Étude de la transition vitreuse au sein de polymères selon une approche atomistique en utilisant la dynamique moléculaire
266
0
0
Texte intégral
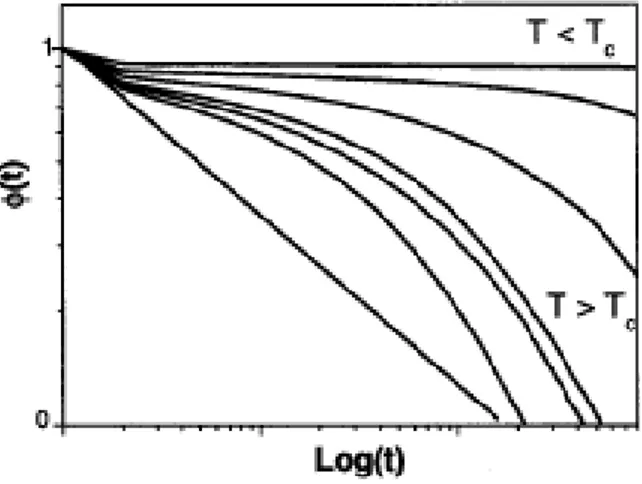
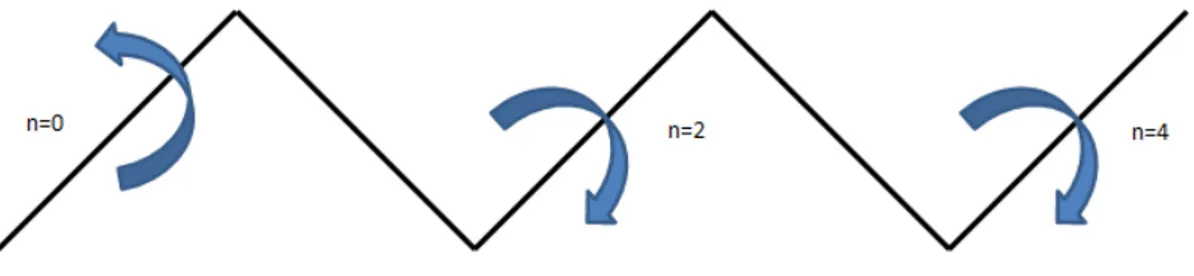
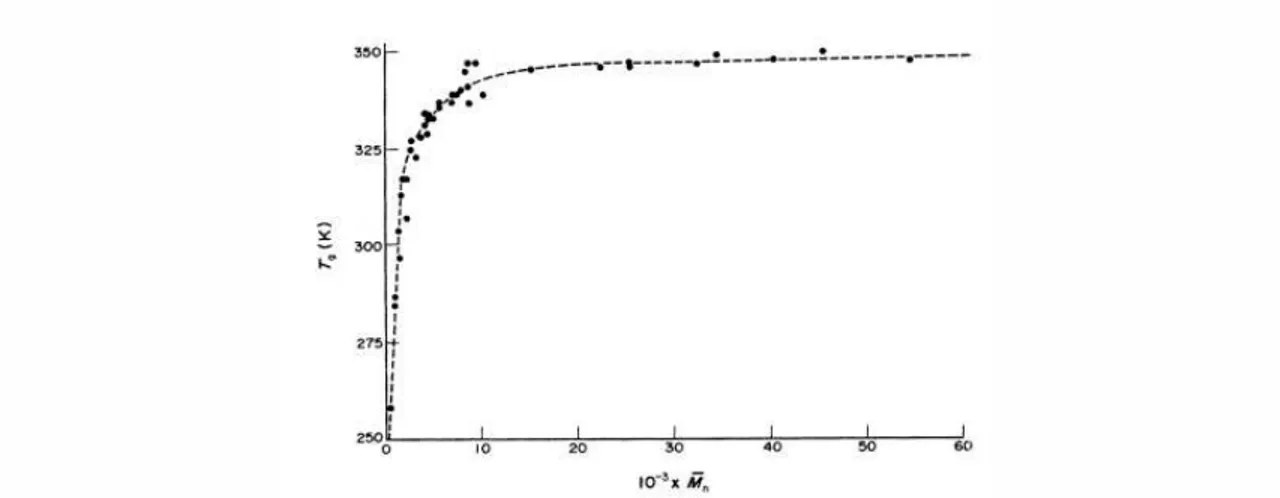
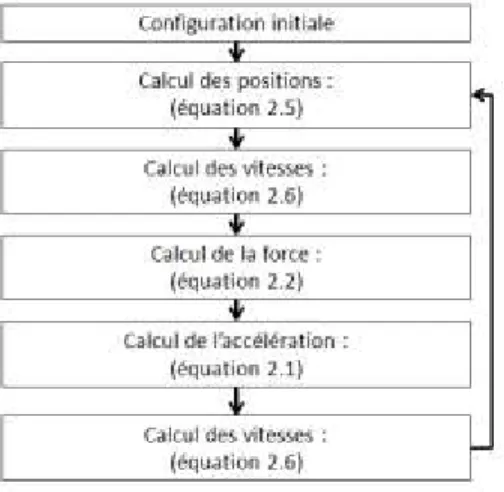
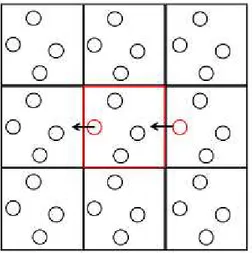
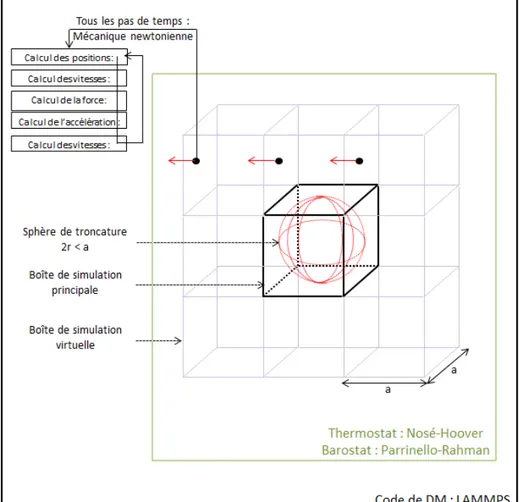
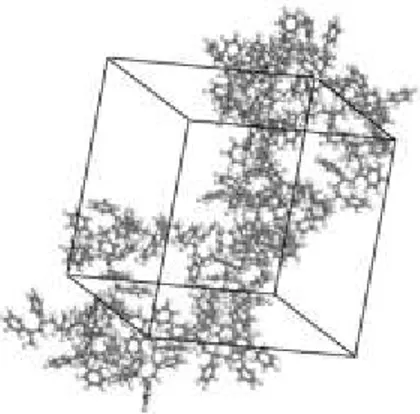
Figure

+7


Documents relatifs
