA et AAAAAA sont les motifs optimaux de l’élément PE (Guo et Sherman, 1995). Finalement, deux éléments riches en T ont été identifiés de part et d’autre du site de clivage (Dichtl et Keller, 2001, Graber et al., 1999). Cette organisation d’éléments en cis chez la levure (EE-PE-T-p(A)-T) rappelle celle présente chez les mammifères (T-hexamère-T-p(A)-T-G). Les éléments en cis chez la levure sont toutefois plus sujets à être dégénérés (Graber et al., 1999, Guo et Sherman, 1995).
Chez S. pombe, les éléments en cis ont été principalement caractérisés au niveau du gène ura4. Le PAS du gène ura4 est complexe et s’étend sur plusieurs centaines de nucléotides. Il comporte un élément SDE (pour site-determining element), positionnée en amont du site de clivage. L’élément SDE possède notamment une séquence agissant comme le motif PE de chez S. cerevisiae ou le motif hexamérique de mammifères. Un deuxième élément, l’élément EE (pour efficiency element), agit de manière à augmenter considérablement l’efficacité du processus de clivage (Humphrey et al., 1994). On trouve également un élément DSE (pour downstream sequence element) en aval du site de coupure. L’ensemble de ces éléments sont notamment nécessaires afin d’induire un état de pause au niveau du complexe d’élongation de l’ARNPII de manière à favoriser la maturation 3’ de l’ARN et la terminaison de la transcription (Aranda et Proudfoot, 1999, Birse et al., 1997). En général, les éléments en cis chez S. pombe exhibent une organisation semblable à celle observée chez S. cerevisiae et les mammifères. Ces éléments sont également fonctionnellement similaires dans la mesure où un PAS de chez S. pombe est reconnu par la machinerie de clivage et de polyadénylation présente chez S. cerevisiae et vice versa (Humphrey et al., 1991). Une telle interchangeabilité n’est toutefois pas observable avec les PAS des mammifères et suggère une reconnaissance
différentielle des motifs en cis par les facteurs protéiques impliqués dans la maturation 3’ entre les levures et les mammifères.
4.2. La reconnaissance des PAS par des facteurs protéiques chez les mammifères
La maturation 3’ d’un ARNm est accomplie par des actions coopératives entre plusieurs complexes protéiques. Chez les mammifères, 4 complexes multiprotéiques forment l’unité de base de la machinerie de clivage et de polyadénylation soit les complexes CPSF (pour cleavage and polyadenylation specificity factor), CstF (pour cleavage stimulation factor), CFI et CFII (pour cleavage factor I et II respectivement) (Takagaki et al., 1989). Tous ces complexes sont indispensables pour une réaction de clivage efficace. Ces 4 complexes interagissent également avec plusieurs autres protéines. Des quelques 85 protéines identifiées lors d’une étude protéomique effectuée chez des cellules humaines, plusieurs sont des composantes intégrales de la machinerie de maturation 3’, telles que l’ARNPII, PABPN1 ou encore la PAP, alors que d’autres ont plutôt des fonctions de régulation ou servent d’intermédiaires entre la maturation 3’ et d’autres processus cellulaires (Shi et al., 2009).
4.2.1. Le complexe CPSF
CPSF est un complexe multiprotéique recruté au niveau du domaine CTD de l’ARNPII et est composé des sous-unités CPSF-160, CPSF-100, CPSF-73, CPSF-30, Wdr33 et Fip1 (Bienroth et al., 1991, Kaufmann et al., 2004, McCracken et al., 1997, Murthy et Manley, 1992, Shi et al., 2009). Deux de ces protéines, Wdr33 et CPSF-30, lient directement, d’après des études in vitro et in vivo, une étroite région d’ARN couvrant le motif hexamérique du PAS (Chan et al., 2014, Schonemann et al., 2014). Ces interactions permettent ainsi d’ancrer le complexe CPSF à l’ARN naissant et de fournir une spécificité à la réaction de maturation 3’. Outre son affinité pour l’ARN, CPSF-30 interagit également avec l’ARNPII influençant l’association du complexe CPSF à l’ARNPII durant l’événement transcriptionnel (Nag et al., 2007). Tout comme CPSF-30, la plus imposante sous-unité du complexe CPSF, CPSF-160, est également impliquée dans des interactions type protéine-ARN et protéine-protéine. CPSF-160 interagit notamment avec l’ARN sur une région diffuse en amont du site de
clivage (Gilmartin et al., 1995, Martin et al., 2012, Murthy et Manley, 1995) de manière à stabiliser le complexe CPSF au niveau du pré-ARNm. CPSF-160 établie de plus des interactions avec la composante CstF-77 du complexe CstF ainsi qu’avec la PAP (Murthy et Manley, 1995). Fip1 contribue également à la liaison du complexe CPSF au PAS. L’interaction de Fip1 avec le pré-ARNm s’effectue notamment au niveau de séquences riches en U situées en amont du motif hexamérique (Kaufmann et al., 2004, Lackford et al., 2014, Martin et al., 2012). Fip1 établie aussi des interactions avec d’autres composantes de la machinerie de clivage et de polyadénylation, interagissant avec CPSF-30, CPSF-160, CstF-77 ainsi qu’avec la PAP (Kaufmann et al., 2004, Murthy et Manley, 1995). Au-delà de la liaison au PAS, le complexe CPSF catalyse le clivage du pré-ARNm au site d’addition de la queue poly(A) par le biais de sa sous-unité CPSF-73 (Mandel et al., 2006, Ryan et al., 2004), une protéine appartenant à la famille d’hydrolase métal-dépendante à domaine β-CASP (Callebaut et al., 2002). CPSF-100, quant à elle, possède une séquence protéique hautement similaire à celle de CPSF-73 (Callebaut et al., 2002). Tout comme CPSF-73, CPSF-100 possède un domaine β-CASP. Toutefois, les résidus du domaine β-CASP impliqués dans la liaison aux ions métalliques sont absents chez CPSF-100 privant celle-ci de fonction enzymatique (Callebaut et al., 2002, Mandel et al., 2006). CPSF-100 possède également un domaine à son extrémité carboxy permettant son hétérodimérisation avec CPSF-73 (Dominski et al., 2005). Le rôle de CPSF-100 dans le contexte de la machinerie de clivage et de polyadénylation semble ainsi plus lié à une fonction de régulation de l’activité enzymatique de CPSF-73 via son interaction avec cette dernière.
4.2.2. Le complexe CstF
CstF est un complexe formé de trois sous-unités: CstF-77, CstF-64 et CstF-50 (Gilmartin et Nevins, 1991, Takagaki et al., 1990). Son rôle au niveau de la maturation 3’ est le reflet à la fois de son interaction avec le domaine CTD de l’ARNPII (Fong et Bentley, 2001, McCracken et al., 1997), le complexe CPSF (Gilmartin et Nevins, 1991, Wilusz et al., 1990) ainsi qu’avec le pré-ARNm via sa liaison aux éléments DSEs. L’interaction du complexe CstF avec le pré-ARNm s’effectue via le domaine de liaison à l’ARN (RRM : RNA recognition motif) de CstF-64. Celui-ci lie préférentiellement des séquences riches en U, mais
peu également lier des séquences riches en GU (MacDonald et al., 1994, Martin et al., 2012, Takagaki et Manley, 1997). Pour sa part, CstF-77 permet d’unir CstF-50 et CstF-64 au sein d’un même complexe (Takagaki et Manley, 2000). Quant à la sous-unité CstF-50, celle-ci est uniquement présente chez les mammifères et ses fonctions à l’intérieur du complexe CstF sont peu connues.
4.2.3. Le complexe CFI
Le complexe CFI existe sous la forme d’un hétérotétramère formé de l’agencement de deux dimères à la composition protéique variable. Chaque dimère se compose d’une sous-unité invariable, CFI-25, à laquelle une de deux protéines hautement similaires CFI-68 ou CFI-59 vient se lier (Ruegsegger et al., 1996). Ce complexe est présent uniquement chez les métazoaires. Des expériences de sélection in vitro de substrat d’ARN (SELEX) ont permis d’identifier comme motif de liaison pour le complexe CFI la séquence UGUAN (Brown et Gilmartin, 2003), une séquence qui a depuis été validée par des approches structurales et de liaisons in vivo (Martin et al., 2012, Yang et al., 2010). Au niveau de la reconnaissance globale du PAS, CFI joue un rôle important en favorisant le recrutement et la stabilisation du complexe CPSF sur le pré-ARNm (Ruegsegger et al., 1996, Venkataraman et al., 2005).
4.2.4. Le complexe CFII
Parmi tous les facteurs formant la machinerie de base de clivage et polyadénylation chez les mammifères, le complexe CFII est sans contredit le complexe multiprotéique le moins bien caractérisé. Deux protéines composent le complexe CFII chez les mammifères : Pcf11 et Clp1 (de Vries et al., 2000). CFII interagit toutefois avec plusieurs autres protéines faisant de ce complexe un point central pour la coordination de nombreux événements cellulaires (de Vries et al., 2000). Pcf11 a un rôle au niveau de la promotion de la terminaison de la transcription tandis que Clp1 a un rôle d’échafaudage en permettant l’interaction aux complexes CPSF et CFI (de Vries et al., 2000, West et Proudfoot, 2008).
4.3. La reconnaissance des PAS par des facteurs protéiques chez la levure
La grande majorité des composantes protéiques formant la machinerie de clivage et de polyadénylation chez les mammifères est présente chez la levure. L’arrangement de ces facteurs en structures multiprotéiques est toutefois légèrement différent. Chez S. cerevisiae, la machinerie de maturation 3’ est formée des complexes CPF (pour cleavage and polyadenylation factor), CFIA et CFIB (pour cleavage factor A/B). Le complexe CPF est lui-même formé de deux sous-complexes, CFII (pour cleavage factor II) et PFI (pour polyadenylation factor I). Le complexe CFII contient notamment les sous-unités orthologues aux facteurs trouvés au sein du complexe CPSF des mammifères (160/Cft1, CPSF-100/Cft2, CPSF-73/Ysh1) à l’exception de CPSF-30 (Yth1), Fip1 (Fip1) et Wdr33 (Psf2), lesquels font partis du complexe PFI. Pour sa part, CFIA est composé de facteurs orthologues à ceux des complexes CstF (CstF77/Rna15, CstF-64/Rna14) et CFII des mammifères (Pcf11/Pcf11 et Clp1/Clp1). CFIB, formé uniquement de Hrp1, est, quant à lui, unique aux levures (Mandel et al., 2008, Xiang et al., 2014). Plusieurs de ces protéines sont connus pour lier l’ARN et le domaine CTD de l’ARNPII (Barilla et al., 2001, Dichtl et al., 2002, Kyburz et al., 2003) et ont une fonction équivalente à leur contrepartie mammifère. Ysh1 est notamment responsable du clivage endonucléolytique du pré-ARNm (Ryan et al., 2004). Cft1 et Cft2 lient toutes deux l’ARN à proximité du site de coupure via des séquences riches en U (Dichtl et al., 2002, Dichtl et Keller, 2001). Une telle préférence de liaison est également observée pour Yth1 (Barabino et al., 2000). Toutefois, une étude d’identification de site de liaison à l’ARN in vivo indique un patron de liaison distinctif pour Yth1 par rapport à d’autres sous-unités du complexe CPF (Baejen et al., 2014). En se liant environ 17 nucléotides avant le site de coupure, Yth1 lierait l’environnement immédiat de l’élément PE ce qui serait en accord avec sa fonction connue chez les mammifères. Fip1 des levures, mammifères ou d’autres espèces partagent une similarité en ce qui attrait à l’organisation de leurs domaines protéiques. Toutefois, chez S. cerevisiae, Fip1 ne possède pas le domaine RD ni le domaine riche en arginine présent à l’extrémité carboxy de la version mammifère lesquels sont nécessaires à la liaison à l’ARN (Kaufmann et al., 2004). Fip1 semble donc avoir évoluée de manière à perdre sa capacité à lier l’ARN au profit d’interaction protéine-protéine. Or, Fip1 fut notamment identifiée suite à son interaction avec la PAP (Preker et al., 1995). La fonction
primaire de Fip1 semble donc être de positionner la PAP dans l’environnement immédiat du site de coupure. Pour sa part, Psf2 participe à l’assemblage de la machinerie de clivage et de polyadénylation en établissant des interactions avec plusieurs sous-unités (Ohnacker et al., 2000). Le complexe CFIA lie, de par sa sous-unité Rna15, des séquences riches en U situées en aval du site de coupure (Baejen et al., 2014, Mayer et al., 2012), un positionnement également observé chez les mammifères (Martin et al., 2012). Finalement, CFIB s’associe à l’élément EE (Kessler et al., 1997).
Beaucoup moins d’informations sont actuellement disponibles sur la composition de la machinerie de clivage et de polyadénylation chez S. pombe. Une certaine conservation semble toutefois se dégager des études protéomiques effectuées (Roguev et al., 2004, Vanoosthuyse et al., 2014). Cette conservation est également observable fonctionnellement. Ainsi, une version mutante de Pfs2 et de Rna14 engendre des défauts de maturation 3’ et de terminaison de la transcription (Sonkar et al., 2016, Wang et al., 2005). L’analyse détaillée des sous-complexes formant la machinerie de clivage et de polyadénylation reste cependant à être établie de même que le réseau d’interaction protéine-ARN.
5. Les protéines liant les queues poly(A) (PABP)
Une fois le clivage endonucléolytique du pré-ARNm effectué, une queue poly(A) est ajoutée à l’extrémité 3’ du produit de clivage en amont. Les queues poly(A) sont présentes à l’extrémité 3’ de la quasi-totalité des ARNms contribuant à leur stabilité et leur traduction (Chen et Shyu, 2011, Preiss et Hentze, 1998). L’influence sur ces processus ne dépend toutefois pas de la seule présence de la queue poly(A), mais requiert la participation de protéines qui viennent s’y lier avec une haute affinité et spécificité. Ces PABPs sont généralement conservées chez les eucaryotes, alors qu’elles sont absentes chez les procaryotes. Ces protéines sont divisées en deux grandes familles sur la base de leur localisation nucléaire (PABPN) ou cytoplasmique (PABPC). Parmi les différentes protéines liant les queues poly(A), PAPBN1 et PABPC1 sont les représentants nucléaires et cytoplasmiques les plus étudiés et les mieux caractérisés (Kuhn et Wahle, 2004, Mangus et
al., 2003). Cela dit, à des fins de limitation et de pertinence, seul PABPN1 sera subséquemment abordée.
5.1. Les domaines protéiques de PABPN1
PABPN1 est largement conservée chez les eucaryotes à l’exception de chez S. cerevisiae (Wigington et al., 2014, Winstall et al., 2000). PABPN1 possède de nombreux domaines protéiques fonctionnels (Nemeth et al., 1995) (Figure 2). PABPN1 comprend, en autre, un domaine N-terminal acidique caractérisé par la présence d’une suite de dix alanines suivant la méthionine initiatrice. Cette particularité est importante dans la mesure où une extension de cette séquence d’alanine est retrouvée chez des individus atteints de la dystrophie musculaire occulopharyngée (OPMD : occulopharyngeal muscular dystrophy) (Brais et al., 1998). Le domaine N-terminale comprend également un motif structural de type coiled-coil essentiel pour la stimulation de l’activité de polymérisation de la poly(A) polymérase (Kerwitz et al., 2003, Kuhn et al., 2003). La liaison spécifique et de haute affinité à des séquences homopolymériques d’adénosines s’effectue, quant à elle, par le biais d’un RRM unique positionné entre les domaines N- et terminaux (Kuhn et al., 2003). Le domaine C-terminale est, pour sa part, basique et ne contient aucun motif structural apparent, mais est néanmoins fonctionnellement important dans la mesure où il contribue à la fois à la liaison à l’ARN (Kuhn et al., 2003) et à la poly(A) polymérase (Kerwitz et al., 2003) en plus d’influencer la capacité d’oligomérisation de molécules de PABPN1 (Fan et al., 2001, Kuhn et al., 2003). C’est également au niveau de ce domaine que se situe le signal de localisation nucléaire (NLS : nuclear localization signal) (Calado et al., 2000, Mallet et Bachand, 2013). Finalement, le domaine C-terminal est sujet à l’activité de multiples protéines arginine méthyle-transférases de type I (Fronz et al., 2008, Smith et al., 1999) ce qui a pour effet d’affaiblir l’interaction entre PABPN1 et transportin (karyophérine β2/Kapβ2), son récepteur d’import nucléaire (Fronz et al., 2011). Cela dit, il appert toutefois que le mécanisme d’import nucléaire de PABPN1 est largement indépendamment de Kapβ2 (Mallet et Bachand, 2013). L’importance fonctionnelle de la di-méthylation asymétrique des arginines sur les propriétés de PABPN1 est donc très énigmatique surtout considérant que
ces modifications n’affectent en rien sa capacité à lier un ARN polyadénylé (Kuhn et al., 2003).
Figure 2. Schéma des domaines protéiques présents chez PABPN1 et Pab2.
Quatre domaines peuvent être discernés chez PABPN1: une suite de 10 alanines positionnée à l’extrémité amino(N)-terminale, un domaine coiled-coil, un domaine de reconnaissance à l’ARN (RRM) ainsi qu’un domaine carboxy/(C)-terminale. Pab2 est l’orthologue présumé de PABPN1 chez S. pombe. Pab2 diffère de PABPN1 en regard de sa taille, mais également par l’absence de la suite d’alanines en N-terminal. Par contre, les trois autres domaines sont relativement bien conservés conférant au final un pourcentage d’identité et de similarité en respect de PABPN1 de 47 et 66% respectivement. Les domaines protéiques sont positionnés approximativement et ne sont pas à l’échelle.
5.2. Fonction nucléaire canonique de PABPN1
L’addition d’une queue poly(A) à l’extrémité 3’ des ARNms s’effectue via l’activité enzymatique de poly(A) polymérases suivant le clivage du pré-ARNm naissant. Parmi les PAPs nucléaires, la PAP dite canonique est la plus étudiée et est conservée des levures aux mammifères (Lingner et al., 1991, Raabe et al., 1991, Wahle et al., 1991). Chez ces derniers, la majorité des ARNms possèdent, à l’équilibre, une queue poly(A) d’une longueur d’environ 50-100 nucléotides (Chang et al., 2014, Subtelny et al., 2014) contrastant avec les quelques 250-300 nucléotides originellement caractérisés (Sheets et Wickens, 1989, Wahle et al., 1991). Ces données proviennent cependant de contextes expérimentaux forts différents. Alors que la taille des queues poly(A) fut initialement caractérisée à partir de réactions de polyadénylation in vitro utilisant soit un extrait nucléaire ou des facteurs protéiques purifiés
et un substrat d’ARN synthétique, l’actuelle mesure provient plutôt du séquençage massif d’ARNs polyadénylés isolés de lignées cellulaires. Considérant l’influence de diverses déadénylases cytoplasmiques sur la taille des queues poly(A) des ARNms (Yamashita et al., 2005), la divergence observée provient vraisemblablement de l’absence de facteurs protéiques modulant négativement l’étendue des queues poly(A) lors des essais de polyadénylation in vitro. La taille moyenne des queues poly(A) varie également d’organisme en organisme, étant d’environ 30 nucléotides chez les levures S. pombe et S. cerevisiae (Subtelny et al., 2014).
In vitro, le processus de polyadénylation ne requiert que trois composantes: la PAP, le complexe CPSF ainsi que PABPN1. Lorsque combiné, une réaction de polyadénylation efficace s’enclenche menant à l’obtention de queues poly(A) d’une longueur moyenne d’environ 250 nucléotides (Bienroth et al., 1993, Wahle et al., 1991). La présence conjointe de ces trois composantes est requise dans la mesure où la PAP est hautement inefficace et peu spécifique. Seule, PAP possède une faible affinité pour ses substrats d’ARN, ce qui résulte en une fréquence d’association/dissociation au substrat élevée engendrant une polyadénylation lente (Wahle, 1991). Cette affinité est grandement augmentée par la présence du complexe CPSF et de PABPN1 qui, par des interactions directes, stabilisent la liaison entre la PAP et l’ARN (Kerwitz et al., 2003, Murthy et Manley, 1995). Ces interactions modulent l’activité de la PAP faisant passer la réaction de polyadénylation d’un mode lent à un mode rapide (Bienroth et al., 1993). La spécificité du processus de polyadénylation est également favorisée dans la mesure où le complexe CPSF recrute la PAP au niveau d’un substrat d’ARN pourvu d’un motif hexamérique AAUAAA (Keller et al., 1991) tandis que PABPN1 fait de même en présence d’un substrat doté d’une répétition de quelques adénosines à son extrémité 3’ (Wahle et al., 1991). Cette stimulation synergique de la PAP est toutefois transitoire car passé ≈ 250 nucléotides, l’activité de la PAP redevient lente (Wahle, 1995). La synthèse des queues poly(A) peut donc être considérée comme un processus biphasique dynamiquement régulé. Cette transition de phase implique un remodelage des interactions entre le complexe CPSF, PABPN1 et la PAP (Figure 3). Durant la phase initiale de synthèse, un contact continu est présent entre ces trois facteurs. L’établissement de ce réseau d’interaction a lieu dès l’ajout par la PAP de 11-14 adénosines,
une longueur suffisante pour la liaison d’une première molécule de PABPN1 (Meyer et al., 2002). Selon les données actuelles, le revêtement de la queue poly(A) en élongation par des molécules de PABPN1 mène à la formation d’une particule sphérique permettant à l’ARN d’adopter un repliement faisant en sorte que le complexe CPSF, PABPN1 et la PAP soit en communication continue (Keller et al., 2000). Cependant, passé ≈ 250 nucléotides, cette particule sphérique ne permet plus d’accommoder des molécules de PABPN1 brisant ainsi l’interaction synergique entre la PAP, le complexe CPSF et PABPN1. Se faisant, toute élongation subséquente par la PAP, médiée uniquement par PABPN1, devient lente ce qui correspond à la deuxième phase du processus de polyadénylation (Kuhn et al., 2009). PABPN1 peut donc être considérée comme un déterminant critique régissant la taille des queues poly(A) au noyau. La/les raison(s) derrière(s) l’accommodation d’un nombre défini de molécules de PABPN1 au sein de la structure sphérique est/sont toutefois incomprise(s).
Figure 3. Schéma du mécanisme de polyadénylation PABPN1-dépendant.
Le recrutement de la poly(A) polymérase (PAP) à l’extrémité 3’ d’un ARN clivé est favorisé par l’interaction entre celle-ci et le complexe CPSF (1). Cette interaction permet à PAP de synthétiser une courte queue poly(A) permettant le recrutement d’une première molécule de PABPN1 (2). La présence de PABPN1 mène à un accroissement de la cinétique de synthèse de la queue poly(A). Cette influence positive est toutefois abrogée lorsque la queue poly(A) atteint ≈ 250 nucléotides (3). À ce moment, l’interaction de la PAP avec le complexe CPSF et PABPN1 est déstabilisée menant à l’interaction de la PAP avec uniquement PABPN1 (4).
5.3. Pab2: l’orthologue putatif de PABPN1 chez S. pombe
Chez S. pombe, Pab2 est l’orthologue présumée de PABPN1 (Perreault et al., 2007). Pab2 partage 47% d’identité et 66% de similarité au niveau de sa séquence en acide aminé et possède des domaines protéiques hautement similaires (Figure 2). Pab2 est cependant passablement plus petite que sa contrepartie humaine conséquence de l’absence d’un domaine N-terminale acidique. De manière similaire à PABPN1, Pab2 est di-méthylée asymétriquement et possède un NLS au niveau de son domaine C-terminal riche en arginine. La méthylation de Pab2 n’est toutefois pas requise pour sa localisation nucléaire laquelle requiert la participation de Kap104, l’orthologue chez S. pombe de la Kapβ2 chez l’humain (Mallet et Bachand, 2013). Tout comme PABPN1, Pab2 possède une spécificité de liaison pour des ARNs polyadénylés et cette liaison est indépendante de son statut de méthylation. En fait, la seule fonction connue à ce jour pour cette modification post-traductionnelle est dans la limitation de l’oligomérisation de molécules de Pab2 (Perreault et al., 2007). Curieusement, Pab2 ne constitue pas une protéine essentielle. Son absence n’est toutefois pas sans conséquence alors que des cellules pab2 affichent un retard de croissance à basse température (Perreault et al., 2007). L’absence de Pab2 engendre également l’expression d’ARNs hyperadénylés indiquant une fonction pour Pab2 dans le métabolisme des queues poly(A) (Perreault et al., 2007). La détection de ces ARNs hyperadénylés indique toutefois que, contrairement à PABPN1, Pab2 n’est nécessaire ni au recrutement de la PAP au substrat d’ARN ni à l’établissement d’une réaction de polyadénylation processive. Une fonction de Pab2 dans le contrôle de la taille des queues poly(A), telle que définie pour PABPN1, est également peu probable compte tenu que le phénotype d’hyperadénylation est limité à une sous-population d’ARNs. Ainsi, en respect des connaissances actuelles, le rôle de Pab2 au niveau du métabolisme des queues poly(A) est à être éclairci.
6. La terminaison de la transcription
La terminaison de la transcription est le processus par lequel l’ARNP se dissocie de la matrice d’ADN à la fin d’un évènement de transcription. Fonctionnellement, la terminaison de la transcription est nécessaire à la partition du génome en définissant la frontière terminale