Université de Sherbrooke
Propriétés structurales et fonctionnelles du récepteur AT
1de
l’angiotensine II
Par Ivana Domazet
Département de pharmacologie
Thèse présenté(e) à la Faculté de médecine et des sciences de la santé en vue de l‟obtention du grade de philosophiae doctor (Ph.D.)
en pharmacologie
Sherbrooke, Québec, Canada mars, 2015
Membres du jury d‟évaluation
Dr.Gaétan Guillemette, département de pharmacologie Dr.Emanuel Escher, département de pharmacologie Dr. Michel Grandbois, département de pharmacologie Dr. Marek Rola-Pleszczynski , département de pédiatrie Dr. François Marceau, département de médecine, Université de Laval
RÉSUMÉ
Propriétés structurales et fonctionnelles du récepteur AT1 de l’angiotensine II Par
Ivana Domazet
Programmes de pharmacologie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l‟obtention du diplôme de philosophiae doctor (Ph.D.) en pharmacologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 L‟angiotensine II (Ang II), une hormone jouant un rôle important dans l‟homéostasie cardiovasculaire, produit la majorité de ses effets en activant le récepteur AT1 appartenantà
la grande famille des GPCRs. Les sept domaines transmembranaires (TM) des GPCRs contribuent à former la pochette de liaison du ligand. Afin d‟identifier les acides aminés du TM2 et du TM5 impliqués dans la formation de la pochette de liaison du récepteur AT1,
nous avons utilisé l‟approche SCAM qui consiste à évaluer les propriétés de liaison du récepteur suite à sa réaction avec le MTSEA. Le MTSEA alkyle les cystéines endogènes ou introduites par mutagénèse dirigée, causant ainsi un encombrement stérique qui interfère avec la liaison du ligand. Une série de mutants ont été produits en remplaçant successivement par une cystéine les résidus 70 à 94 du TM2 ainsi que les résidus 190 à 217 du TM5 du récepteur AT1 et de son mutant constitutivement actif N111G-AT1. Après le
prétraitement avec le MTSEA, les mutants D74C, L81C, L83C, A85C, A89C ont montré une diminution significative d‟affinité pour le ligand 125I-Sar1,Ile8Ang II, suggérant que ces résidus sont orientés dans la pochette de liaison du récepteur AT1. Le mutant
D74C-N111G est devenu insensible au MTSEA, alors que la sensibilité de L81C-D74C-N111G fut diminuée. Par contre, le mutant V86C-N111G s‟est avéré sensible au MTSEA. Ces résultats suggèrent que l‟activation constitutive du récepteur AT1 implique un mouvement
de pivot du TM2, favorisant le rapprochement du haut du TM2 vers la pochette de liaison. Pour le TM5,après le prétraitement avec le MTSEA, les mutants L197, N200, I201, G203 et F204 ont montré une diminution significative d‟affinité pour le ligand 125I-Sar1,Ile8Ang II, suggérant que ces résidus sont orientés dans la pochette de liaison du récepteur AT1. Le
mutant I201C-N111G est devenu plus sensible au MTSEA, alors que la sensibilité de G203C-N111G fut diminuée. Ces résultats suggèrent que l‟activation constitutive du récepteur AT1 implique un mouvement de rotation du TM5 dans le sens anti-horaire. Le
récepteur AT1 est connu pour coupler préférentiellement à la protéine Gq et les propriétés
fonctionnelles de ce récepteur ont surtout été évaluées en fonction de sa capacité à induire la production des inositol phosphates et à mobiliser le Ca2+ intracellulaire. Par contre, le récepteur AT1 interagit avec d‟autres protéines G (Gi et G12/13) et active également des voies
de signalisation indépendantes des protéines G (MAPK). Nous avons évalué une série d‟analogues de l‟Ang II pour leur capacité à inhiber ou activer plus ou moins sélectivement les diverses voies de signalisation en aval du récepteur. C‟est la notion de sélectivité fonctionnelle. Nos résultats démontrent que les substitutions à la position 1 ne confèrent pas de séléctivité fonctionnelle, alors que les substitutions à la position 4 montrent un biais vers la signalisation la MAPK et que les substitutions à la position 8 montrent un biais pour le recrutement des β-arrestines.
Mots clés : récepteur AT1, angiotensine II, pochette de liaison, SCAM, sélectivité
Table des matières
Résumé ... iv
Liste des figures ... ix
Liste des tableaux ... xi
Liste des abréviations ... xii
Introduction ... 1
Les récepteurs couplés à une protéine G (GPCRs) ... 1
Système rénine-angiotensine (RAS) ... 2
Effets physiologiques de l’Ang II:... 3
Récepteurs de l’Ang II : ... 4
Structure générale du récepteur AT1 de l’Ang II ... 4
Nomenclature des résidus situés dans TM ... 5
Ang II ... 6
Structure du récepteur AT1 de l’Ang II ... 6
Pochette de liaison du récepteur AT1 ... 6
Méthode SCAM : identification de la pochette de liaison du récepteur AT1 ... 8
Utilisation du SCAM afin de délimiter la pochette de liaison des GPCRs ... 9
État basal versus état actif du récepteur ... 10
Activation constitutive du récepteur AT1 ... 11
Activation des récepteurs couplés à une protéine G (GPCRs) ... 12
Mécanisme moléculaires impliqués dans l’activation des GPCRs ... 12
Mécanisme d’activation du récepteur AT1 de l’Ang II ... 12
Sélectivité fonctionnelle ... 14
Concept de sélectivité fonctionnelle ... 14
Pourquoi faire des études de sélectivité fonctionnelle? ... 15
La signalisation du récepteur AT1 ... 15
Voies de signalisation impliquées dans l’activité du récepteur AT1 ... 15
Activation de la voie de MAPK/ERK par le récepteur AT1 ... 17
Méthodes utilisés pour mesurer l’activation de différentes voies de signalisation .... 19
Production des inositols phosphates ... 19
Activation de la voie des ERKs ... 19
Recrutement de l’arrestine et l’activation de la voie G12 ... 20
Problématique ... 22
Objectifs ... 23
Article 1 ... 24
The second transmembrane domain of the human type 1 angiotensin II receptor participates in the formation of the ligand binding pocket and undergoes integral pivoting movement during the process of receptor activation ... 24
Résumé ... 25 Introduction ... 27 EXPERIMENTAL PROCEDURES ... 29 RESULTS ... 34 DISCUSSION ... 38 FOOTNOTES ... 46 REFERENCES ... 46 Article 2 ... 48
The fifth transmembrane domain of angiotensin II Type 1 receptor participates in the formation of the ligand-binding pocket and undergoes a counterclockwise rotation upon receptor activation ... 48
Résumé ... 48 INTRODUCTION ... 50 EXPERIMENTAL PROCEDURES ... 52 RESULTS ... 54 DISCUSSION ... 64 REFERENCES ... 70 Article 3 ... 73
Characterization of Angiotensin II molecular determinants involved in AT1 receptor functional selectivity ... 73
Résumé ... 73
INTRODUCTION ... 75
MATERIALS AND METHODS ... 76
RESULTS ... 80
DISCUSSION ... 98
REFERENCES ... 102
Discussion ... 114 CONCLUSIONS ... 133 Liste des références ... 136
LISTE DES FIGURES
INTRODUCTION
Figure 1 Le système rénine-angiotensine………3
Figure 2 Représentation schématique du récepteur AT1 ... 5
Figure 3 Arrangement des domaines transmembranaires autour de la pochette de liaison du récepteur AT1 ... 8
Figure 4 Substituted Cysteine Accessibility Method (SCAM) ... 10
Figure 5 Sélectivité fonctionnelle ... 15
Figure 6 Les différentes voies de signalisation du récepteur AT1 ... 17
ARTICLE 1 Figure 1 Schematic representation of the human AT1 receptor ... 30
Figure 2 MTSEA treatment of the wild-type AT1 receptor and sensitive reporter cysteine-bearing mutant receptors ... 32
Figure 3 Effects of MTSEA on different mutant AT1 receptors bearing a reporter cysteine in TMD2 ... 33
Figure 4 MTSEA treatment of the N111G-AT1 receptor and sensitive reporter cysteine-bearing mutant N111G-AT1 receptors ... 36
Figure 5 Effect of MTSEA on different mutant N111G-AT1 receptors bearing a reporter cysteine in TMD2 ... 39
Figure 6 [Sar1,Ile8]Ang II protection of MTSEA-sensitive mutant receptors ... 40
Figure 7 Basal levels of inositol phosphates in cells expressing the wild type and mutant AT1 receptors ... 42
Figure 8 Helical wheel representation of TMD2 reporter cysteines and their pattern of reactivity to MTSEA ... 43
ARTICLE 2 Figure 1 Schematic representation of the human AT1 receptor ... 57
Figure 2 MTSEA treatment of the wild-type AT1 receptor and sensitive reporter
cysteine-bearing mutant receptors ... 58
Figure 3 Effects of MTSEA on mutant AT1 receptors bearing a reporter cysteine in TMD5 ... 59
Figure 4 MTSEA treatment of the N111G-AT1 receptor and sensitive reporter cysteine-bearing mutant N111G-AT1 receptors ... 62
Figure 5 Effect of MTSEA on N111G-AT1 mutant receptors bearing a reporter cysteine in TMD5 ... 63
Figure 6 [Sar1,Ile8]Ang II protection of MTSEA-sensitive mutant receptors ... 65
Figure 7 Basal levels of inositol phosphates in cells expressing the wild-type (WT) and mutant AT1 receptors ... 66
Figure 8 Helical wheel representation of TMD5 reporter cysteines and their pattern of reactivity to MTSEA ... 68
ARTICLE 3 Figure 1 Inositol1-phosphate production induced by Ang II analogs………81
Figure 2 βarrestin1 recrutement to the AT1 receptor by Ang II analogs ... 84
Figure 3 βarrestin2 recrutement to the AT1 receptor by Ang II analogs ... 85
Figure 4 G12 activation by Ang II analogs……….87
Figure 5 Ang II induced ERK activation ... 88
Figure 6 EGFR-dependent ERK activation by Ang II analogs ... 90
Figure 7 PKC-dependent ERK activation by Ang II analogs ... 92
Figure 8 Atypical PKC-dependent ERK activation ... 94
LISTE DES TABLEAUX
ARTICLE 1
Tableau 1 Binding Properties of [Sar1,Ile8]Ang II to cysteine-substitued hAT1 mutant
receptors………....29
Tableau 2 Binding Properties of [Sar1,Ile8]Ang II to cysteine-substitued hAT1 mutant receptors bearing the N111G mutation……….35
ARTICLE 2 Tableau 1 Binding Properties of [Sar1,Ile8] Ang II to Cysteine-Substitued hAT1 Mutant Receptors……….55-56 Tableau 2 Binding Properties of [Sar1,Ile8] Ang II to Cysteine-Substitued hAT1 Mutant Receptors Bearing the Asn111Gly Mutation……….60-61 ARTICLE 3 Tableau 1 Binding Properties of AT1 receptor ligands ………...107
Tableau 2 Activation of Gq, βarrestin1, βarrestin2 and G12 by AT1 receptor ligands……….108
Tableau 3 Activation of Gq, PKC-ERK and EGFR-ERK by AT1 receptor ligands ………...109
Tableau 4 Transduction ratios of AT1 receptor ligands………..…………110
Tableau 5 Biais factor of AT1 receptor ligands modified at position 1……….112
Tableau 6 Biais factor of AT1 receptor ligands modified at position 4……….113
LISTE DES ABRÉVIATIONS
AT1AT2
Ang II
Récepteur à l‟angiotensine II type 1 Récepteur à l‟angiotensine II type 2 angiotensine II
Ang III Ang IV
angiotensine III angiotensine IV
AMPc adénosine monophosphate 3', 5'-cyclique ACE enzyme de conversion de l‟angiotensine II
Bpa para-benzoyl-L-phenylalanine
Ca2+ calcium
CAM mutant constitutivement actif Cys cystéine DAG EGF EGFR ERK1/2 GDP GPCR GRK Diacylglycérol
Facteur de croissance épidermique
Récepteur du facteur de croissance épidermique Extracellular signal-regulated kinases
Guanosine diphosphate
Récepteur couplé à une protéine G
Kinase des récepteurs couplés à une protéine G GTP
JAK
JNK3
Guanosine triphosphate Janus kinase
c-Jun N-terminal kinase IP3 inositol 1,4, 5-trisphosphate
IP3R Récepteur à inositol 1,4, 5-trisphosphate
PDGF platelet-derived growth factor
PIP2 phosphatidylinositol 4,5-bisphosphate
PKC protéine kinase C
RAF RAF kinase
IGFR insulin-like growth factor 1 receptor PIP phosphatidylinositol 4,5-bisphosphate
PKC protéine kinase C PLC phospholipase C
MAPK mitogen-activated kinase
MEK MAP kinase kinase
MMP métalloprotéinase
MPA methionine proximity assay MTS méthanethiosulfonate
MTSEA méthanethiosulfonate-ethylammonium MTSES méthanethiosulfonate-éthylsulfonate TM domaine transmembranaire
SCAM substituted cysteine accessibility method Src proto-oncogene tyrosine-protein kinase
INTRODUCTION
Les récepteurs couplés à une protéine G (GPCRs)
Les récepteurs situés à la surface cellulaire permettent de capter un stimulus venant de l‟extérieur afin de le traduire en une réponse appropriée à l‟intérieur de la cellule. Il existe quatre grandes catégories de récepteurs : les récepteurs nucléaires, les récepteurs canaux, les récepteurs à activité enzymatique et les récepteurs couplés à une protéine G. Les récepteurs couplés à une protéine G sont les protéines transmembranaires les plus nombreuses et les plus diversifiées impliquées dans la transduction du signal intracellulaire. Plus de 2% des gènes de notre organisme codent pour des récepteurs couplés à une protéine G (GPCRs).
Les GPCRs sont impliqués dans l‟homéostasie de divers systèmes. Ces récepteurs sont impliqués dans le contrôle d‟une multitude de processus biologiques (vision, métabolisme, neurotransmission, olfaction, réponse immunitaire, réponse inflammatoire) et pathophysiologiques (maladies cardiovasculaires, cancer, diabète). Ils représentent donc des cibles très intéressantes pour le traitement de différentes maladies telles que les maladies cardiaques, certains désordres inflammatoires et autres (Lebon and Tate, 2012). Aujourd‟hui, les GPCRs représentent la cible la plus importante de tous les médicaments sur le marché. Environ 40% des médicaments actuellement disponibles sur le marché ont leurs effets thérapeutiques via un ou des GPCRs (Ma and Zemmel, 2002), d‟où l‟importance de bien comprendre leur structure et leur activation.
Des stimuli de nature très variée peuvent activer les GPCR (photons, ions, amines, acides aminés, peptides, protéines, lipides, nucléotides, glycoprotéines et phospholipides) (Kroeze, et al., 2003).
L‟unité de signalisation de base d‟un GPCR comprend un récepteur, une protéine G hétérotrimérique et un effecteur. Suite à la liaison du ligand, le GPCR subit un changement conformationnel qui permet l‟activation de la protéine G. Les GPCRs doivent leur nom à leur mécanisme de transduction le mieux connu, soit l‟activation de protéines G hétérotrimériques. Les protéines G hétérotrimériques servent d‟intermédiaires dans l‟activation d‟effecteurs à l‟intérieur de la cellule tels que les enzymes, les canaux ioniques et autres. Les protéines G sont composés de trois sous-unités, soit la sous-unité Gα, Gβ et Gγ. Les sous-unités Gβ et Gγ forment un dimère stable. Suite à l‟activation, les GPCRs catalysent l‟échange d‟une molécule de GDP pour une molécule de GTP à l‟intérieur de la sous-unité Gα de la protéine G. Cet
échange cause l‟activation et la dissociation de la sous-unité Gα du dimère Gβγ. Après dissociation, Gα et Gβγ peuvent chacune activer leurs effecteurs respectifs (Pierce, et al., 2002).
On peut classer la protéine Gα en fonction de sa capacité à activer certaines voies de signalisation. Par exemple, la protéine Gαs active l‟adenylate cyclase, et favorise
l‟augmentation de l‟AMP cyclique. À l‟inverse, la protéine Gαi inhibe l‟adénylate cyclase et
cause une baisse de l‟AMP cyclique. La protéine Gαq active la phospholipase C (PLCβ) qui
clive le phosphatidyl inositol 4,5-biphosphate (PIP2) en diacylglycérol et en inositol
1,4,5-triphosphate (IP3). La protéine Gα12∕13, quant à elle, active la Rho kinase impliquée dans la
réorganisation du cytosquelette (Pierce, et al., 2002).
Même si les protéines G sont responsables de la majorité des effets suite à l‟activation des GPCRs, il est bien connu que les GPCRs sont capables d‟activer d‟autres voies de signalisation indépendantes de la protéine G (Kenakin, 2011) telles que le recrutement d‟arrestine, la transactivation du récepteur de l‟EGF, ainsi que l‟activation de certaines kinases (Src).
Système rénine-angiotensine (RAS)
Le récepteur de l‟Ang II (récepteur AT1) est un GPCR et il constitue l‟élément central
du système rénine-angiotensine. Le système rénine-angiotensine joue un rôle clé dans la régulation de la pression artérielle. Suite à une baisse de la concentration de sodium, ou à une baisse du volume sanguin ou encore à une baisse de la pression artérielle, les reins sécrètent la rénine. La rénine est une enzyme qui clive son unique substrat, l‟angiotensinogène, une protéine plasmatique produite par le foie. Le clivage de l‟angiotensinogène produit l‟angiotensine I (Ang I), un décapeptide qui ne possède aucune fonction biologique connue mais qui est converti en Ang II) par l‟enzyme de conversion de l‟angiotensine (ACE). L‟Ang II est la molécule active du système rénine-angiotensine; c‟est l‟agoniste du récepteur AT1
(figure 1). L‟Ang II a une demi-vie très courte et elle est rapidement métabolisée en angiotensine III (Ang III) par l‟aminopeptidase A et cette dernière en angiotensine IV (Ang IV) par l‟aminopeptidase N (Hunyady and Catt, 2006) .
FIGURE 1. Le système rénine-angiotensine. Représentation schématique de la production de
l‟angiotensine II et ses effets physiologiques.
Effets physiologiques de l‟Ang II:
L‟Ang II favorise une élévation de la pression artérielle par une variété d‟actions. Elle stimule la sécrétion d‟aldostérone par le cortex de la glande surrénale qui favorise la réabsorption de sodium (Na+) et de l‟eau par le tubule rénal. L‟Ang II est aussi un puissant vasoconstricteur qui augmente la résistance vasculaire périphérique. De plus, l‟Ang II stimule la croissance et la prolifération cellulaire. Également, l‟Ang II favorise la sécrétion de la vasopressine afin d‟augmenter la réabsorption d‟eau. L‟Ang II agit aussi dans le cerveau, pour augmenter la soif pour favoriser l‟absorption d‟eau, ce qui augmente le volume sanguin et par conséquent augmente la pression artérielle (de Gasparo, et al., 2000; Bader, 2010).
Afin de réduire les effets physiologiques de l‟Ang II, il existe plusieurs classes de médicaments sur le marché. Les inhibiteurs d‟ACE bloquent l‟enzyme de conversion et empêchent la production d‟Ang II, abolissant ses principales fonctions : la vasoconstriction et la libération d‟aldostérone. Ce type de médicament est utilisé pour traiter l‟hypertension artérielle et l‟insuffisance cardiaque et le premier inhibiteur de cette classe a été le captopril. Il existe aussi des antagonistes du récepteur AT qui bloquent son activation par l‟Ang II. Les
antagonistes du récepteur AT1 tel que le losartan agissent en diminuant l‟effet
vasoconstricteur et l‟effet mitogènique de l‟Ang II. Enfin, les inhibiteurs de la rénine empêchent la formation de l‟Ang I à partir de l‟angiotensinogène. L‟aliskiren est un exemple de la nouvelle génération d‟inhibiteurs de la rénine qui sont utilisés en clinique et qui peuvent être administrés par la voie orale (Burnier, 2001; Paulis and Unger, 2010).
Récepteurs de l‟Ang II :
Deux types de récepteurs ont été identifiés pour l‟Ang II : le récepteur AT1 et le
récepteur AT2 (de Gasparo, et al., 2000). Le récepteur AT2 est composé de 363 acides aminés
et possède une homologie de séquence de 34 % avec le récepteur AT1. Il s‟exprime surtout
durant la vie fœtale. Le rôle physiologique du récepteur AT2 est encore relativement obscur
bien que certaines études suggèrent que ce récepteur aurait des actions d‟antagoniste physiologique du récepteur AT1, ayant des effets antiprolifératifs et pro-apoptotiques
(Steckelings, et al., 2005). La majorité des effets physiologiques connus de l‟Ang II passent par le récepteur AT1 (Burnier, 2001; Miura, et al., 2003a).
Structure générale du récepteur AT1 de l‟Ang II
Le récepteur AT1 est présent dans plusieurs organes tels que le foie, la glande surrénale,
le cerveau, les poumons, le rein, le cœur et les vaisseaux sanguins. Composé de 359 acides aminés, le récepteur AT1 fait partie de la classe A des GPCRs, ayant une forte homologie avec
le récepteur de la rhodopsine. Ce récepteur comme tous les autres GPCRs est composé de sept domaines transmembranaires α-hélicaux (TM) ainsi que d‟un domaine N-terminal extracellulaire et un domaine C-terminal intracellulaire. Le domaine extracellulaire de l‟AT1
se caractérise par la présence de trois sites consensus de glycosylation (Asn4, Asn176, Asn188) ainsi que quatre résidus cystéine impliqués dans la formation de deux ponts disulfures essentiels pour la liaison de l‟Ang II (figure 2) (Yamano, et al., 1992; Lanctot, et al., 1999; Lanctot, et al., 2005).
Le domaine intracellulaire quant à lui est composé de 3 boucles intracellulaires et d‟une queue C-terminale. Il contient plusieurs résidus sérine/thréonine pouvant être phosphorylés par les kinases de récepteurs couplés à une protéine G (GRK) afin de recruter l‟arrestine et initier le processus de désensibilisation et d‟internalisation du récepteur (Mehta and Griendling, 2007).
Nomenclature des résidus situés dans TM
Pour faciliter la comparaison des résidus situés dans les domaines transmembranaires (TM) de différentes GPCR de la classe A, plusieurs nomenclatures ont été proposées. Dans cette thèse, j‟ai retenu la nomenclature de Ballesteros et Weinstein (Ballesteros, J.A. 1995). La nomenclature est basée sur le fait que dans chaque TM le résidu le plus hautement conservé est désigné le résidu X.50 où X représente le numéro du TM. Les autres résidus sont numérotés relativement à leur position par rapport au résidu le plus conservés. Par exemple, le résidu le plus hautement conservé dans le 2e TM est un aspartate qu‟on va désigner Asp-74(2.50). Le résidu situé avant l‟aspartate est une alanine qui sera donc identifié comme Ala-73(2.49). Par contre, le résidu situé après l‟aspartate 74 est une leucine et sera désigné en tant que Leu-75(2.51).
FIGURE 2 : Représentation schématique du récepteur AT1. Les numéros indiquent les
positions des résidus dans le récepteur. Les cercles gris représentent les cystéines qui forment les ponts disulfures et les cercles noirs représentent les cystéines qui ne sont pas impliquées dans la formation de ponts disulfures. Les sites de glycosylation potentiels (Asn-4, Asn-176 et Asn-188) sont aussi identifiés. L‟Asn-111 dans le TM3 est aussi indiquée en gris.
Ang II
L‟octapeptide Ang II (Asp1-Arg-Val-Tyr-Ile-His-Pro-Phe8) est le médiateur principal du système rénine-angiotensine (de Gasparo, et al., 2000). Les différentes études effectuées in
vivo et in vitro avec les analogues de l‟Ang II ont permis de déterminer les résidus impliqués
dans la liaison et dans l‟activation du récepteur AT1 (Khosla, et al., 1974; Regoli, et al., 1974).
L‟activité biologique de l‟Ang II dépend en grande partie du résidu aromatique Phe8 et de son carboxylate dans la partie C-terminale du peptide (de Gasparo, et al., 2000). Les chaines latérales aromatiques en position 4 et 6 (Tyr4 et His6) semblent également impliquées dans l‟activation du récepteur (Khosla, et al., 1974). Par exemple, en remplaçant les résidus aromatiques Tyr4 et Phe8 par des résidus aliphatiques, on génère des antagonistes du récepteur AT1. Il faut cependant mentionner que le résidu Tyr-4 cause principalement une
importante baisse d‟affinité pour le récepteur, ce qui le rend moins intéressant du point de vue structure-fonction (Guillemette, et al., 84). Les résidus situés dans la partie N-terminale sont importants pour la liaison au récepteur ainsi que pour la durée d‟action du ligand. L‟Ang III (Ang-2-8) formée par la délétion de l‟aspartate en position 1 est aussi puissante que l‟Ang II. L‟Ang IV (Ang-3-8) garde une faible activité biologique et se comporte comme un agoniste partiel ayant une faible affinité pour le récepteur AT1 (de Gasparo, et al., 2000).
Structure du récepteur AT1 de l‟Ang II
Pochette de liaison du récepteur AT1
Les GPCRs de la classe A possèdent une cavité hydrophile au centre de la protéine appelée la pochette de liaison. La pochette de liaison est délimitée par les sept domaines transmembranaires et sert à accommoder le ligand au sein du récepteur (voir figure 3). Il est connu que dans le cas des GPCRs ayant des ligands plus volumineux comme les peptides (Ang II), le site de liaison se situe en partie dans les domaines transmembranaires et en partie dans les régions extracellulaires (Schwartz, et al., 2006).
Différentes études utilisant des analogues de l‟Ang II en combinaison avec de la mutagénèse dirigée ont permis l‟identification des déterminants moléculaires importants pour la liaison de l‟Ang II. Les acides aminés essentiels à la liaison de l‟Ang II incluent les résidus cystéines qui forment les ponts disulfures ainsi que plusieurs autres résidus situés dans la partie supérieure du récepteur. Par exemple, les résidus chargés situés dans les domaines transmembranaires, tels que la Lys-102(3.26) dans le haut du TM3 ainsi que la Lys-199(5.42)
dans le haut du TM5, participent à la liaison du peptide (Yamano, et al., 1995; de Gasparo, et al., 2000).
Initialement, on a formulé l‟hypothèse selon laquelle le groupement carboxyle en C-terminal de l‟Ang II, de même que les chaines latérales Arg2, Tyr4 et His6 sont importants pour la liaison et l‟activité du peptide (Khosla, et al., 1974; Hsieh and Marshall, 1986). Il a été également démontré que le groupement C-terminal de la Phe8 forme une liaison ionique avec la chaine latérale du résidu Lys-199(5.42) du récepteur AT1 (Monnot, et al., 1996), alors que la
chaine latérale de la Tyr4 du peptide lierait le résidu N111 du récepteur AT1 (Feng, et al.,
1998). De plus, les chaines latérales des résidus His6 et Phe8 de l‟Ang II se retrouvent à proximité des résidus His-256(6.51) et Phe-259(6.54) du récepteur AT1 (Noda, et al., 1995).
Des études de marquage par photoaffinité ont permis d‟identifier de façon plus directe les points de contact entre l‟Ang II et son récepteur. Notamment, l‟Asp1 du ligand entre en contact avec la 2ième boucle extracellulaire du récepteur AT1 (Laporte, et al., 1999), alors que
la Val3 de l‟Ang II est à proximité du résidu Ile-172 dans la 2ième boucle extracellulaire (Boucard, et al., 2000), et que la Phe8 de l‟AngII est à proximité des résidus Phe-293(7.44) et Asn-294(7.45) situé dans le 7ième domaine transmembranaire (Perodin, et al., 2002).
Avec une autre approche de photomarquage par affinité qui exploite la préférence marquée du groupement photosensible para-benzoyl-L-phenylalanine (Bpa) pour le résidu méthionine (Methionine Proximity Assay (MPA)), tous les résidus qui sont à une proximité de 8Å de la position 8 de l‟Ang II et qui sont donc situés dans la pochette de liaison du récepteur AT1 ont été identifiés (Clement, et al., 2005; Clement, et al., 2009; Fillion, et al.,
2009; Fillion, et al., 2010; Fillion, et al., 2013). Parmi ces résidus, on retrouve Phe77(2.53) situé dans 2e domaine transmembranaire, Leu112(3.36), Tyr113(3.37) situés dans 3e domaine transmembranaire, Asn200(5.43) dans 5e domaine transmembranaire, Phe249(6.44), Trp253(6.48), His256(6.51),Thr260(6.55) situés dans 6e domaine transmembranaire, Phe293(7.44), Asn294(7.45), Asn295(7.46), Cys296(7.47) et Leu297(7.48) situés dans 7e domaine transmembranaire (Fillion, et al., 2010).
En ce qui concerne le récepteur AT1, on peut déduire que l‟Ang II est capable de
pénétrer profondément dans la pochette de liaison constituée à la fois des domaines transmembranaires et des domaines extracellulaires.
FIGURE 3 : Pochette de liaison du récepteur AT1 : Arrangement des domaines
transmembranaires autour de la pochette de liaison du récepteur AT1.
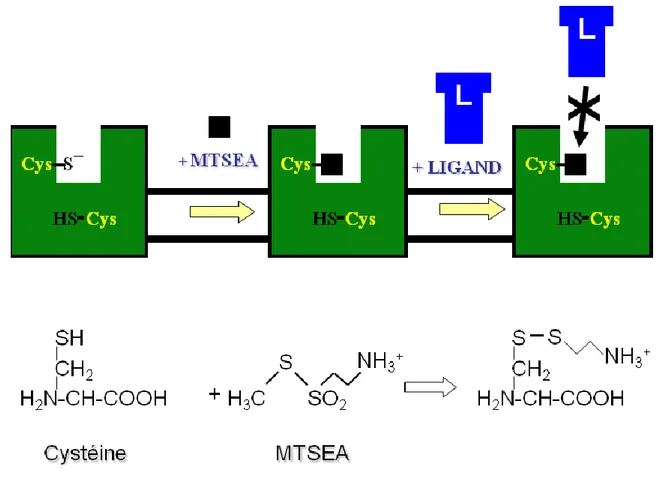
Méthode SCAM : identification de la pochette de liaison du récepteur AT1
L‟acide aminé cystéine qui possède un souffre dans sa chaîne latérale est souvent utilisé pour étudier les relations entre la structure et la fonction d‟une protéine. En effet, la cystéine est capable de former un lien disulfure avec toute autre molécule possédant un groupement thiol libre. Cette propriété peut être exploitée pour modifier les cibles de façon covalente, ce qui a mené au développement des agents capables de réagir spécifiquement avec les cystéines. La cystéine demeure un bon choix pour substituer un résidu natif dans une protéine puisque sa taille est intermédiaire et elle n‟a pas tendance à former des structures secondaires et donc ne change pas la structure globale de la protéine.
Ainsi, Roberts et collaborateurs (Roberts, et al., 1986) ont développé des composés méthanthiosulfonates (MTS), hautement séléctifs au groupement thiol permettant de modifier de façon spécifique les cystéines. Il existe des MTS contenant un groupement éthylammonium (MTSEA) ou éthylsulfonate chargé négativement (MTSES). Ces composés peuvent modifier des protéines (cibles) contenant des cystéines pour y introduire une charge, ainsi qu‟un certain encombrement stérique. L‟approche SCAM (Substituted Cysteine Accessibility Method) consiste à remplacer tous les acides aminés, un à la fois, par une cystéine et ensuite d‟évaluer l‟effet d‟un MTS sur les propriétés de liaison du récepteur mutant.
Dans un premier temps, le SCAM a été utilisé pour caractériser les canaux ioniques (Akabas, et al., 1992). Les résidus constituant le pore du canal forment une pochette
hydrophile par laquelle passent les ions et ainsi la modification de ces résidus par un composé MTS bloque l‟accès aux ions, modifiant ainsi la fonctionnalité du canal. En absence de modification des propriétés fonctionnelles, il y a deux explications possibles : soit la cystéine introduite n‟a pas pu réagir avec le réactif MTS, soit la modification de la cystéine ne cause pas de changement détectable de la fonctionnalité de la protéine.
Utilisation du SCAM afin de délimiter la pochette de liaison des GPCRs
Afin de mieux comprendre les propriétés moléculaires du récepteur AT1, nous avons
utilisé l‟approche SCAM (Akabas, et al., 1992; Javitch, et al., 1994; Javitch, et al., 1995) qui permet d‟identifier les acides aminés faisant partie de la pochette de liaison du récepteur en plus de déterminer l‟orientation des domaines transmembranaires lors de l‟activation du récepteur. Cette approche est basée sur la réactivité de dérivés des méthanethiosulfonates (MTS) ayant une spécificité pour les groupements sulfhydryls des cystéines (Cys). Parmi les différents MTS disponibles sur le marché, le MTSEA (2-aminoethyl méthanethiosulfonates) est un réactif chargé et hydrophile. Il réagit un milliard de fois plus rapidement avec les thiols ionisés (RS¯) qu‟avec les thiols non-ionisés (RSH). Ainsi, seulement les cystéines présentes dans le compartiment aqueux sont ionisées et peuvent réagir avec le MTSEA. La méthode de SCAM consiste à vérifier l‟accessibilité du ligand dans la pochette de liaison suite à la réaction entre le réactif MTS et les cystéines introduites de façon systématique par mutagénèse dirigée dans les domaines transmembranaires.
La première étape consiste donc à remplacer de façon successive un par un chacun des résidus dans un domaine transmembranaire par une cystéine. Ces récepteurs mutants sont par la suite traités avec le réactif MTSEA et leur propriété de liaison est évaluée. La cystéine située dans la pochette de liaison sera alkylée par le MTSEA, causant ainsi l‟encombrement stérique qui interférera avec la liaison du ligand (figure 4). Par contre, si l‟alkylation n‟affecte pas les propriétés de liaison du récepteur mutant, on déduit que la cystéine substituante n‟est pas située dans la pochette de liaison.
FIGURE 4 : Substituted Cysteine Accessibility Method (SCAM). Le MTSEA réagit
spécifiquement avec les cystéines libres. Il réagit un milliard de fois plus rapidement avec les thiols ionisés (RS-) qu‟avec les thiols non-ionisés (RSH). Ainsi, seulement les cystéines dans le compartiment aqueux sont ionisées et réagiront avec le MTSEA. La cystéine située dans la pochette de liaison sera alkylée par le MTSEA, causant ainsi un encombrement stérique qui interférera avec la liaison du ligand.
État basal versus état actif du récepteur
Les GPCRs sont des protéines dynamiques capables de transition entre de multiples conformations (Lefkowitz, 2007). Ces diverses conformations sont en équilibre dynamique avec les effecteurs intracellulaires et les ligands extracellulaires. Le modèle ternaire cubique a été formulé pour décrire l‟interaction dynamique entre le récepteur, son ligand et sa protéine G effectrice (Schwartz, et al., 2006).
De façon générale, la forme basale d‟un GPCR est la plus énergétiquement favorable en absence de ligand. La liaison d‟un agoniste complet stabilise une conformation active du récepteur, (augmente la population des récepteurs de conformation active aux dépens de la population étant dans la conformation inactive) ce qui augmente l‟affinité du récepteur pour
une protéine effectrice intracellulaire et mène à une signalisation maximale. Pour leur part, les agonistes partiels stabilisent une conformation active du récepteur de façon moins efficace que les agonistes complets, sans toutefois produire une réponse maximale. Les antagonistes neutres occupent le site de liaison du récepteur, mais n‟ont aucune influence sur l‟adoption des diverses conformations du récepteur. En fait, les antagonistes ne font que bloquer l‟accès de l‟agoniste au récepteur. Les agonistes inverses stabilisent la conformation inactive du récepteur. La notion de l‟agoniste inverse a été proposée récemment, suite à la découverte de l‟activité constitutive de certains récepteurs, qui peuvent donc adopter une conformation active en absence d‟agoniste (Seifert and Wenzel-Seifert, 2002).
En absence du ligand, la majorité des GPCRs possèdent un faible niveau d‟activité constitutive (Seifert and Wenzel-Seifert, 2002) (une sous-population des récepteurs ayant assez d‟énergie pour être dans leur conformation active). Certaines mutations peuvent déstabiliser la forme inactive d‟un GPCR et favoriser les conformations plus actives : ce sont des mutants constitutivement actifs (CAM). Les CAM favorisent le déplacement de l‟état inactif vers l‟état actif des GPCRs et par ce fait, offrent un précieux outil dans l‟identification des résidus impliqués dans l‟activation des GPCRs. En plus des résidus identifiés à l‟aide des mutants constitutivement actifs (CAM), on croit que les résidus hautement conservés dans les GPCRs jouent un rôle essentiel dans le mécanisme d‟activation (Gether and Kobilka, 1998).
Dans les études visant à identifier et caractériser les mouvements des domaines transmembranaires qui accompagnent l‟activation d‟un GPCR, il est nécessaire de discriminer entre les conformations basales et actives du récepteur. Dans le cas du récepteur AT1,
l‟utilisation du mutant constitutivement actif s‟avère donc très appropriée pour représenter la conformation active du récepteur, alors que l‟utilisation du récepteur de type sauvage est un bon modèle de la conformation basale.
Activation constitutive du récepteur AT1
La découverte d‟une activité constitutive chez certains GPCRs qui peuvent activer leur protéine G en absence d‟agoniste fut une avancée importante. Il est connu que certaines mutations favorisent l‟augmentation de l‟activité constitutive.
Le récepteur AT1 fait partie d‟un large groupe de GPCRs démontrant une certaine
activité constitutive. Une légère activité constitutive du récepteur AT1 a été observée lors de
sa surexpression dans les cellules COS-1, causant ainsi une augmentation de l‟activité basale de la PLCβ (Noda, et al., 1996). Ensuite, d‟autres études indépendantes ont elles aussi
résidu Asn111(3.35) situé dans le 3e domaine transmembranaire est remplacé par des résidus moins encombrants tels que l‟alanine ou la glycine (Noda, et al., 1996; Balmforth, et al., 1997; Groblewski, et al., 1997).
Il a été suggéré que les GPCRs dans leur état basal sont contraints dans une conformation inactive favorisé par certaines interactions intramoléculaires. Suite à la liaison d‟un agoniste ou sous la présence d‟une mutation spécifique, cette contrainte est libérée ce qui favorise la conformation active d‟un récepteur. Il est bien connu que le résidu Asn-111 est hautement mais non strictement conservé dans la classe A des GPCRs.
Activation des récepteurs couplés à une protéine G (GPCRs) Mécanisme moléculaires impliqués dans l‟activation des GPCRs
Afin de propager un signal vers l‟intérieur de la cellule, un GPCR doit adopter une conformation active. Dans le modèle le plus simple de l‟activation des GPCRs, un ligand agoniste complet stabilise une conformation active du récepteur, ce qui augmente l‟affinité de ce dernier pour un ou des effecteurs intracellulaires et mène à une signalisation maximale.
Les GPCRs possèdent tous une structure commune dictée par l‟arrangement des sept domaines transmembranaires dans la membrane plasmique (Palczewski, et al., 2000; Park, et al., 2008; Rasmussen, et al., 2007; Rasmussen, et al., 2011). Plusieurs résidus et motifs (D/ERY, NPxxY) conservés pour les GPCRs de la classe A ont été impliqués dans leur processus d‟activation (Katritch, et al., 2013; Rosenbaum, et al., 2009).
L‟ensemble des informations disponibles suggère que tous les GPCRs pourraient partager un mécanisme d‟activation commun (Rosenbaum, et al., 2009). En dépit de toutes les informations disponibles, il reste encore beaucoup de travail à effectuer afin de déterminer les différentes conformations qu‟adoptent les GPCRs au cours de leur transition de l‟état inactivé à l‟état activé.
Mécanisme d‟activation du récepteur AT1 de l‟Ang II
Chez plusieurs GPCRs, une étape clé de l‟activation est un éloignement entre le TM3 et le TM6 du côté cytoplasmique du récepteur (Gether, et al., 1997; Ballesteros, et al., 2001). Si on bloque ce mouvement, on empêche l‟activation de la protéine G. Dans le cas du récepteur AT1, il y a des évidences qui montrent qu‟une interaction forte entre le résidu Tyr-113(3.37)
situé dans le 3e domaine transmembranaire et His-256(6.51) situé dans le 6e domaine transmembranaire favorise l‟état inactif du récepteur et empêche l‟activation de la protéine Gq
(production des inositols phosphates) (Miura, et al., 2006). Il a été démontré qu‟un mouvement vers l‟extérieur du côté cytoplasmique du 6e domaine transmembranaire favorise la flexibilité de la 3e boucle intracellulaire, ce qui favorise l‟adoption d‟une conformation active, présentant un lieu d‟ancrage pour la protéine G (Martin, et al., 2007; Conchon, et al., 1997).
Il existe différents motifs impliqués dans l‟activation des GPCRs. Un de ces motifs, le motif D/ERY est celui qui a été le plus étudié. Les trois résidus composant ce motif sont une Asp ou un Glu en position 3.49, une arginine en position 3.50 et une tyrosine en position 3.51. Ce motif est situé sur le côté cytoplasmique du 3e domaine transmembranaire, à l‟interface de la 2e boucle intracellulaire. Plusieurs études montrent que ce motif joue un rôle central dans la régulation de l‟état conformationnel des GPCRs. L‟arginine(3.50) est impliquée dans une interaction ionique avec l‟aspartate (3.49) adjacente ainsi qu‟avec un résidu chargé situé en position 6.30 dans le TM6 afin de former ce qu‟on appelle la serrure ionique (« ionic lock »). L‟interaction intramoléculaire entre ces 3 résidus serait importante pour le maintien du récepteur dans sa forme inactive et cette interaction doit être brisée lors de l‟activation. Les structures cristallines de la rhodopsine (Palczewski, et al., 2000) ainsi que du récepteur β2
-adrénergique (Yao, et al., 2006) semblent appuyer cette hypothèse. Dans le cas du récepteur AT1, il n‟y a pas de résidu chargé négativement à la position 6.30 mais le motif DRY semble
jouer un rôle dans l‟activation de la protéine G et dans le recrutement de l‟arrestine. Par exemple, en substituant l‟aspartate (3.49) par des résidus plus petits (alanine ou glycine), on empêche l‟activation de la protéine G mais on permet encore une signalisation indépendante de la protéine G (Wei, et al., 2003; Seta, et al., 2002; Gaborik, et al., 2003).
Des études suggèrent que lors de l‟activation du récepteur de l‟urotensine, les résidus du motif DRY sont impliqués dans une interaction directe avec la protéine G. En effet, une mutation à l‟intérieur du motif DRY conduit souvent à une forme inactivable du récepteur par rapport à la protéine G (Proulx, et al., 2008).
Un autre motif impliqué dans l‟activation des GPCRs est le motif NPxxY situé dans le 7e domaine transmembranaire. Il a été suggéré que ce motif joue le rôle d‟un interrupteur moléculaire qui serait important dans la transition du récepteur de la conformation basale vers la conformation active. Selon cette hypothèse, le résidu Asn(7.49) est orienté vers le 6e domaine transmembranaire dans la conformation inactive du récepteur et suite à l‟activation, ce résidu se réoriente de façon à interagir avec l‟Asp-74(2.50) du 2e domaine transmembranaire. Plusieurs études montrent l‟importance de l‟Asp-74 (2.50) (Bihoreau, et al., 1993) ainsi que de plusieurs autres résidus situés dans le 7e domaine transmembranaire (Tyr-292(7.43),
Asn-294(7.45), Asn-298(7.49), Tyr-302(7.52)) (Perodin, et al., 2002; Clement, et al., 2005; Balmforth, et al., 1997; Miura, et al., 2003b; Laporte, et al., 1996) pour l‟activation du récepteur AT1.
L‟activation du récepteur AT1 dans cette section fait référence à l‟activation du
récepteur via la protéine G mais récemment il a été mis en évidence que le récepteur AT1 est
également capable d‟activer des voies de signalisation indépendantes de la protéine G, d‟où l‟importance du concept de sélectivité fonctionnelle.
Sélectivité fonctionnelle
Concept de sélectivité fonctionnelle
L‟interaction d‟un ligand avec un récepteur est l‟un des fondements de la pharmacologie. Cette interaction (ligand-récepteur) est à l‟origine des principaux paramètres pharmacologiques que sont la capacité du ligand à lier le récepteur (affinité) et la capacité à produire la réponse biologique (efficacité). Jusqu‟à tout récemment, on évaluait l‟efficacité d‟un récepteur par sa capacité à activer une seule voie de signalisation. Il est maintenant bien connu que le récepteur AT1 active plusieurs voies de signalisation en interagissant avec
différents effecteurs responsables de la variété d‟actions de l‟Ang II. Il est donc logique de penser que divers ligands peuvent induire une variété de conformations d‟un GPCR et ainsi inhiber ou activer plus ou moins sélectivement les diverses voies de signalisation en aval du récepteur. C‟est le concept de sélectivité fonctionnelle (Galandrin, et al., 2007)(Kenakin, 2011; Violin and Lefkowitz, 2007; Rajagopal, et al., 2010) (Figure 5). Par exemple, une certaine conformation du récepteur peut coupler plus efficacement à une voie de signalisation X et moins bien à la voie Y. À l‟inverse, une autre conformation adoptée par le récepteur activera plus efficacement la voie Y et moins bien la voie X.
FIGURE 5 : Sélectivité fonctionnelle. Représentation schématique de divers ligands pouvant
induire diverses conformations d‟un même récepteur et pouvant activer ainsi diverses voies de signalisation engendrant différentes conséquences fonctionnelles.
Pourquoi faire des études de sélectivité fonctionnelle?
L‟importance de la sélectivité fonctionnelle est qu‟elle permet de concevoir des ligands plus sélectifs qui favoriseront un effet voulu au détriment des effets indésirables. La sélectivité fonctionnelle trouve son importance dans le développement des nouveaux médicaments que l‟on veut plus sélectifs. Comment y parvenir? En testant plusieurs ligands ayant une structure chimique différente, il sera possible de déterminer les voies d‟activation privilégiées par ces derniers. Dans un premier temps, à partir de ces données, il sera possible de tirer les conclusions sur l‟influence de la structure chimique des ligands sur la voie d‟activation privilégiée par le récepteur AT1. Par la suite, en faisant du modelage moléculaire,
il sera possible de déduire la conformation du récepteur efficace à activer l‟une ou l‟autre voie de signalisation. Finalement, l‟étape ultime sera de designer des super ligands agonistes ou antagonistes pour chacune des voies de signalisation.
La signalisation du récepteur AT1
Voies de signalisation impliquées dans l‟activité du récepteur AT1
La voie de signalisation classique du récepteur AT1 passe par la protéine Gq qui active
la phospholipase Cβ (PLCβ). La PLCβ hydrolyse le phosphatidyl inositol 4,5-bisphosphate (PIP2) menant à la production de deux seconds messagers : l‟inositol 1,4,5-trisphosphate (IP3)
amplifient d‟autres cascades de signalisation. L‟IP3 active un récepteur/canal, l‟IP3R, dans le
réticulum endoplasmique et provoque une relâche du Ca2+ intracellulaire (Ferris, et al., 1992). Le DAG active la protéine kinase C (PKC), une sérine/thréonine kinase qui phosphoryle des substrats dans la cellule et ainsi module leur activité (Kanashiro and Khalil, 1998). Les connaissances actuelles suggèrent que le récepteur AT1 interagit aussi avec d‟autres protéines
G hétérotrimériques telles que la protéine Gi responsable de l‟inhibition de l‟adenylyl cyclase
et la protéine G12/13 impliquée dans le remodelage du cytosquelette (Hunyady and Catt,
2006b; Mehta and Griendling, 2007).
L‟effet hypertrophique de l‟Ang II met en jeu l‟activation de plusieurs protéines kinases. L‟Ang II régule l‟activité des kinases de la famille JAK (Ali, et al., 1997; Ali, et al., 1998; Mascareno and Siddiqui, 2000) et de la famille Src (Seta, et al., 2002; Sayeski, et al., 1998; Sayeski and Ali, 2003). Les kinases de la famille JAK stimulent la phosphorylation et l‟activité de facteurs de transcription tels que les STATs (Signal Transducers and Activators of Transcription) (Liang, et al., 1999; McWhinney, et al., 1998). Les kinases de la famille Src (cSrc) peuvent phosphoryler de nombreuses protéines et mener à l‟activation de MAP kinases (mitogen activated protein) telles que les ERK1/2 (extracellular signal-regulated kinases) (Seta, et al., 2002; Kim, et al., 2009). L‟Ang II est aussi capable de réguler l‟activité de certains récepteurs de facteurs de croissance (PDGF, EGFR, IGFR) (Mehta and Griendling, 2007; Heeneman, et al., 2000; Shah and Catt, 2003; Folli, et al., 1997).
Le récepteur AT1 serait également capable d‟activer des voies n‟impliquant pas les
protéines G (figure 6). Par exemple, le récepteur AT1 peut lier l‟arrestine, une protéine
d‟échafaudage qui favorise la désensibilisation et l‟internalisation du récepteur via les puits tapissés de clathrine (Gaborik, et al., 2001; Hunyady, et al., 2000). En inhibant une voie de signalisation, l‟arrestine peut également en activer d‟autres. Par exemple, l‟arrestine permet la formation des complexes pouvant activer la voie des MAP kinases (JNK3 et ERK1/2) (Wei, et al., 2003; McDonald, et al., 2000; Wei, et al., 2004). En plus d‟activer les MAP kinases via les arrestines (Wei, et al., 2003), il est aussi connu que le récepteur AT1 peut activer les MAP
kinases via la protéine kinase C (Tian, et al., 1998) et via la transactivation du récepteur à l‟EGF (epidermal growth factor) (Shah, et al., 2004).
FIGURE 6 : Les différentes voies de signalisation du récepteur AT1 : Récepteur AT1 de l‟Ang
II peut activer plus d‟une voie de signalisation en interagissant avec différents effecteurs. Récepteur AT1 peut activer les voies de signalisation dépendante ou indépendante de la
protéine G.
Activation de la voie de MAPK/ERK par le récepteur AT1
Les voies de signalisation impliquant les MAP kinases contrôlent des processus cellulaires importants tels que la croissance, la différenciation et la prolifération cellulaire. Ce sont des protéines kinases qui catalysent la phosphorylation d‟autres protéines (enzymes, facteurs de transcription) afin de les activer. L‟activation des MAP kinases se fait en cascade : la protéine sérine/thréonine MAPK kinase kinase (Raf) phosphoryle MAPK kinase (MEK1/2) et cette cascade de phosphorylation se poursuit par l‟activation des MAPK (ERK1/2) (Rozengurt, 2007).
Comme déjà mentionné, le récepteur AT1 peut activer les ERK1/2, une des voies des
MAP kinases, de trois façons différentes : par une voie dépendante de la protéine Gq et
L‟activation des ERK1/2 passant par la voie de la protéine Gq met en jeu la PKC. Cette
dernière est responsable de la phosphorylation de cibles en amont de la cascade de signalisation des ERK1/2. La PKC active une petite protéine G, la protéine Ras, qui interagit avec la kinase Raf initiant l‟activation de la cascade de signalisation impliquant Raf, Mek1/2 et ERK1/2 (Tian, et al., 1998).
L‟activation de la voie des ERK1/2 n‟impliquant pas la protéine G passe par les arrestines. Les arrestines favorisent le recrutement des complexes protéiques menant vers l‟activation de MAP kinases (Wei, et al., 2003; Wei, et al., 2004).
L‟Ang II peut aussi activer les ERK1/2 via la transactivation du récepteur de l‟EGFR. La transactivation de l‟EGFR est initiée par une augmentation du Ca2+ intracellulaire, l‟activation de la PKC et de la kinase Src. Src active une métalloprotéinase (MMP) relâchant le ligand EGF (epidermal growth factor) lié à l‟héparine (HB-EGF). La liaison du ligand EGF entraîne un changement conformationnel de l‟EGFR et sa dimérisation. La dimérisation favorise une autophosphorylation au niveau des résidus tyrosine dans la queue cytoplasmique du récepteur. Cette phosphorylation crée des sites d‟ancrage pour d‟autres protéines adaptatrices possédant les domaines SH2, et recrute ensuite le complexe Shc-Grb2-Sos. Ceci permet l‟activation de la petite protéine G Ras menant à l‟activation des MAP kinases impliquant Raf, Mek1/2 et ERK1/2 (Hunyady and Catt, 2006b; Mehta and Griendling, 2007). La transactivation de l‟EGFR par le récepteur AT1 a été démontrée dans plusieurs tissus tels
que le foie, le rein, la prostate, le cœur et les cellules musculaires lisses vasculaires (Thomas, et al., 2004).
Des études récentes montrent que l‟activation de ERK1/2 par la voie dépendante de la protéine G (PKC) possède un profil cinétique très différent de l‟activation de ERK1/2 par la voie indépendante de la protéine G (les arrestines) (Ahn, et al., 2004). L‟activation d‟ERK1/2 par la PKC est un phénomène rapide et transitoire favorisant la translocation des ERKs dans le noyau et la transcription du gène c-Fos. Par contre l‟activation des ERKs par les arrestines est un phénomène beaucoup plus lent et plus soutenu dans le temps et ERK activé reste dans les compartiments cytosoliques (Wei, et al., 2003; Wei, et al., 2003; Ahn, et al., 2004; Ahn, et al., 2004; Aplin, et al., 2007; Tohgo, et al., 2002).
Les conséquences moléculaires semblent aussi différer en fonction de la voie d‟activation de ERK1/2. L‟activation des ERKs par les arrestines estassociée aux phénotypes cardioprotecteurs tels que la survie cellulaire (Ahn, et al., 2009b), la régulation de la contraction cellulaire (Rajagopal, et al., 2006) et la migration (Hunton, et al., 2005). Dans les études faites au niveau du cœur et de cardiomyocytes isolés, l‟activation de la voie des
arrestines favorise la croissance et l‟hypertrophie (Zhai, et al., 2005), la prolifération des cardiomyocytes (Aplin, et al., 2007; Hansen, et al., 2008), et une augmentation au niveau de leur contractilité (Rajagopal, et al., 2006; Violin, et al., 2010). Dans les cellules musculaires lisses vasculaires, la signalisation via les arrestines augmente la synthèse protéique en interagissant avec la MAP kinase interacting kinase 1 (Mnk-1) (DeWire, et al., 2008) et protège contre l‟apoptose par la phosphorylation de BAD (Ahn, et al., 2009)
Méthodes utilisés pour mesurer l‟activation de différentes voies de signalisation
Production des inositols phosphates
Nous avons mesuré l‟activation de la voie Gq/PLC/IP3/Ca2 par un immunoessai
compétitif IPone HTRF (homogeneous time resolved fluorescence) développé par la compagnie Cisbio.Cet immunoessai mesure l‟accumulation de l‟IP1 en présence de LiCl qui
bloque la conversion de l‟IP1 en myoinositol. Le signal spécifique est généré entre l‟anticorps
dirigé contre l‟IP1 et l‟IP1 synthétique couplé à l‟accepteur fluorophore. Suite à la stimulation
du récepteur, l‟IP1 provenant de la dégradation de l‟IP3 rentre en compétition avec l‟IP1
synthétique pour la liaison à l‟anticorps ce qui produit une baisse du signal de fluorescence. Donc, le transfert de l‟énergie est inversement proportionnel à la concentration de l‟IP1
produit suite à la stimulation du récepteur dans le lysat cellulaire.
Activation de la voie des ERKs
Afin de vérifier l‟état de l‟activation des ERK1/ 2, nous avons utilisé l‟essai Alpha Screen Sure Fire (Amplified Luminescent Proximity Homogenous Assay). Dans cet essai, un premier anticorps, reconnaissant la forme phosphorylée de ERK, est capturé par une bille enrobée de protéine A (bille accepteuse). Un deuxième anticorps, reconnaissant ERK, est biotinylé et capturé par une bille enrobée de streptavidine (bille donneuse). Les deux billes sont à proximité seulement s‟il y a présence de ERK phosphorylé. Lorsque les deux billes sont à proximité, l‟excitation de la bille donneuse par AlphaScreen libère le radical libre O2
qui excite la bille accepteuse, et cette dernière émet alors de la lumière. L‟activation de ERK1/2 est proportionnelle à la quantité de la lumière émise par la bille accepteuse.
Recrutement de l‟arrestine et l‟activation de la voie G12
Afin de vérifier l‟activation de la voie G12 et le recrutement de l‟arrestine (β1-arrestine
et β2-arrestine), nous avons utilisé l‟essai BRET. L‟essai BRET est fondé sur le transfert
d‟énergie entre un donneur bioluminescent et un accepteur fluorescent. Pour qu‟il y ait un transfert d‟énergie, le donneur et l‟accepteur doivent être situé à la proximité (10-100Å). La méthode BRET utilise une luciférase RLuc dont le substrat est la coelenterazine. En présence de l‟oxygène, la luciférase favorise la transformation du substrat coelenterazine en coelenteramide en émettant une légère lumière visible dans le bleu (395nm). Lorsqu‟un possible accepteur est situé à proximité de cette source lumineuse, la lumière bleu est captée par l‟accepteur, dans ce cas la protéine GFP10. L‟excitation de cette dernière (GFP10) entraîne l‟émission d‟une lumière verte à 510nm. L‟efficacité du transfert d‟énergie du complexe Rluc à la GFP10 est déterminée par le rapport entre l‟intensité d‟émission de l‟accepteur (lumière verte) sur l‟intensité d‟émission du donneur (lumière bleu). Pour le recrutement des arrestines, nous avons utilisé la construction RLuc fusionné avec les arrestines (β1 ou β2 arrestine) et la protéine GFP10 fusionnée avec le récepteur AT1. Pour
l‟activation de la voie G12, nous avons utilisé la construction de RLuc fusionnée avec la
protéine G12 et la protéine GFP10 fusionnée avec la sous-unité gamma de la protéine G
(Ggamma).
Calcul du biais de signalisation
Dans l‟objectif d‟identifier efficacement la signalisation biaisée de différents analogues de l‟Ang II et de faciliter la compréhension des données, nous avons utilisé la méthode proposée par le groupe de Kenakin nous permettant de calculer le biais de signalisation (Kenakin, et al., 2012). Les ratios de transduction log(τ/Ka) ont été calculé en utilisant le
logiciel GraphPad Prism. Le ratio de transduction représente une estimation de l‟effet (puissance et efficacité) d‟un analogue sur la conformation du récepteur et par la suite sur l‟interaction du complexe ligand-récepteur avec les effecteurs intracellulaires. Afin d‟évaluer le biais à l‟intérieur d‟une voie de signalisation donnée, il faut comparer le comportement d‟un ligand avec le ligand de référence. L‟Ang II a été capable de produire une réponse maximale ayant des puissances semblables pour toutes les voies de signalisation que nous avons testées, et nous l‟avons choisi pour être notre ligand de référence. En comparant le log(τ/Ka) du ligand de référence (Ang II) à la valeur du log(τ/Ka) de chaque analogue testé pour une voie de signalisation, nous avons pu établir le biais à l‟intérieur d‟une voie de
signalisation donnée, soit le Δlog(τ/Ka). Afin d‟évaluer le biais entre les différentes voies de signalisation pour chaque analogue, nous avons calculé le ΔΔlog(τ/Ka) et le facteur biaisé. ΔΔ(τ/Ka) a été calculé en comparant la valeur du Δlog(τ/Ka) pour une voie de signalisation versus la valeur du Δlog(τ/Ka) d‟une autre voie de signalisation. Le facteur biaisé représente la base de 10 de la valeur calculée du ΔΔlog(τ/Ka).
Importance de la sélectivité fonctionnelle du récepteur AT1
Le récepteur AT1 fut un des premiers récepteurs pour lequel l‟importance de la
signalisation biaisée a été démontrée. Le récepteur AT1 joue un rôle important dans la
régulation de la pression sanguine et une suractivité peut mener à plusieurs pathologies telles que l‟hypertension, la dysfonction rénale ainsi que l‟insuffisance cardiaque.
Les études de mutagénèse sur le récepteur AT1 montrent qu‟en bloquant sa capacité à
activer la protéine Gq, on n‟affecte pas sa phosphorylation, son internalisation, son
recrutement ainsi que sa signalisation via l‟arrestine (Wei, et al., 2003; Gaborik, et al., 2003). Il a été démontré que l‟activation de la signalisation β-arrestine via le récepteur AT1 favorise
les effets cardioprotecteur et anti-apoptotique (Ahn, et al., 2009b; Rakesh, et al., 2010). Suite à une délétion du récepteur AT1 ou des arrestines, le stress mécanique au niveau du cœur
induit l‟apoptose, suggérant que la signalisation via les β-arrestines par le récepteur AT1 est
cardioprotectrice (Rakesh, et al., 2010).
Les propriétés de l‟analogue [Sar1,Ile4,Ile8]Ang II (SII) qui active la voie des ERKs en absence de production d‟inositol phosphate l‟ont fait reconnaître comme l‟un des premiers ligands biaisés (Holloway, et al., 2002). D‟autres travaux montrent que SII améliore la contractilité des cardiomyocytes chez la souris via la signalisation dépendante de la β-arrestine et ceci en absence de la signalisation dépendante de la protéine Gq (Rajagopal, et al.,
2006). D‟autres expériences faites avec le SII montrent qu‟il cause une diminution de l‟apoptose, qu‟il stimule la chimiotaxie, qu‟il augmente la contractilité cardiaque, la croissance et la prolifération cellulaire (Aplin, et al., 2007; Ahn, et al., 2009b; Rajagopal, et al., 2006; Hunton, et al., 2005; DeWire, et al., 2008). Il a également été suggéré que le ligand biaisé SIIet l‟Ang II stabilisent différentes conformations actives du récepteur AT1 (Sauliere,
et al., 2012).
Sadoshiwa et al ont généré des souris transgéniques qui surexpriment le récepteur AT1
de type sauvage et un mutant du récepteur AT1 incapable de coupler à la protéine Gq. Bien
stimulation chronique par l‟Ang II, il y a eu moins d‟apoptose et de fibrose au niveau du myocarde des souris exprimant le récepteur AT1 incapable de coupler à la protéine Gq (Zhai,
et al., 2005). Ceci porte à croire que ces deux aspects (fibrose et apoptose) impliqués dans la progression de la maladie sont potentiellement reliés à l‟activité de la voie de signalisation dépendante de la protéine Gq.
Les antagonistes du récepteur AT1 tel le losartan, utilisé couramment en clinique pour
traiter l‟hypertension, bloquent toutes les voies de signalisation en aval du récepteur. Par contre, il serait intéressant de développer une nouvelle génération d‟antagonistes capables de bloquer sélectivement une voie de signalisation et n‟influençant pas l‟activation des autres voies de signalisation. Par ce fait même, on pourrait espérer développer des médicaments pouvant contrôler sélectivement certains aspects de la fonction du récepteur où le contrôle de la pression artérielle serait influencé indépendamment de la croissance pathologique des cardiomyocytes ou des cellules musculaires lisses vasculaires (VSMC).
Problématique
Une connaissance approfondie de la structure moléculaire du récepteur, en particulier de la topologie de la pochette de liaison ainsi que des changements structuraux associés à l‟activation du récepteur est requise pour le développement de nouveaux outils thérapeutiques. L‟élucidation de la pochette de liaison d‟un GPCR fournit des informations importantes sur les mécanismes de reconnaissance moléculaire entre une hormone et son récepteur. L‟interaction spécifique entre l‟hormone et son récepteur se fait au niveau de la pochette de liaison. Les sept TM des GPCRs contribuent à former la pochette de liaison du ligand. Il existe différentes approches biochimiques pour étudier la structure moléculaire d‟un GPCR. Nous proposons d‟utiliser l‟approche SCAM (Substituted Cysteine Accessibility Method) qui nous permettra de caractériser la pochette de liaison en identifiant les résidus de chaque domaine transmembranaire impliqués dans sa formation. De plus, en utilisant le mutant constitutivement actif N111G-AT1, l‟approche SCAM nous aidera à déterminer la
position relative de chaque domaine transmembranaire suite à l‟activation du récepteur. Ces résultats seront utiles pour élaborer un modèle moléculaire de la pochette de liaison du récepteur AT1 de l‟Ang II. Afin d‟améliorer ce modèle, nous allons étudier les différents
changements conformationels se produisant lors de l‟activation du récepteur. Au cours des dernières années, il est devenu de plus en plus évident que le récepteur AT1 peut activer plus
d‟une voie de signalisation en interagissant avec différents effecteurs intracellulaires. Le concept de sélectivité fonctionnelle suggère que différents ligands peuvent induire différentes conformations du récepteur et ainsi favoriser l‟activation ou l‟inhibition de certaines voies de signalisation. Nous proposons donc de tester les propriétés fonctionnelles d‟une série d‟analogues de l‟Ang II possédant des structures chimiques différentes et possiblement des propriétés pharmacologiques distinctes. Ces ligands nous donneront l‟information par rapport à la sélectivité fonctionnelle en identifiant les voies de signalisation préférentiellement activées ou inhibées par ces derniers. Les données obtenues serviront à améliorer notre modèle moléculaire du récepteur AT1.
Objectifs
1ère et 2ième publication
1. Déterminer la pochette de liaison du récepteur AT1 avec l‟approche de SCAM
2. Identifier les résidus du TM2 et du TM5 qui participent dans la formation de la pochette de liaison du récepteur AT1 et de son mutant constitutivement actif AT1-N111G
3. Déterminer le type de mouvement qui se produit lorsque le récepteur passe de la forme inactive vers la forme active
3ième publication
1.Étudier la sélectivité fonctionnelle de différents ligands analogues de l‟Ang II
2. Évaluer la puissance (EC50) et l‟efficacité (Emax) des différents analogues de l‟Ang II à
activer sélectivement une voie de signalisation. 1. Gq/PLC
2. G12
3. Le recrutement des arrestines
ARTICLE 1
The second transmembrane domain of the human type 1 angiotensin II receptor participates in the formation of the ligand binding pocket and undergoes integral pivoting movement during the process of receptor activation
Ivana Domazet, Brian J. Holleran, Stéphane S. Martin, Pierre Lavigne, Richard Leduc, Emanuel Escher and Gaétan Guillemette.
J. Biol. Chem, 284: 11922-11929 (2009).
Statut de l’article: publié dans Journal of Biological Chemistry .
Référence : Domazet I., Holleran B.J., Martin S.S, Lavigne P., Leduc R., Escher E. &
Guillemette G. (2009). The second transmembrane domain of the human type 1 angiotensin II receptor participates in the formation of the ligand binding pocket and undergoes integral pivoting movement during the process of receptor activation. J. Biol. Chem. 284 (18): 11922-9.
Avant-propos: J‟ai dirigé cette étude et produit toutes les manipulations de ce manuscrit. J‟ai
Résumé : L‟angiotensine II, une hormone jouant un rôle important dans l‟homéostasie cardiovasculaire, produit la majorité de ses effets en activant le récepteur AT1 qui appartientà
la grande famille des GPCRs. Comme tous les autres GPCRs, le récepteur AT1 possède sept
domaines transmembranaires (TM) qui contribuent à former la pochette de liaison du ligand. Afin d‟identifier les acides aminés du TM2 impliqués dans la formation de la pochette de liaison du récepteur AT1, nous avons utilisé l‟approche SCAM (Substituted Cysteine
Accessibility Method) qui consiste à évaluer les propriétés de liaison du récepteur suite à sa réaction avec le methanethiosulfonate (MTSEA). Le MTSEA alkyle les cystéines endogènes ou introduites par mutagénèse dirigée, causant ainsi un encombrement stérique qui interfèrera avec la liaison du ligand. Une série de mutants ont été produits en remplaçant successivement par une cystéine les résidus 70 à 94 du TM2 du récepteur AT1 et de son mutant
constitutivement actif AT1-N111G. Après le prétraitement avec le MTSEA, les mutants
D74C, L81C, L83C, A85C, A89C ont montré une diminution significative d‟affinité pour le ligand 125ISar1, Ile8AngII, suggérant que ces résidus sont orientés dans la pochette de liaison du récepteur AT1. Des résultats différents ont été obtenus dans le cas du récepteur
N111G-AT1. Le mutant D74C-N111G est devenu insensible au MTSEA, alors que la sensibilité de
L81C-N111G fut grandement diminuée. Par contre, le mutant V86C-N111G s‟est avéré sensible au MTSEA. Ces résultats suggèrent que l‟activation constitutive du récepteur AT1
implique un mouvement de pivot du TM2, favorisant le rapprochement du haut du TM2 vers la pochette de liaison.