Nouvelles approches pour la séparation des actinides
dans les gisements de terres rares
Thèse
Laurence Whitty-Léveillé
Doctorat en chimie
Philosophiæ doctor (Ph. D.)
Nouvelles approches pour la séparation des actinides
dans les gisements de terres rares
Thèse
Laurence Whitty-Léveillé
Sous la direction de :
Dominic Larivière, directeur de recherche
Nicolas Reynier, codirecteur de recherche
Résumé
Récemment, la récupération des éléments de terres rares (ETR) à partir de diverses ressources a suscité l’intérêt de l’industrie minière. Puisque les actinides et les ETR partagent des propriétés chimiques similaires, la séparation de ces deux groupes d’éléments est souvent une préoccupation de l’industrie des ETR afin d’atténuer la contamination radioactive des installations. La séparation des actinides des ETR pose des défis chimiques intéressants. Les actinides et les lanthanides sont souvent dissous par des procédés hydrométallurgiques, ce qui peut entraîner une contamination radioactive pendant le cycle d'extraction.
Les progrès récents dans les techniques d'extraction solide-liquide, y compris l'extraction en phase solide (EPS), ont démontré des performances supérieures avec des facteur de distributions plus élevés, une séparation de phase plus rapide en l'absence d'émulsion et de meilleures récupérations que les méthodes de séparation liquide-liquide d'actinides. L'objectif de cette thèse est de développer de nouvelles techniques pour l'extraction efficace des actinides à partir de minerais et de solutions acides contenant des terres rares.
Le premier projet optimise une procédure de lixiviation séquentielle avec Na2CO3/NaHCO3 et HCl
pour la dissolution rapide et sélective des ETR et des actinides selon un plan expérimental. L’optimisation met l’accent sur les principaux paramètres opérationnels que sont le temps, la température et la concentration, divers types de minéraux (composites et minerais), le rendement maximal de lixiviation et la minimisation de la dissolution d’éléments de la gangue tel que Fe, Si et Ca.
Le deuxième projet porte sur un nouveau type de procédure d’extraction pour extraire l’uranium des solutions acides de terres rares en combinant un support magnétique en phase solide et un ligand à base de Schiff. Nous avons synthétisé et caractérisé trois ligands de base de Schiff (CH3Salen,
H2Salophen et MeOSalophen). La base de Schiff magnétique avec le MeOSalophen a montré une
grande sélectivité envers l’U(VI) par rapport à plusieurs métaux dans une solution de lixiviation des ETR.
Enfin, les derniers projets concernent la conception et le développement de supports solides fonctionnalisés (tels que la silice et des nanoparticules cœur-coquille) utilisant la base de Schiff MeOSalophen pour l'extraction de l'uranium. Différents types de greffage ont été évalués (attachement unilatéral ou bilatéral) et une cinétique d'extraction rapide a été obtenue, ainsi qu'un haut degré de réutilisabilité dans les conditions testées.
Abstract
Recently, the recovery of rare earth elements (REEs) from a variety of resources has gained interest in the mining industry. As naturally occurring actinides and REEs share similar chemical properties, the separation of those two groups of elements is often a concern in the rare earth industry to mitigate radioactive contamination of facilities and workers. Separating naturally occurring actinides from REEs poses interesting chemical challenges. Actinides and lanthanides are often co-dissolved in hydrometallurgical processes, potentially resulting in radioactive contamination of extraction cycle.
Recent developments in solid-liquid extraction techniques, including solid-phase extraction (SPE), have demonstrated superior performances with higher enrichment factors, faster phase separation in the absence of emulsion, and better recoveries than liquid-liquid methods for isolating actinides. The goal of this thesis is to develop new extraction techniques for the efficient extraction of actinides from rare earths-bearing ores and leachates.
The first project optimizes a sequential leaching procedure with Na2CO3/NaHCO3 and HCl
for the rapid and selective dissolution of REEs and actinides through experimental design. Optimization emphases on the main operational parameters which are time, temperature and concentration, various types of minerals (REE composites and ores), maximum leaching yield, and minimization of gangue metal dissolution like Fe, Si, and Ca.
The second project focusses on a new type of extraction procedure for the removal of uranium from rare earth leachates by combining a magnetic solid-phase support and a Schiff base
ligand. We synthesized and characterized three Schiff base ligands (CH3Salen, H2Salophen
and MeOSalophen) and the magnetic MeOSalophen Schiff base shown a high selectivity towards U(VI) over several metals in a real rare earth leaching solution.
Finally, the last projects concern the design and development of functionalized solid supports (i.e. silica and core-shell nanoparticles) using tethered MeOSalophen Schiff base ligand for uranium extraction. Different grafting approached were evaluated (one-side or two-side tethering) and a fast extraction kinetic was obtained, as well as a high degree of reusability in the conditions tested.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... viii
Liste des tableaux ... xii
Liste des schémas ... xiii
Abréviations ... xiv
Remerciements ... xvii
Avant-propos ... xx
Introduction ... 1
1.1 Généralités ... 1
1.1.1 Chimie des lanthanides et actinides ... 3
1.1.2 Coordination et complexes organométalliques ... 5
1.2 Techniques de dissolution du minerais ... 6
1.2.1 Dissolution hydrométallurgique – Lixiviation acide ... 6
1.2.2 Dissolution hydrométallurgique – Lixiviation basique ... 11
1.2.3 Précipitation chimique ... 14
1.2.4 Autres techniques de dissolution ... 15
1.3 Techniques de séparation hydrométallurgique ... 17
1.3.1 Ligands couramment utilisés ... 18
1.3.2 Extraction solide-liquide ... 26
1.4 Objectifs de la thèse ... 33
Chapitre 2. Lixiviation rapide et sélective des actinides et des éléments de terres rares contenus dans des minéraux porteurs de terres rares et des minerais ... 35
2.1 Résumé ... 36
2.2 Abstract ... 36
2.3 Introduction ... 37
2.4 Material and methods ... 39
2.4.1 Experimental ... 39
2.4.2 Statistical modelling ... 41
2.5 Results and discussion ... 42
2.5.2 Optimization of leaching parameters for rare earth minerals with HCl ... 44
2.5.3 Two-step leaching process for a rare earth mineral composite ... 50
2.5.4 Model predictability ... 53
2.5.5 Sequential leaching of ores ... 54
2.6 Conclusions ... 56
2.7 Acknowledgments ... 57
2.8 Supporting Information ... 57
2.8.1 Instrumental parameters for the analysis of REE ... 57
2.8.2 Additional figures and tables ... 58
Chapitre 3. Extraction sélective de l'uranium dans des solutions acides de terres rares par extraction magnétique en phase solide à l'aide de ligands de base de Schiff ... 81
3.1 Résumé ... 82 3.2 Abstract ... 82 3.3 Introduction ... 83 3.4 Experimental ... 85 3.4.1 Preparation of samples ... 85 3.4.2 Synthesis of Fe3O4 nanoparticles ... 86
3.4.3 Synthesis of Bis(2-hydroxyacetophenone) ethylenediimine (CH3Salen) ... 87
3.4.4 Synthesis of Bis(salicyla1dehyde) o-phenylenediimine (H2Salophen) ... 87
3.4.5 Synthesis of N,N’-bis(3-methoxylsalicylidene)-1,2-phenylenediamine (MeOSalophen) ... 87
3.4.6 Preparation of the magnetic Schiff base ... 88
3.4.7 Adsorption experiments ... 88
3.4.8 Characterization and analysis ... 89
3.5 Results and discussion ... 89
3.5.1 Selection of ligand ... 89
3.5.2 Characterization of the magnetic Schiff base ... 91
3.5.3 Effect of solution pH in various media ... 93
3.5.4 Effect of sulfate concentration ... 95
3.5.5 Effect of contact time and adsorption mechanisms ... 96
3.5.6 Adsorption isotherms of uranium ... 97
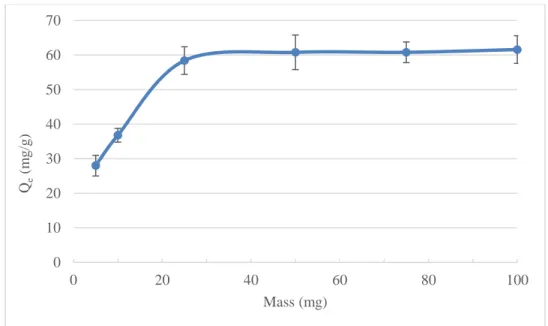
3.5.7 Effect of the amount of magnetic Schiff base ... 99
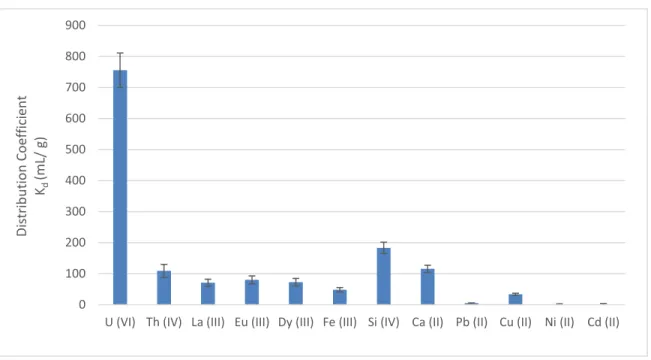
3.5.8 Selectivity of magnetic Schiff base for actinides in solution ... 100
3.7 Acknowledgments ... 104
3.8 Supporting Information ... 104
3.8.1 Additional Figures ... 105
Chapitre 4. Conception d'un ligand de base de Schiff greffé sur un support de silice comme technique très efficace pour l’élimination de l'uranium et du thorium en solution acide ... 115
4.1 Résumé ... 116
4.2 Abstract ... 116
4.3 Introduction ... 117
4.4 Experimental ... 119
4.4.1 Preparation of surrogate and samples ... 119
4.4.2 Synthesis of Bis(3-methoxysalicylaldehyde) o-phenylenediimine (MeOSalophen) ... 120
4.4.3 Grafting approaches for the modified silica supports ... 121
4.4.4 Adsorption experiments ... 123
4.4.5 Material characterization ... 124
4.5 Results and discussion ... 125
4.5.1 Surface Characterization ... 125
4.5.2 Extraction Studies ... 128
4.6 Conclusions ... 139
4.7 Acknowledgments ... 139
4.8 Annexe 1 : Grafting MeOSalophen on mesoporous silica ... 140
4.9 Supporting Information ... 142
4.9.1 Additional Figures and Tables ... 142
Chapitre 5. Nanoparticules cœur-coquille portant une base de Schiff pour l’adsorption efficace des actinides et une séparation sélective en milieu aqueux ... 147
5.1 Context ... 147
5.2 Experimental section ... 149
5.2.1 Preparation of samples ... 149
5.2.2 Synthesis of the silica-coated magnetic nanoparticles (Fe3O4@SiO2) ... 150
5.2.3 Synthesis of Bis(3-methoxysalicylaldehyde) o-phenylenediimine (MeOSalophen) ... 151
5.2.4 Two-Step Functionalization (Fe3O4@SiO2-MeO) ... 152
5.2.5 Adsorption experiments ... 153
5.2.6 Material characterization ... 154
5.3 Results and discussion ... 155
5.3.2 Effect of pH ... 157
5.3.3 Kinetic study ... 158
5.3.4 Adsorption isotherms of uranium ... 161
5.3.5 Selectivity test ... 162
5.3.6 Reusability test ... 165
5.4 Partial conclusion ... 166
Conclusions et travaux futurs ... 167
6.1 Retour sur les objectifs ... 167
6.1.1 Étude de la séparation hydrométallurgique des actinides avec différents minéraux ... 167
6.1.2 Développement d’un nouveau ligand organique sélectif aux actinides ... 168
6.1.3 Développement d’une nouvelle méthode d’extraction sur support solide ... 168
6.2 Perspectives ... 169
6.2.1 Mise en place d’une usine pilote ... 170
6.2.2 Application des bases de Schiff à divers systèmes ... 170
6.2.3 Intégration des bases de Schiff dans l’économie circulaire minière ... 172
Liste des figures
Figure 1.1. Rayons ioniques des lanthanides et actinides selon leur degré d’oxydation. ... 4
Figure 1.2. Structure du N,N'-diméthyl-N,N'-dibutyl-tétradécylmalonamide (DMDBTDMA). ... 21
Figure 1.3. Coordination bidentate des An par des ligands de la famille des malonamide. . 21
Figure 1.4. Représentation schématique de: A) HC301, B) HC302 et C) HC272. ... 23
Figure 1.5. Structure de la 1,2,4-triazine pyridine et la position respective des atomes d’azote. ... 23
Figure 1.6. Structure générale des ligands de la famille des bases de Schiff. Les substituants (R1-R4, R1’-R4’) sont facilement modifiables et peuvent être utilisés pour contrôler aisément les propriétés du ligand. ... 25
Figure 1.7. Comparaison des méthodes d’extraction liquide-liquide (ELL) et solide-liquide (ESL).124 ... 26
Figure 1.8. Structure chimique de la résine Diphonix ... 29
Figure 1.9. Procédé pour l'extraction magnétique en phase solide... 31
Figure 1.10. Structure chimique de la quercétine utilisée pour la sorption de l'uranium ... 32
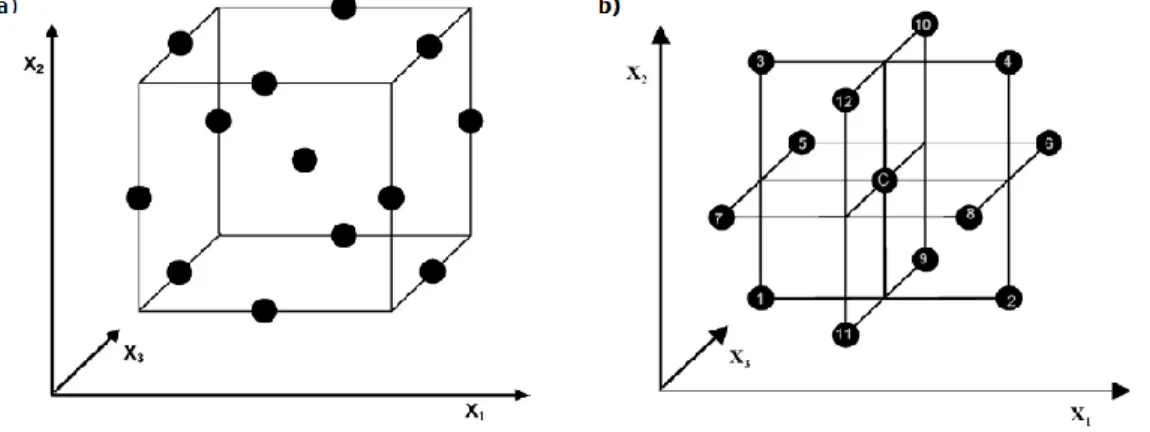
Figure 2.1. The cube design for a) BBD and b) three interlocking 22 factorial design. (Adapted from Ferreira et al.)197... 41
Figure 2.2. X-ray diffraction analysis of the MBX sample ... 43
Figure 2.3. Surface response area in monazite for Th (A-C) and LREEs (D-F) using HCl as leaching agent (initial pulp density 1% (w/w) solids) ... 45
Figure 2.4. Surface response area in bastnasite for Th (A-C) and LREEs (D-F) using HCl as leaching agent (initial pulp density 1% (w/w) solids) ... 47
Figure 2.5. Surface response area in xenotime for Th (A-C) and LREE (D-F) using HCl as leaching agent (initial pulp density 1% (w/w) solids) ... 49
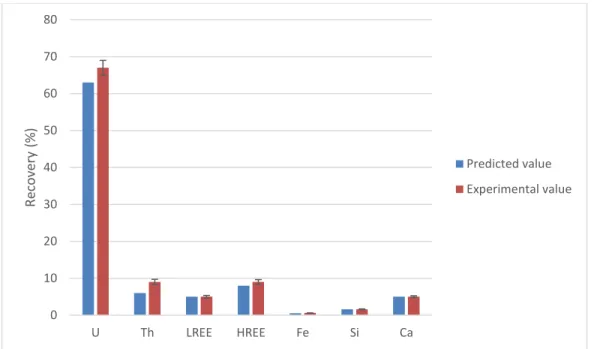
Figure 2.6. Comparison between predicted and experimental dissolution efficiency for the carbonate pre-leaching step on the MBX composite sample (n=3) ... 53
Figure 2.7. Comparison between predicted and experimental leaching recovery for the hydrochloric acid leaching performed on the residues obtained after carbonate pre-leaching step on the MBX composite sample (n=3) ... 54
Figure 3.1. Effect of structure on the adsorption capacity of U(VI) ions by 3 crystalline Schiff bases. Initial uranium concentration 100 mg L-1, pH 6, temperature 20°C, adsorbent mass 0.08 mmol, stirring time 24 h. ... 91
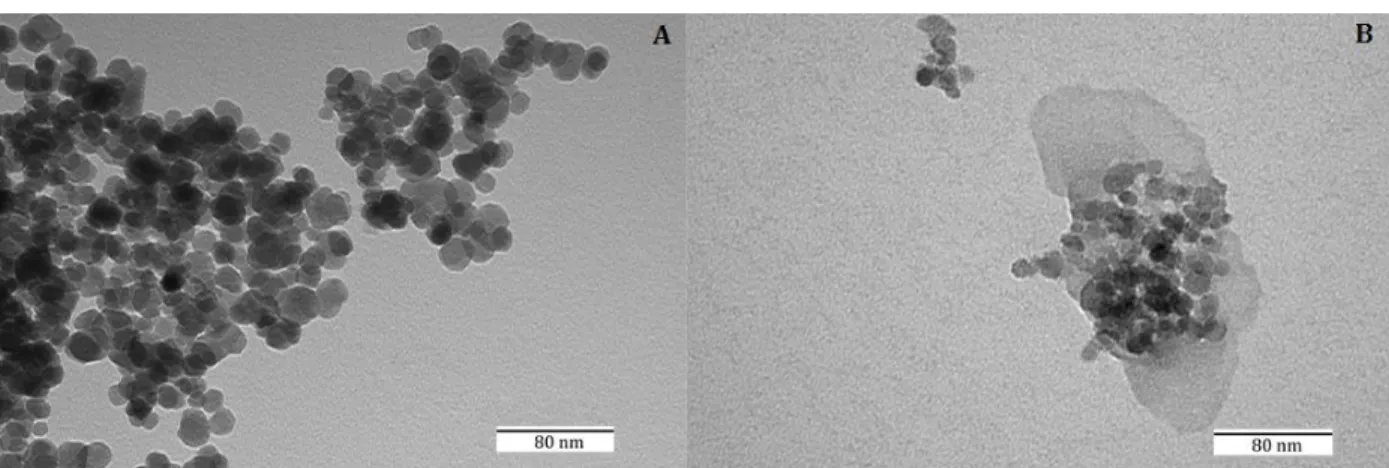
Figure 3.2. A) TEM image of the Fe3O4 nanoparticles. B) TEM image of the magnetic Schiff base. ... 92
Figure 3.3. ATR spectra of MSB before extraction (dotted line) and after extraction (full line) of a solution containing 100 mg L-1 of U(VI). ... 93
Figure 3.4. Impact of various media on the removal of U(VI) ions by MSB. Initial uranium
concentration 100 mg L-1, anion concentration 4,000 mg L-1, temperature 20°C, adsorbent
mass 0.025 g, stirring time 24 h... 94 Figure 3.5. Effect of the sulfate concentration on the removal of uranyl(VI) ion by magnetic
Schiff base. Initial uranium concentration 100 mg L-1, pH 6, temperature 20 °C, adsorbent
mass 0.025 g, stirring time 24 h... 95 Figure 3.6. Adsorption kinetic of U (VI). Initial uranium concentration in sulfate media 100
mg L-1, pH 6.0, temperature of 20 °C, amount of magnetic Schiff base 0.025 g. ... 96
Figure 3.7. Effect of magnetic Schiff base mass on the uptake of U(VI) in sulfate media.
Initial uranium concentration 100 mg L-1, pH 6, temperature 20 °C, adsorbent mass 0.025 g,
stirring time 24 h. ... 100 Figure 3.8. Selectivity results of the magnetic Schiff base in a sulfate surrogate solution.
Initial concentrations of U and REEs were1 mg L-1, initial concentration of Th was 10 mg
L-1, pH 6.0, temperature 20°C, mass of adsorbent 0.025 g, stirring time 24 h. ... 101
Figure 3.9. Competitive adsorption of concurrent ions on magnetic Schiff base, magnetic nanoparticles and MeOSalophen in real leaching solution. pH 1.82, temperature 20 °C, mass of adsorbent 0.025 g, stirring time 24 h. ... 103
Figure 4.1 Solid state A) 13C CP NMR and B) 29Si MSA NMR ... 126
Figure 4.2. IR spectra for the different functionalized silica materials ... 127 Figure 4.3. Thermogravimetric analysis curves of the MeOsalophen-modified silica
samples ... 128 Figure 4.4. Impact on the distribution coefficient of the different silica materials. Initial
uranium concentration 100 mg L-1, adsorbent mass 25 mg, pH 6, temperature 20 °C,
stirring time 24 h. ... 129
Figure 4.5. Effect of the presence of various anions on uranium sorption on SiO2-MeO-2.
Initial uranium concentration 100 mg L-1, anion concentration 4,000 mg L-1, adsorbent
mass 25 mg, pH 6.0, temperature 20 °C, stirring time 24 h. ... 130
Figure 4.6. Effect of pH on sorption of uranium by SiO2-MeO-2. Initial uranium
concentration in sulfate media 100 mg L-1, adsorbent mass 25 mg, temperature 20 °C,
stirring time 24 h. ... 131
Figure 4.7. Adsorption kinetics of U (VI) on SiO2-MeO-2. Initial uranium concentration in
sulfate media 100 mg L-1, adsorbent mass 25 mg, pH 6.0, temperature 20 °C. ... 132
Figure 4.8. Competitive distribution coefficients (Kd) values for the functionalized silica
bead support for actinides in the presence of REEs. The initial concentrations of U and
REEs were 1 mg L-1, initial concentration of Th was 10 mg L-1, adsorbent mass 25 mg, pH
6.0, temperature 20 °C, stirring time 6 h. ... 136 Figure 4.9. Competitive adsorption of concurrent ions on Schiff base functionalized silica in leach liquors. Adsorbent mass 25 mg, pH 1.82, temperature 20 °C, stirring time 6 h. .. 137
Figure 4.10. Reusability tests on SiO2-MeO-2. Loading with initial uranium concentration
in sulfate media 100 mg L-1, adsorbent mass 25 mg, pH 6.0, temperature 20 °C, stirring
time 6 h. ... 139 Figure 4.11. Adsorption capacities of functionalized materials on different silica supports.
Initial concentration of uranium 100 mg L-1, mass 25 mg, pH 6, temperature 20 °C, stirring
time 24 h. ... 140
Figure S4.1. Carbon and nitrogen XPS spectra of SiO2-MeO-1 ... 142
Figure S4.2. Carbon and nitrogen XPS spectra of SiO2-MeO-2 ... 143
Figure S4.3. Pseudo-second-order plot for the removal of U(VI) by magnetic Schiff base.
Initial uranium concentration 100 mg L-1, sulfate concentration 4000 mg L-1, pH 6.0,
temperature 20 °C, mass of sorbent 0.025 g ... 144 Figure S4.4. Pseudo-first order plot for the removal of U(VI) by magnetic Schiff base.
Initial uranium concentration 100 mg L-1, sulfate concentration 4000 mg L-1, pH 6.0,
temperature 20 °C, mass of sorbent 0.025 g ... 144 Figure S4.5. A) Langmuir plot for the removal of U(VI) by grafted Schiff base and B)
Linear fit of the Langmuir model. Initial uranium concentration 100 mg L-1, sulfate
concentration 4000 mg L-1, pH 6.0, temperature 20 °C, mass of sorbent base 0.020 g ... 145
Figure S4.6. Freundlich plot for the removal of U(VI) by grafted Schiff base. Initial
uranium concentration 100 mg L-1, sulfate concentration 4000 mg L-1, pH 6.0, temperature
20 °C, mass of sorbent base 0.020 g ... 146 Figure 5.1 TEM images of A) Fe3O4 and B) Fe3O4@SiO2 nanoparticles. ... 155
Figure 5.2. Size distribution histogram of Fe3O4 nanoparticles from the TEM images
analysis ... 156
Figure 5.3. Distribution of the hydrodynamic diameter of A) Fe3O4 and B) silica-coated
Fe3O4 nanoparticles according to NTA ... 157
Figure 5.4. Effect of pH on sorption of uranium by Fe3O4@SiO2-MeO. Initial U(VI)
concentration in sulfate media 100 mg L-1, adsorbent mass 25 mg, temperature 20°C,
stirring time 24 h. ... 158
Figure 5.5. Adsorption kinetic of U(VI) by Fe3O4@SiO2-MeO. Initial U(VI) concentration
in sulfate media 100 mg L-1, pH 6.0, temperature 20 °C, amount of grafted magnetic
support 0.025 g. ... 159 Figure 5.6. A) Pseudo-second-order and B) pseudo-first-order plot for the removal of U(VI) by Fe3O4@SiO2-MeO. ... 160
Figure 5.7. Langmuir plot for the removal of U(VI) by Fe3O4@SiO2-MeO. ... 162
Figure 5.8. Competitive distribution coefficients (Kd) values for the functionalized
core-shell nanoparticle for actinides in the presence of REEs. Initial concentrations of U and
REEs were 1 mg L-1, initial concentration of Th was 10 mg L-1, adsorbent mass 25 mg, pH
6.0, temperature 20°C, stirring time 6 h. ... 163 Figure 5.9. Competitive adsorption of concurrent ions on magnetic Schiff base in leach liquors. Adsorbent mass 25 mg, pH 1.82, temperature 20 °C, stirring time 6 h. ... 164
Figure 5.10. Reusability tests on Fe3O4@SiO2-MeO. Loading with initial U(VI)
concentration in sulfate media 100 mg L-1, adsorbent mass 25 mg, pH 6.0, temperature
20°C, stirring time 6 h. ... 166 Figure 6.5.11. A) Base de Schiff traditionnelle et B) Base de Schiff plus large ... 171
Liste des tableaux
Tableau 1.1. Tableau récapitulatif de quelques résultats de lixiviation acide ... 11 Tableau 1.2. Tableau récapitulatif de certains ligands ou famille de ligands utilisés pour la séparation des actinides et des lanthanides. ... 19 Table 2.1. Concentrations of metal species in rare earth-bearing minerals and ores measured by ICP-MS ... 43 Table 2.2. Optimized leaching conditions in HCl and the predicted recovery (Rq, %) for various mineral phases... 50 Table 2.3 Recovery (%) of actinides, REEs and gangue metals from the MBX composite using NaHCO3/Na2CO3 solutions (initial pulp density 1% w/w solids) ... 51
Table 2.4. Recovery (%) of actinides, REEs and gangue metals from MBX composite using HCl solution after a pre-leach step using carbonates (initial pulp density 1% w/w solids) . 52 Table 2.5. Recovery (%) of actinides, REEs and gangue metals of different Canadian ores using NaHCO3/Na2CO3 solutions (initial pulp density 1% w/w solids) ... 55
Table 2.6. Recovery (%) of actinides, REEs and gangue metals of different Canadian ores using HCl solution after a pre-leach step using carbonates (initial pulp density 1% w/w solids) ... 55 Table 3.1 Composition of the major anions and cations present ... 86
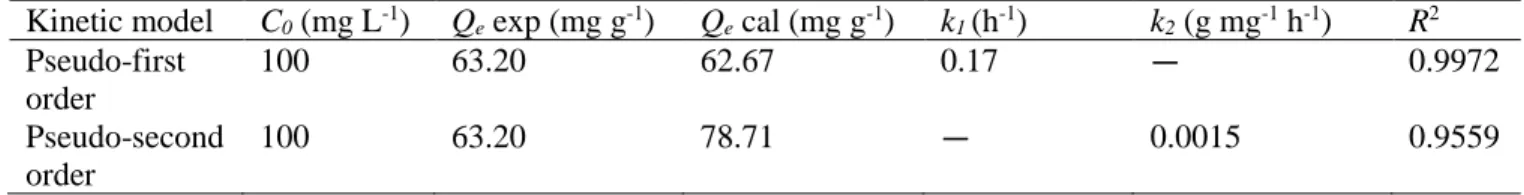
Table 3.2. Pseudo-first and pseudo-second-order constants and values of R2 for magnetic
Schiff base ... 97 Table 3.3. Parameters determined using Langmuir and Freundlich models ... 99
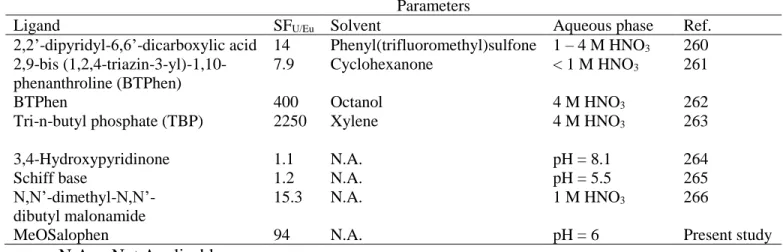
Table 3.4. Comparison of different SFU/Ln values for ligands in various aqueous solutions
... 101
Table 3.5. Effect of the Schiff base and Fe3O4 nanoparticles component on the separation
factors of magnetic Schiff base ligand in the mining leachate ... 103 Table 4.1. Composition of the major anions and cations present in the leach liquor ... 120 Table 4.2. Pseudo-second order model regression values for two-step functionalized silica support ... 133 Table 4.3. Parameters determined using Langmuir and Freundlich models ... 134 Table 4.4. Error functions parameters calculated for the Langmuir and Freundlich
adsorption isotherm ... 135 Table 5.1 Composition of the major anions and cations present in the leach liquor ... 150 Table 5.2. Pseudo-second order model regression values for two-step functionalized silica support ... 161 Table 5.3. Langmuir model parameters calculated for the different functionalized silica materials ... 162
Liste des schémas
Scheme 3.1. Structures of the Schiff base ligands synthesize and tested. A) CH3Salen, B)
H2Salophen, C) MeOSalophen ... 90
Scheme 3.2. General structure of the Schiff base family ligands and its coordination of the uranyl ion. Substituents on the phenyl rings (R1-R4, R1’-R4’) are freely modified and may be used for easy control over the ligand properties. ... 90 Scheme 4.1. Detailed synthesis of compound 2 ... Erreur ! Signet non défini. Scheme 4.2. One-step and two-step modification of the surface of silica to generate the uranium-selective sorbent ... Erreur ! Signet non défini. Scheme 4.3. Synthesis of modified MeOSalophen for the one-step grafting. Erreur ! Signet
non défini.
Scheme 5.1. Detailed synthesis of compounds 1 and 2 ... 152 Scheme 5.2. Modification steps of the silica shell to generate the uranium-selective
Abréviations
APTS (3-aminopropyl)triethoxysilane
ATR attenuated total reflectance
DFT théorie de la fonctionnelle de densité
DMF N,N-diméthylformamide
DMSO dimethylsulfoxide
EDCI 1-éthyl-3-(3-diméthylaminopropyl)carbodiimide
EMPS extraction magnétique en phase solide
ETR éléments de terres rares
HOBt hydroxybenzotriazole
IX ionic exchange
MAS magic-angle spinning
MNP magnetic nanoparticle
MSB magnetic Schiff base
MSPE magnetic solid-phase extraction
NMR nuclear magnetic resonance
NORM naturally occurring radioactive materials
NPM nanoparticule magnétique
NTA nanoparticle tracking analysis
REE rare earth element
SDS dodécylsulfate de sodium
SEC size exclusion chromatography
SEM scanning electron microscope
SPE solid-phase extraction
TEA trimethylamine
TEM transmission electron microscopy
TEOS tetraethyl orthosilicate
TGA thermogravimetric analysis
XPS X-ray photoelectron spectroscopy
À maman et papa, les tout premiers ayant cru en moi
Remerciements
J’ai souvent affirmé que mon doctorat était un projet collaboratif; je n’aurais pas pu réaliser toutes ces expériences sans l’aide de personnes dévouées et passionnées. J’aimerais ainsi leur exprimer ma sincère et plus grande gratitude.
J’aimerais d’abord remercier mon directeur et mentor des huit dernières années, Dominic Larivière. Tu m’as fait grandir autant sur le plan scientifique que sur le plan personnel. Tu avais, et tu as toujours, cette façon de poser les questions auxquelles je ne voulais pas répondre, en soulignant les limites de mon travail que je tentais de survoler et d’oublier. Tu m’as fourni des conseils et des orientations différentes pour le projet, mais tu ne m’as pas surprotégé. Tu as été mon superviseur pendant les huit dernières années, et pourtant je sentais que plus j’avançais dans mes études graduées, plus tu me traitais comme une collègue. Tu as donné de la valeur à mon opinion, puis tu me disais gentiment pourquoi, parfois, c'était mal orienté, me montrant une meilleure solution à la place. En rétrospective, tu étais simplement une personne formidable et je suis profondément reconnaissante d’avoir été ton étudiante durant toutes ces années. Un doctorat est difficile. Mais un bon directeur facilite beaucoup les choses.
Un immense merci à mon co-directeur, Nicolas Reynier. Tu m’as fait confiance dès le début avec ce projet de doctorat, et cette confiance n’a jamais failli au cours des dernières années. Sans ton support constant face à la machine gouvernementale, je n’aurais pas accompli ce défi incroyable que de finir un doctorat en trois ans. Tu m’as poussée à me dépasser et à ne pas avoir peur de l’opinion, parfois vieillotte, de chercheurs plus expérimentés. D’étudiante anxieuse en 2016, tu m’as aidée à devenir une chercheuse aguerrie et confiante. Je ne saurais t’en être plus reconnaissante.
My most sincere acknowledgements to my colleagues from NRCan: Cheryl, Jacob, André, Alain and Jean-François. You were the most supportive colleagues! You were always coming to all of my presentation practices even though they were in French and always helping me out in the lab, whether it was for the preparation of samples or just fixing a screw. My time at Canmet would not have been as amusing if it wasn’t of you. Thank you for the Halloween candies, the Friday’s cookies and the coffees. Thank you for being such amazing coworkers.
Un merci incroyable à mes collègues et collaborateurs de la dernière année, Audrey et Cyril. Une grande partie de cette thèse n’aurait pas pu être possible sans vous. Vous avez donné de votre temps, sans compter, et vous avez toujours été enthousiastes aux différentes idées du projet de greffage. Merci d’avoir été patients par rapport à mes connaissances un brin rouillées en chimie organique et de m’avoir aussi bien épaulée dans ce projet. Je suis fière du travail que nous avons accompli et je suis fière que divers horizons de la chimie aient donné naissance à un si beau projet!
Merci du fond du cœur à mes amis qui m’ont supporté dans cette thèse. Merci à ma sœur et meilleure amie, Myriam, pour ta patience et ton soutien indéfectible. Nos appels quotidiens m’ont fait réaliser que la distance qui nous séparait n’était que physique, et tes nombreuses visites à Ottawa m’ont fait tellement de bien! Tu sais toujours me ramener sur terre quand j’en ai grandement besoin, tout comme tu sais me faire sortir de ma tête quand tu me sens renfrognée. Merci d’avoir toujours cru en moi et de continuer à m’inspirer quotidiennement. Merci à Mathilde et Mylène, mes fidèles amies depuis près de huit ans. Vos conseils m’ont grandement aidée lors de ma thèse, tout comme nos nombreux fous rires. Un nouveau chapitre s’amorce pour nous trois en 2019 et je ne pourrais être plus heureuse de vivre toutes ces aventures avec vous. Un gros merci à Maxime. De très bon collègue, tu es rapidement devenu un de mes meilleurs amis. Ta présence et ton soutien journalier m’ont aidé plus que tu ne le crois à donner le meilleur de moi-même. Tu es une source inépuisable de bonne humeur et d’énergie, et je suis très chanceuse d’avoir pu te côtoyer quotidiennement.
Un merci tout particulier à mes parents, Josette et Jean. Au cours de mes nombreuses années d’études, vous m’avez toujours encouragée à réaliser mon plein potentiel et m’avez toujours apporté votre soutien inconditionnel. Je sais que j’étais plutôt dissipée étant jeune. Toutefois, vous avez décelé, derrière cet air rebelle, un potentiel que je ne connaissais même pas. Au cours de mes très nombreuses années d’études, vos efforts, votre présence et votre amour n’ont jamais failli, me donnant la détermination de poursuivre des études universitaires graduées. Je vous serai éternellement reconnaissante de tout ce que vous m’avez offert et des sacrifices que vous avez faits pour me permettre de me rendre au doctorat. Je n’aurais pu rêver de meilleurs parents! Je vous aime si fort!
Finalement, merci au plus important d’entre tous, Pier Alexandre. Que ce soit de Los Angeles, d’Ottawa ou de Newark, ton soutien, ton aide et ton amour n’ont jamais faibli. Tes encouragements dans mes moments de doutes m’ont permis d’affronter de trop nombreuses épreuves de remises en question; pour cela, je te remercie de ta patience légendaire! Tu m’inspires à chaque jour à donner le meilleur de moi-même et je ne serais pas où j’en suis aujourd’hui sans toi. De nouveaux défis m’attendent à la sortie de l’université, mais je me sens tout à fait prête à les affronter, puisque je sais que nous serons ensemble. Je t’aime!
Avant-propos
La thèse qui suit est divisée en six chapitres, dont quatre principaux qui décrivent de la recherche originale. Pour les chapitres tirés d’articles scientifiques, le texte et les figures ont été recopiés sans modifications, sauf bien sûr pour adapter la numérotation des tableaux, figures et schémas.
Le chapitre 2 est basé sur un article scientifique paru dans le journal Hydrometallurgy et officiellement publié en ligne le 24 mars 2018. J’ai réalisé la totalité des expériences et j’ai écrit l’article en tant que première auteur, avec l’aide de mon directeur Dominic Larivière ainsi que mon co-directeur Nicolas Reynier.
Le chapitre 3 est basé sur une publication parue dans le journal Industrial & Engineering
Chemistry Research qui a été publiée en ligne le 6 décembre 2018. J’ai planifié et réalisé les
expériences avec le soutien de mes directeurs. Avec leurs directives, j’ai aussi écrit l’article à titre d’auteure principale et regroupé tout le document d’informations supplémentaires. Le chapitre 4 est une publication soumise au journal Separation and Purification Technology en date du 5 février 2019. Il représente les travaux d’une collaboration avec le groupe de recherche du Prof. Jean-François Morin à l’Université Laval. Cyril Aumaître, chercheur postdoctoral au sein du groupe de Prof. Morin, a fait la caractérisation complète des différents composés organiques. Quant à moi, j’ai réalisé les différentes synthèses organiques menant au greffage, fait les diverses expériences analytiques et écrit l’article avec le soutien de mon collègue et de mes directeurs. J’ai également écrit le document d’informations supplémentaires de l’article.
Le chapitre 5 est une addition d’expériences d’extraction sur un support magnétique que j’ai réalisées avec l’aide d’Audrey Picard-Lafond, étudiante au doctorat au sein du groupe du Prof. Denis Boudreau de l’Université Laval. Audrey a fait la synthèse des nanoparticules cœur-coquille magnétiques et leur caractérisation complète. Pour ma part, j’ai fait le greffage du ligand et les différentes expériences d’extraction. Bien que le chapitre n’ait pas encore été publié, les résultats qu’il contient sont très pertinents pour la thèse.
En fonction de ce qui a été dit ci-haut, la présente thèse est écrite à première personne du pluriel. En effet, la recherche qui y est présentée a toujours été un travail d’équipe, même lorsque j’étais seule pour réaliser les expériences.
Introduction
1.1 Généralités
Le terme « éléments de terres rares » (ETR) réfère à la période du tableau périodique commençant par le lanthane et se concluant par le lutécium; période également appelée famille des lanthanides (Ln). Le scandium et l'yttrium sont souvent inclus dans cette famille d’éléments puisque leurs propriétés chimiques sont très semblables à celles des lanthanides. En dépit de leur nom, l'abondance des ETR dans la croûte terrestre est plus importante que
les éléments du groupe du platine, du mercure ou du plomb.1,2 Par exemple, l'abondance
terrestre du scandium est de 2,2x10-3% massique comparativement à 5x10-7% massique pour
l'or.3
Les ETR possèdent des propriétés physicochimiques uniques qui en font des éléments
essentiels pour de nombreuses composantes de hautes technologies.4 Ils peuvent être extrait
dans un grand nombre de minéraux de terres rares, mais seuls trois d’entre eux sont majoritairement utilisés pour la production d’oxydes de terres rares, à savoir la bastnasite
((ETR)(FCO3)), la monazite ((ETR)PO4) et le xénotime (YPO4).5 Les ETR possèdent des
propriétés chimiques similaires à celles de U et de Th. Il n’est donc pas rare que ces actinides (An) et leurs produits de filiations se retrouvent dans les minéraux d’ETR par substitution au niveau de la maille cristalline, ce qui ajoute une composante radioactive dans le traitement du minerai. Par conséquent, la radioactivité est une préoccupation constante lors du traitement des terres rares.6
Des concentrations allant jusqu'à 5% d'uranium et 1% de thorium se trouvent couramment dans le minéral de xénotime. De grandes quantités de thorium, allant jusqu'à 20%, se trouvent fréquemment dans la monazite. Des traces d'uranium peuvent également être retrouvées dans ce minéral, bien que des concentrations allant jusqu'à 16% aient été déjà été rapportées dans
la littérature.7 La bastnasite contient généralement des quantités plus faibles d’uranium
les gisements d’ETR, comme l’uranpyrochlore (U,Ca,Ce)2(Nb,Ta)2O6(OH,F) et la thorite
((Th,U)SiO4), sont également une source de préoccupation en raison de leur degré significatif
de radioactivité. Ainsi, il est important de mettre au point des méthodes appropriées pour séparer les actinides des minerais d’ETR afin de réduire et d’éliminer la contamination
radioactive des concentrés de terres rares.8 L’extraction et le traitement des gisements d’ETR
sont donc des procédés compliqués, coûteux et pouvant avoir des effets néfastes sur l’environnement. Les mines sont la principale source de rejet anthropique de deux principaux
contaminants: les radionucléides et les fines poussières.9
Une fois extrait de la croûte terrestre, le minerai d’ETR doit subir un traitement physique ou physicochimique avant d’être envoyé à l’étape de lixiviation. Ainsi, des étapes de broyage et concassage sont nécessaires afin de libérer les grains des phases minérales présentes dans le minerai et d’en permettre la séparation. Ces grains de minéraux d’ETR étant habituellement fins, ce traitement minéralurgique requiert une réduction de la taille des particules souvent sous les 80 μm. Des procédés conventionnels de séparation magnétique, gravimétrique, électrostatique et par flottation sont ensuite utilisés pour séparer les grains des différents
minéraux et obtenir un concentré d’ETR.10
En règle générale, les actinides sont partiellement séparés des ETR lors du processus de flottation et une fraction d'entre eux sont rejetés sous forme de résidus. Le procédé de flottation consiste à rendre hydrophobes les particules des minéraux visés (ici les minéraux contenant des actinides) par l’addition d’un réactif appelé collecteur, puis à injecter des bulles
d’air auxquelles ces particules hydrophobes s’accrocheront pour remonter à la surface.10 Les
particules seront ensuite récupérées. Un ou plusieurs autres réactifs appelés déprimants, ajoutés avant le collecteur, peuvent aussi être requis afin de maintenir le caractère hydrophile
des particules des minéraux non désirés hydrophiles, les empêchant donc de flotter.10 La
fraction non collectée entre dans la transformation avec le concentré et, au terme d’un long procédé, est ensuite rejetée sous forme de résidus. Une tonne de déchets radioactifs est générée par tonne de terres rares, pouvant occasionner différents effets néfastes sur
l'environnement.11 Il est alors primordial de vouloir séparer les actinides promptement dans
les étapes de traitement et de les valoriser d’une manière qui se veut respectueuse de l'environnement.
Une compréhension accrue des propriétés physicochimiques liant les lanthanides et actinides permettra de mieux mettre en lumière la difficulté de la séparation des deux groupes d’éléments. Puis, les détails de la séparation physique des minéraux contenant des ETR et des actinides pour produire un concentré exempt de composés radioactifs seront le cœur de la présente thèse.
1.1.1 Chimie des lanthanides et actinides
La configuration électronique de la couche de valence des lanthanides est 6s25d14f n-1 ou
6s24f n. À l'état fondamental, les électrons des orbitales 4f et 5d ont des énergies similaires.
Les lanthanides sont des métaux électropositifs qui existent classiquement à l'état d'oxydation
trivalent (Ln3+). D'autres états d'oxydation sont atteints lorsque l'ion contient une sous-couche
f vide (f 0), à moitié pleine (f 7) ou remplie (f 14). De ce fait, les éléments Sm, Eu, Tm et Yb peuvent se retrouver sous forme bivalente (+2), alors que la forme quadrivalente (+4) est accessible pour les éléments Ce et Tb. L’yttrium possède des propriétés très similaires aux ETR, bien qu'il apparaisse dans la cinquième période du tableau périodique avec sa
configuration électronique 4d15s2, et non pas dans celle des lanthanides; ses propriétés
chimiques se rapprochant plus de Lu que de La.12,13 Les électrons f sont étroitement retenus
par le noyau, une fois que les électrons s et d sont enlevés.
Les rayons atomiques des lanthanides diminuent avec l’augmentation du numéro atomique,
allant de 103 pm pour La3+ à 86 pm pour Lu3+ (Figure 1.1). Cet effet est communément
appelé « contraction des lanthanides ». Puisque les orbitales 4f ont de mauvaises propriétés de protection, les électrons de valence sont plus susceptibles d’être attirés par la charge
nucléaire.14 La répulsion entre électrons, alors que le nombre d’électrons de valence
augmente à travers le tableau périodique, ne compense pas l’augmentation de la charge
nucléaire. En conséquence, la charge nucléaire effective (Zeff) est augmentée à travers le
Figure 1.1. Rayons ioniques des lanthanides et actinides selon leur degré d’oxydation.
Les actinides, de leur côté, regroupent les éléments ayant un nombre atomique entre 89 et 103, soit d’actinium à lawrencium. La configuration électronique de leurs couches de
valence, similaires à celle des lanthanides, est décrite par 7s26d15f n-1 ou 7s25f n. Alors que
les lanthanides ne possèdent généralement qu’un nombre d’oxydation +3, les actinides
bénéficient d’une plage plus large allant de +3 à +6.15 Résultant de ces variations de l’état
d’oxydation, les actinides ne se ressemblent pas entre eux autant que les lanthanides, qui sont très chimiquement similaires. Toutefois, les rayons ioniques et une réactivité similaire entre An et Ln (Figure 1.1), résultant de la contraction des lanthanides, rendent leur séparation particulièrement difficile.
En effet, l’actinium ressemble grandement au lanthane. Le thorium, protactinium et uranium sont davantage similaires à leurs congénères verticaux (Hf, Ta et W) qu’au cérium,
praséodyme et néodyme, respectivement.13 À compter du neptunium, la ressemblance avec
les lanthanides augmente doucement et, à partir de l’américium, les actinides se comportent
chimiquement de la même manière et présentent un degré d'oxydation +3 prédominant.13 Ces
nombreux phénomènes sont observés dû au fait que les orbitales 7s, 6d et 5f sont beaucoup plus proches en énergie que les niveaux 6s, 5d et 4f.
1.1.2 Coordination et complexes organométalliques
Le comportement de liaison des lanthanides et des actinides, reconnus pour être des acides de Lewis durs, fait l’objet de débats depuis près de 50 ans, car ils présentent une réactivité
comparable,16 mais s’expliquent en partie par le principe de Pearson d’acide dur et mou (hard
and soft acid and base, HSAB), introduit en 1963.17 Ce concept a été largement utilisé puisque sa déclaration audacieuse sur les acides/bases de Lewis, selon laquelle « le mou aime le mou, le dur aime le dur» est facilement comprise et directement appliquée.
Les lanthanides présentent une variété de nombres et d'environnements de coordination, dû au fait que les électrons f sont enfouis dans la couche électronique et n'ont pas d'influence stéréochimique significative. En conséquence, les ligands adoptent des positions de
coordination qui minimisent les répulsions ligand-ligand.12 Comme les orbitales 5d sont
vides et que les orbitales 4f sont dissimulées, le mode de liaison organométallique des lanthanides est limité. Tel que mentionné précédemment, les ions lanthanides trivalents sont des acides de Lewis durs qui préfèrent se lier à des ligands portant des atomes donneurs durs, très souvent de l'oxygène. Les nombres de coordination des ions lanthanides vont normalement de 6 à 9, mais sont souvent plus élevés avec des petits ligands bidentés, tels que
les nitrates.18 Les ligands monodentates (F-, H
2O, Cl-, etc.) ont généralement une coordination
maximale de 9 pour les lanthanides, alors que les ligands bidentés forment normalement des chélates de 6, 7 ou 8 coordinations, comme LnX3, LnX3·H2O, LnX4-, où X est un ligand
bidentate.19
Les actinides, en contrepartie, ont une plus grande tendance à former des complexes que les lanthanides. En effet, leurs orbitales 5f ont une plus grande extension spatiale puisqu’elles ne sont pas protégées par des sous-couches 6s et 6p remplies, contrairement aux orbitales 4f des
lanthanides (protégées par les sous-coquilles 5s et 5p correspondantes).20 Par ailleurs, l’écart
énergétique entre les configurations 5f n7s2 et 5f n-16d7s2 est inférieur à celui des lanthanides correspondants. Les orbitales 5f sont moins des « orbitales internes », dans la même mesure que le sont les orbitales 4f des lanthanides qui sont donc plus perturbées dans la liaison. En plus, la quasi-dégénérescence des électrons 5f, 6d et 7s signifie qu'une plus grande partie des
électrons de la couche externe peuvent être impliqués dans la formation des complexes.21
Lors de la complexation, les actinides se lieront facilement aux anions monovalents (F-, NO3⁻,
Cl-, etc.) et divalents (CO32-, C2O42-, SO42-). Une stabilité accrue peut également être obtenue
par l’ajout de groupements chélatant tels que le α-hydroxycarboxylate,22 l’acétylacétonate23
ou l’éthyldiaminetétraacétate.24 Évidemment, de par leur caractère d’acide de Lewis dur, les
actinides ont également une forte capacité de complexation avec les ligands porteurs
d’oxygène; les oxydes de carbamoylméthylphosphine,25 malonamides26 et
diglycolamides27,28 étant les ligands les plus couramment utilisés.
1.2 Techniques de dissolution du minerai
Généralement, le processus d’extraction des terres rares est divisé en trois étapes: i) extraction minière et broyage; ii) procédés d'enrichissement du minerai comme la flottation et la séparation gravimétrique et magnétiques afin de générer un concentré d'ETR, et iii) méthodes hydrométallurgiques pour extraire les composés d’ETR. Ce dernier se divise également, ici en deux étapes: i) les processus de lixiviation, de neutralisation et/ou de précipitation, et ii) techniques de séparation et de purification, telles que l’extraction par
solvant ou échange ionique.29,30
Dans un premier temps, nous nous attarderons particulièrement aux différents procédés de lixiviation retrouvés dans la littérature. Deux types de lixiviation sont présents, soit la lixiviation acide et la lixiviation basique. Chaque procédé possède ses avantages et inconvénients, et c’est ce que nous mettrons en lumière dans la prochaine section.
1.2.1 Dissolution hydrométallurgique – Lixiviation acide
1.2.1.1 Traitement à l’acide sulfurique
L'acide sulfurique est l’acide le plus largement utilisé pour la lixiviation des minéraux d’ETR,
en raison de son faible coût, de son caractère d’acide fort et puisque l'ion sulfate du H2SO4
les phosphates et fluorocarbonates de terres rares et de thorium réagissent avec l'acide sulfurique pour former une boue grise foncée contenant des sulfates de terres rares et de
thorium (Équations 1.1 à 1.3).32
2𝐸𝑇𝑅(𝐹𝐶𝑂3) + 3𝐻2𝑆𝑂4→ 𝐸𝑇𝑅2(𝑆𝑂4)3+ 2𝐻𝐹 + 2𝐻2𝑂 + 2𝐶𝑂2 Équation 1.1
2𝐸𝑇𝑅(𝑃𝑂4) + 3𝐻2𝑆𝑂4→ 𝐸𝑇𝑅2(𝑆𝑂4)3+ 6𝐻+(𝑎𝑞)+ 2𝑃𝑂43−(𝑎𝑞) Équation 1.2
𝑇ℎ3(𝑃𝑂4)4+ 6𝐻2𝑆𝑂4→ 3𝑇ℎ(𝑆𝑂4)2+ 12𝐻+(𝑎𝑞)+ 4𝑃𝑂43−(𝑎𝑞) Équation 1.3
Lors de la méthode de digestion à l'acide sulfurique, le minerai est finement broyé afin d’accroitre la surface de contact pour favoriser la décomposition pendant l'attaque chimique, puis mis en contact avec de l'acide sulfurique concentré pendant un maximum de huit
heures.33 Passé ce délai, le groupe de Li ne constatait plus de dissolution significative des
ETR. Tel que démontré à l’Équation 1.1, la présence de fluorocarbonate dans le minerai entraîne une vive réaction entre le carbonate et l’acide provocant la formation de mousse. En conséquence, l'addition d'acide pendant la lixiviation doit être contrôlée et les réservoirs doivent être de taille adéquate.
L’avantage d’avoir un minerai riche en bastnasite, cependant, est la grande solubilité des fluorocarbonate. Des recherches sur la lixiviation à température ambiante ont d’ailleurs été
menées sur un concentré contenant 10% de bastnasite.34 Une récupération d’environ 50% des
ETR a été obtenue après lixiviation du minerai avec 1,5 tonne de H2SO4/tonne de minerai
pendant trois heures. Dix ans plus tard, le groupe de Topkaya a réussi à se surpasser en
employant moins d’une tonne de H2SO4/tonne de minerais.35 Sur un minerai concentré en
bastnasite contenant 28% d’ETR, et en utilisant uniquement 890 kg de H2SO4/tonne de
minerais, les chercheurs ont pu récupérer 90% des lanthanides en seulement une heure. Pour ce faire, ils ont remplacé l’étape de lixiviation acide classique par une étape de cuisson acide (acid baking) à 200 °C durant 2h, puis ont procédé à une lixiviation à l’eau d’une durée de 30 min.
Des températures oscillantes entre 200-230 °C, et un rapport entre l’acide sulfurique et le
minerai compris entre 2:1 et 3:1 (ou une addition d'acide de 2 à 3 tonnes de H2SO4/tonne de
(160 °C)38 et de plus faibles additions d'acide (1,5 tonne H2SO4/tonne de minerai)39 ont
également été recensées dans la littérature sur des minerais contenant peu de fluorocarbonates. Dans une étude menée par Takeuchi, un temps de réaction très court (30 minutes) a été utilisé pour digérer le minerai, mais la température de la réaction a été
augmentée significativement à 250-300 °C.40 Le chercheur a constaté que l'addition d'acide
sulfurique et l'augmentation de la température augmentaient le temps de réaction requis et réduisaient la récupération du thorium en raison de la formation de pyrophosphate de thorium insoluble dans l'eau.38 L'énergie d'activation de la réaction entre l’acide sulfurique et les
minéraux était d'environ 4,2 kJ mol-1, ce qui suggère que la vitesse de réaction était contrôlée
par la diffusion du H2SO4 à travers le produit de réaction formé à la surface des particules de
monazite, bastnasite et xénotime.38
L'utilisation d'acide sulfurique dans diverses industries pour traiter le minerai entraîne un certain nombre d'inconvénients, notamment une faible solubilité des sulfates formés nécessitant de grandes dilutions de la liqueur de lixiviation et une séparation inefficace du
thorium et des ETR.41 Le phosphate doit également être séparé de la liqueur en aval.42 De
plus, la nature corrosive des liqueurs contenant le mélange acide sulfurique et phosphorique
entraîne des coûts de maintenance élevés pour les usines de traitement.43
1.2.1.2 Traitement à l’acide chlorhydrique
Jusqu'en 2002, dans le procédé de la mine de Mountain Pass, CA, exploitée par l’entreprise Molycorp, le concentré d’ETR était préalablement lixivié à l’aide d'acide chlorhydrique dilué
afin d'éliminer les carbonates de calcium et de strontium.44,45 Le résidu était ensuite chauffé
et grillé à l'air à 600-700 °C pour oxyder le cérium et le convertir à l'état tétravalent,
permettant également d'éliminer le CO2 et de concentrer le résidu à 85-90% ETR. Puis, ces
derniers sous forme trivalente étaient lixiviés à l’aide d’HCl, produisant une liqueur de chlorure de terres rares destinée à l’extraction par solvant, et laissant un résidu de Ce(IV)
(Équation 1.4 et 1.5).46
𝐸𝑇𝑅(𝑃𝑂4) + 3𝐻𝐶𝑙 → 𝐸𝑇𝑅𝐶𝑙3+ 𝐻3𝑃𝑂4 Équation 1.5
La lixiviation par HCl d’un minerai principalement composé de bastnasite était également utilisée par l’entreprise Thorium Limited, opérant au Royaume-Uni qui a été en opération
jusqu'en 1976.47 Dans ce procédé, le concentré de bastnasite (70% ETR) était lixivié avec
une masse égale d'acide chlorhydrique concentré pendant six à huit heures à température ambiante. Une quantité supplémentaire d'acide chlorhydrique concentré était ensuite ajoutée et la température de réaction augmentée jusqu’à 100 °C pendant encore quatre à six heures. Pendant ce temps, une petite quantité d'acide sulfurique 50% (v/v) était ajoutée pour précipiter le sulfate de baryum. Pour ce procédé, l'addition d'acide représente environ 1,4 tonne de HCl/tonne de concentré. De plus, les impuretés radioactives étaient majoritairement éliminées en début de procédé par l'étape de précipitation au sulfate de baryum.
En 2013, Kumari et al. ont étudié la lixiviation en deux étapes d’un minerai d’ETR composé
principalement de monazite.48 Une dissolution à l'aide de HCl dilué, d’une température de
350 °C pendant 2 h et d'une densité de pulpe inférieure à 100 g/L, n’a conduit qu'à une dissolution partielle (25%) des ETR. Le concentré, résultant de l’élimination du phosphate grâce à une solution de NaOH 50% (m/v), a été lessivé à l’aide de HCl 6 M à 90 °C pendant 2 h en maintenant une densité de pulpe de l’ordre de 60 g/L, permettant une récupération de 90% des ETR.
Des études concluantes ont également été faites sur la lixiviation à basse température (< 100 °C). En 1965, par exemple, Kruesi et Duker ont rapporté la lixiviation du concentré de flottation de la mine de Mountain Pass avec l’utilisation de 1,8 tonne de HCl/tonne de
minerai à 90 °C pendant quatre heures.49Bien que la température de lixiviation ait entraîné
une perte d'acide, elle était néanmoins nécessaire pour maximiser la décomposition des minéraux. Environ 93% des terres rares ont été extraites par lixiviation à l'acide chlorhydrique, tandis que les terres rares restantes auraient probablement formé des fluorures insolubles. Plus récemment, le groupe de Bian a démontré qu’il était possible de lixivier près de 90% des ETR à l’aide l'acide chlorhydrique concentré à 90 °C et à pression
atmosphérique.50 Leurs expériences de lixiviation ont été menées avec une agitation continue
et une addition d'acide à hauteur de 3,2 tonnes de HCl/tonne de concentré pendant 1,5 heure à 90 °C, après quoi il a été démontré que la dissolution des ETR était complète.
1.2.1.3 Traitement à l’acide nitrique
L’acide nitrique étant beaucoup plus dispendieux que l'acide sulfurique, il n’est pas activement utilisé dans l'industrie minière pour le traitement hydrométallurgique de minéraux. Quelques études intéressantes sur divers minéraux peuvent toutefois être recensées.
En 1994, le groupe de Topkaya présentait une étude sur la bastnasite.51 La lixiviation directe
du minerai non-concentré à l’aide avec du HNO3 a été étudiée pendant trois heures à
température ambiante. Les besoins stœchiométriques en acide pour la lixiviation des ETR
ont été calculés à 1,2 tonne de HNO3/tonne de minerai. La décomposition du minerai a
entraîné la production de nitrates de terres rares, mais les taux de récupération par lixiviation étaient inférieurs à 76%. La réaction proposée pour la réaction de la bastnasite avec l'acide nitrique est présentée ci-contre (Équation 1.6) :
𝐸𝑇𝑅(𝐹𝐶𝑂3) + 3𝐻𝑁𝑂3→ 𝐸𝑇𝑅(𝑁𝑂3)3+ 𝐻𝐹 + 𝐻2𝐶𝑂3 Équation 1.6
L’utilisation de l’acide nitrique sur les minéraux phosphatés a également été rapportée, notamment par Kuzmin et al. en 2012. Ces derniers ont utilisé l'acide nitrique dans des autoclaves rotatifs pour lixivier les métaux d’un minerai d'ETR (gisements de Chuktukon, Sibérie orientale, Russie) contenant 3-7% d'oxydes de terres rares sous forme de phosphate,
des oxydes de fer et manganèse, et 0,5 à 1% d'oxyde de niobium.52 La réaction de lixiviation
de la monazite, le principal minéral phosphaté, avec le HNO3 est indiquée ci-dessous
(Équation 1.7):
𝐸𝑇𝑅(𝑃𝑂4) + 3𝐻𝑁𝑂3→ 𝐸𝑇𝑅(𝑁𝑂3)3+ 𝐻3𝑃𝑂4 Équation 1.7
Au cours de la lixiviation, le fer et le phosphore demeurent dans le résidu en raison de la formation de complexes de phosphates hydroxyferriques, peu solubles dans l'acide nitrique. Des récupérations de terres rares de l’ordre de 87 à 90% ont été obtenues lors de la lixiviation en autoclave pour une plage de températures variant entre 180 et 220 °C.
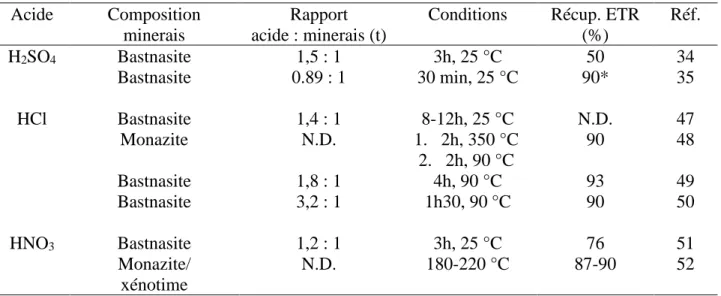
Tableau 1.1. Tableau récapitulatif de quelques résultats de lixiviation acide
Acide Composition
minerais
Rapport acide : minerais (t)
Conditions Récup. ETR
(%) Réf. H2SO4 Bastnasite 1,5 : 1 3h, 25 °C 50 34 Bastnasite 0.89 : 1 30 min, 25 °C 90* 35 HCl Bastnasite 1,4 : 1 8-12h, 25 °C N.D. 47 Monazite N.D. 1. 2h, 350 °C 2. 2h, 90 °C 90 48 Bastnasite 1,8 : 1 4h, 90 °C 93 49 Bastnasite 3,2 : 1 1h30, 90°C 90 50 HNO3 Bastnasite 1,2 : 1 3h, 25 °C 76 51 Monazite/ xénotime N.D. 180-220 °C 87-90 52
*Utilisation de cuisson acide avant l’étape de lixiviation N.D. Non disponible
Bien que la lixiviation acide soit très efficace pour la mise en solution des actinides et ETR, il n’en reste pas moins que ce type de procédé possède plusieurs désavantages. Ainsi, l’utilisation d’acide sulfurique entraîne une perte de teneur en acide phosphorique, une faible solubilité des sulfates d’ETR formés et une mauvaise séparation des Th et des ETR. Du côté de l’acide chlorhydrique, bien qu’il met plus facilement les ETR en solution, son caractère très corrosif entraine des coûts de maintenance très élevés. Finalement, le prix exorbitant de l’acide nitrique en fait un acide peu intéressant pour l’industrie minière compte tenu de la quantité utilisée chaque jour. De ce fait, les inconvénients du procédé de lixiviation acide peuvent rendent la lixiviation basique attirante du point de vue industriel.
1.2.2 Dissolution hydrométallurgique – Lixiviation basique
1.2.2.1 Traitement à l’hydroxyde de sodium
Compte tenu des limites associées au procédé de la lixiviation acide susmentionnées, particulièrement à l’acide sulfurique, des méthodes basiques de traitement du minerai ont été mises au point pour être utilisées en industrie. En particulier, la lixiviation basique s'est avérée une alternative intéressante à la voie acide, puisque les phosphates ou les carbonates pouvaient être récupérés en début du processus comme sous-produit commercial.
Iijima et al. ont étudié la décomposition du gisement de Mountain Pass, CA en utilisant du
NaOH fondu dans un processus continu.53 La fusion a été réalisée à 350 °C en utilisant cinq
tonnes de NaOH/tonne de minerai riche en bastnasite pendant dix minutes afin de réduire la viscosité de la matière fondue. La masse fondue a été dissoute dans de l'eau et ensuite lessivée avec de l'acide chlorhydrique. La décomposition du bastnasite dans l'hydroxyde de sodium fondu à la pression atmosphérique était supérieure à 90% et selon l’équation 1.8.
𝐸𝑇𝑅(𝐹𝐶𝑂3) + 3𝑁𝑎𝑂𝐻 → 𝐸𝑇𝑅(𝑂𝐻)3+ 𝑁𝑎𝐹 + 𝑁𝑎2𝐶𝑂3 Équation 1.8
Amer et al. ont quant à eux démontré le succès de la lixiviation basique sur un concentré de monazite et xénotime avec une solution à 40% d’hydroxyde de sodium pendant quatre heures
à 140 °C.54 Le rapport liquide/solide était de 1,5:1, ce qui correspond à une addition de 600 kg
de NaOH solide/tonne de monazite. Il y a ainsi formation d’hydroxydes de terres rares et
d’un sous-produit de phosphate trisodique (Équation 1.9).55 Le phosphate de thorium présent
dans la monazite subit la même réaction, formant de l'hydroxyde de thorium, tel que présenté
à l’équation 1.10.49
𝐸𝑇𝑅(𝑃𝑂4) + 3𝑁𝑎𝑂𝐻 → 𝐸𝑇𝑅(𝑂𝐻)3+ 𝑁𝑎3𝑃𝑂4 Équation 1.9
𝑇ℎ3(𝑃𝑂4)4+ 12𝑁𝑎𝑂𝐻 → 3𝑇ℎ(𝑂𝐻)4+ 4𝑁𝑎3𝑃𝑂4 Équation 1.10
Dans l’optique de réduire les coûts en NaOH, les chercheurs Miao et Horng ont utilisé une
solution basique à 45% et ont conduit la réaction à 170 °C pour tenter de lixivier les ETR.56
Cette concentration était inférieure à la concentration de NaOH typique (50 à 70%), en raison de la pression plus élevée dans l'autoclave. Ceci a néanmoins permis de maintenir les rendements de lixiviation à environ 90%.
La lixiviation basique à haute température a toutefois des inconvénients, notamment une consommation élevée de NaOH et des difficultés à atteindre une pureté suffisante de phosphate trisodique pour la revente. De plus, il a été observé que des concentrations élevées
de calcium dans la matrice minérale diminuaient l'efficacité de ce type de lixiviation.57 De
plus, cette voie de traitement nécessite l'utilisation d'un équipement résistant à la fois aux températures élevées et aux concentrations élevées de bases. C’est pourquoi certains chercheurs se sont tournés vers des traitements mécaniques. Ainsi, Zhang et Saito ont fait
réagir un gisement riche en bastnasite avec de l'hydroxyde de sodium, en utilisant un
processus de décomposition mécanochimique non thermique.58 Du minerai et du NaOH
(670 kg de NaOH/tonne de minerai) ont été mélangés et broyés pendant un maximum de deux heures, à pression atmosphérique, à l'aide d'un broyeur à boulets. Après cette période, l'hydroxyde de terre rare, le fluorure de sodium et le carbonate de sodium ont été dissous dans de l'eau et la solution a été filtrée. Le degré de décomposition de la bastnasite a été évalué en déterminant par la quantité de fluorure contenue dans le filtrat et il s’est avéré qu’environ 70% des ETR avaient réagi après une heure de broyage.
1.2.2.2 Traitement au carbonate de sodium
Une variation de la lixiviation basique retrouvée dans la littérature implique le carbonate de sodium. La lixiviation au carbonate est particulièrement intéressante dans les cas où des teneurs excessives en minéraux carbonatés rendent l’utilisation de la lixiviation acide peu
rentable.59 Au cours de ce procédé, les fluorures de terres rares ou les phosphates de terres
rares présents dans le minerai sont convertis en carbonates solubles (Équations 1.11 et
1.12),60 puis habituellement lixiviés à l’acide.
2𝐸𝑇𝑅(𝐹𝐶𝑂3) + 𝑁𝑎2𝐶𝑂3→ 𝐸𝑇𝑅2𝑂3+ 3𝐶𝑂2+ 2𝑁𝑎𝐹 Équation 1.11
2𝐸𝑇𝑅(𝑃𝑂4) + 3𝑁𝑎2𝐶𝑂3→ 𝐸𝑇𝑅2𝑂3+ 3𝐶𝑂2+ 2𝑁𝑎3𝑃𝑂4 Équation 1.12
Peu d’études ont été consacrées à la lixiviation des minerais d’ETR à l’aide de carbonates. Les températures requises pour obtenir un rendement de lixiviation satisfaisant sont plus élevées qu’avec le NaOH. Par exemple, dans le procédé mis au point par Chi et al., le concentré de bastnasite du Sichuan (70% d’ETR) a été broyé avec 200 kg de carbonate de sodium/tonne de concentré avant d'être chauffé à 500 °C pendant une heure pour obtenir des
oxydes d’ETR.61
La lixiviation à l’aide de carbonate s’est toutefois révélée être une grande alliée pour la mise
en solution des actinides, principalement l’uranium;62 Eyal et Olander ont même réalisé une
étude en trois volets sur la lixiviation du Th et U de la monazite.63 Ils ont démontré que les
il est possible de ségréger les actinides des autres composants majeurs du minerai. Kim et al.
ont vérifié cette théorie en lixiviant trois minerais contenant de l’uranium.64 À l’aide d’un
mélange de 0,32 M de Na2CO3 et 0,1 M de KOH, ils ont été en mesure de mettre en solution
90% d’U après 3h de lixiviation à 80 °C. Santos et Ladeira ont également testé la lixiviation d’un minerai d’uranium en combinant deux bases, soit le carbonate et le bicarbonate de
sodium.65 Séparément, le carbonate et le bicarbonate se sont révélés être des agents
d'extraction efficaces pour l'uranium; cependant, une combinaison des deux réactifs s'est avérée être une meilleure option que leur utilisation individuelle. Ainsi, des rendements d’extraction d'environ 100% ont pu être obtenus.
La séparation hydrométallurgique utilisant la lixiviation est ainsi apte à extraire les actinides et les ETR de leurs phases minérales. La séparation sélective de ces deux groupes d’éléments est toutefois moins documentée et semble donc plus difficile. Il est alors primordial de se tourner vers la séparation utilisant des ligands organiques tels que l’extraction par solvant, l’échange ionique ou l’extraction sur phase solide pour purifier les métaux désirés.
1.2.3 Précipitation chimique
Avant l'utilisation de méthodes d'extraction par échange d'ions et d'extraction par solvant pour l'extraction et la purification des actinides, des techniques de précipitation étaient utilisées pour les solutions de lixiviation. Deux types de procédés sont employés pour séparer les contaminants des ETR, soit la précipitation des impuretés par neutralisation et la précipitation sélective des ETR sous la forme d’oxalates.
La précipitation par neutralisation utilise le principe selon lequel plusieurs contaminants, tels que le fer, l’aluminium et le thorium, précipitent à un pH plus faible que les ETR. Ainsi, en neutralisant jusqu’à un pH adéquat, il est possible de précipiter sélectivement ces éléments
sous forme d’hydroxydes non solubles, puis de les séparer par filtration.66,67 La neutralisation
présente plusieurs avantages à savoir qu’il s’agit d’un procédé connu de la littérature et dans l’industrie métallurgique. En effet, les coûts des agents neutralisants sont relativement bas, surtout lorsqu’ils peuvent être trouvés à proximité du site minier comme dans le cas du
certains projets d’exploitation canadiens comme celui de Foxtrot, au Labrador.En bref, le
minerai concassé est traité avec du H2SO4 à 200 °C pour rendre les ETR solubles dans l'eau.
Ensuite, l'acide restant est neutralisé avec du carbonate de magnésium (MgCO3), puis du
Na2CO3 est ajouté pour précipiter les ETR sous forme de concentré de ETR2(CO3)3. Ce
concentré est ensuite relavé à l’aide de HCl. Le pH de la solution est finalement ajusté pour rejeter les impuretés, et les terres rares sont précipitées grâce à une solution d'acide
oxalique.69
L’approche par précipitation directe utilisant l’oxalate est toutefois souvent présentée comme une option offrant un meilleur taux de récupération des ETR. Cette précipitation consiste à
ajouter des ions oxalate (C2O42-) à la solution non purifiée jusqu’à précipitation des ETR sous
forme d’oxalates. Puisque ces derniers sont peu solubles,70 ils précipitent sans la majorité des
impuretés ce qui permet une séparation efficace par filtration. Par exemple, à Baotou, en
Chine, le concentré de bastnasite est torréfié au H2SO4 entre 400 et 500 °C pendant plusieurs
heures pour décomposer la matrice de fluorocarbonate et devenir soluble dans la solution de
HCl.57 De l'acide oxalique est ensuite utilisé pour précipiter les ETR sous forme d’oxalate.
Ce type de procédé présente l’avantage d’être simple et d’éviter le recyclage du précipité qui est souvent nécessaire avec la neutralisation pour maintenir des taux de récupérations élevées. Il faut toutefois garder en tête qu’une neutralisation partielle de la solution doit tout de même être réalisée, puisqu’à des pH très bas, comme ceux utilisés en lixiviation, les ajouts
d’oxalates sont plus élevés.71 Ceci entraine par conséquent un coût plus important.
Finalement, les oxalates de thorium n’étant pas solubles, ceux-ci précipitent avec les oxalates
d’ETR, ce qui empêche la séparation des deux groupes d’éléments.55,72
1.2.4 Autres techniques de dissolution
1.2.4.1 Pyrométallurgie
La pyrométallurgie, qui comprend l’incinération, la fusion dans un four à plasma ou un haut fourneau, la formation de scories, le frittage, et les réactions en phase gazeuse à haute température, a été largement utilisée pour le traitement de minerais à haute teneur en métal au cours des derniers siècles. Cette technique présente un facteur de séparation important,
![Table S3.1. Peak list for the XRD analysis of CH 3 Salen Pos. [°2θ] Height [cts] FWHM Left [°2θ] d-spacing [Å] Rel](https://thumb-eu.123doks.com/thumbv2/123doknet/3407368.98717/130.918.106.813.166.794/table-peak-analysis-salen-height-fwhm-left-spacing.webp)
