T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Innovation pharmacologique-Biologie cellulaire
JURY
Thierry LEVADE (Président) Anne COUVELARD (Rapporteur) Annie SCHMID-ALLIANA (Rapporteur)
Laurence TERRAIL (Examinateur)
Ecole doctorale : Biologie-Santé-Biotechnologies Unité de recherche : Inserm U1037
Directeur(s) de Thèse : Catherine SEVA et Audrey FERRAND Rapporteurs : Anne COUVELARD, Annie SCHMIDT-ALLIANA
Présentée et soutenue par Catherine DO Le 25 Octobre 2011
Titre : EXPRESSION DE MARQUEURS PRO-ONCOGENIQUES DANS LES PHASES PRECOCES DE LA CARCINOGENESE COLORECTALE
A notre president de Thèse, Monsieur le Professeur Thierry Levade,
Vous nous faîtes l’honneur de présider cette thèse. Merci de votre confiance et du soutien
que vous nous avez porté. Sachez trouver en ce travail, la traduction de notre reconnaissance.
A Madame le Professeur Anne Couvelard,
Nous vous remercions de l’honneur que vous nous faîtes d’avoir accepté de découvrir et
juger ce travail.
A Madame le Docteur Annie Schmid-Alliana,
Votre présence, le temps et l’attention que vous nous avez accordés à lire ce travail sont
un honneur pour nous.
A Madame le Professeur Laurence Terrail,
Nous vous remercions d’avoir accepté notre invitation en tant que membre de jury à cette
thèse.
A notre directrice de Thèse, Madame le Docteur Catherine Seva,
Nous vous sommes très reconnaissant d’avoir accepté de diriger ce travail de thèse et de
nous avoir permis de le mener à bien. Nous vous remercions pour la confiance que vous nous
avez accordée en accueillant dans votre laboratoire une interne sans expérience en biologie
et aux connaissances limitées dans le domaine de la signalisation cellulaire. Merci également
de nous avoir transmis vos connaissances et partager votre expérience tout au cours de ces
années de master puis de thèse. Votre savoir, votre intégrité et vos qualités humaines font de
vous un modèle que nous espérons atteindre un jour.
A notre directrice de Thèse, Madame le Docteur Audrey Ferrand,
Nous vous remercions pour avoir relu ce travail, pour votre œil expert en détection des
doubles espaces et pour m’avoir montré comment utilisé Adobe Acrobat Pro, à un moment
plus que critique mais surtout pour vos conseils précieux et pertinents de chercheur
expérimenté, et pour votre bonne humeur.
A Madame le Professeur Marie-Bernadette Delisle, Professeur des Universités,
Praticien Hospitalier, Chef du Service d’Anatomie Pathologique de l’Hôpital Rangueil,
Nous vous remercions pour nous avoir permis de réaliser ce projet en collaboration avec
votre service, pour vos conseils et la confiance que vous nous avez accordée, sans lesquels ce
travail n’aurait pu avoir lieu et sommes particulièrement déçue que d’autres obligations vous
aient empêchée de faire partie de ce jury.
REMERCIEMENTS
Au Dr Julien Palasse, Praticien Hospitalier attaché dans le service d’Anatomie
Pathologique du Centre Hospitalo-Universitaire de Limoges et pathologiste dans le
laboratoire de la clinique Saint-Jean de Toulouse, pour avoir relu toutes les lames (le travail
final ne représentant qu’un dixième de tout ce qui a été réalisé, merci pour ces soirées et
weekends passés au microscope du laboratoire), pour son expertise en anatomo-pathologie
digestive mais avant tout pour son soutien sans faille dans les moments les plus difficiles, et
son amitié fidèle, pour avoir été la voix de la raison lorsque des projets insensés germaient
dans ma petite tête ou au contraire de m’avoir encouragée lorsqu’ils n’étaient pas si
incongrus, en résumé d’avoir été mes yeux, mon épaule, ma raison et bien plus, comme par
exemple ma béquille après ce fâcheux accident (causé par « le sauvetage d’un baleineau
échoué sur les rivages des Mascareignes », d’après la version romancée de Sandrine) et enfin
d’avoir toujours été présent.
A Claudine Bertrand pour son amitié, son soutien, ses conseils en matière d’assortiment
des couleurs (je ne mélangerai plus le rouge aloha au rouge fushia), sa compagnie pendant
mes pauses cigarettes (qui n’auront plus lieu puisque j’arrête de fumer… après la
soutenance…sauf en cas de remarques assassines du jury), les inoubliables moments à
Cadaquès, Marciac et Hossgor, autant que pour ces conseils, son expérience et son aide
concernant les analyses immunohistochimiques. C’est pourquoi, je lui adresse cette citation
de Bergson, « Le temps réel échappe aux mathématiques. Son essence étant de passer, aucune
de ses parties n'est encore là quand une autre se présente.», qui fait écho à sa tout aussi
fameuse phrase « Un certain temps ! » qui m’a accompagnée pendant ces quatre années de
thèse. Je lui promets, par ailleurs, que si je dois faire une troisième thèse, le sujet portera sur
les tardigrades ou plus sérieusement, sur la faune encore méconnue de la colline de Rangueil,
parmi laquelle nous avons déjà répertorié plusieurs espèces rares dont le Colubris
Rangueilus et l’Ursus Volans, malheureusement, souvent, respectivement, confondus avec le
papillon-colibri et le bourdon, ces diagnostics différentiels nécessitant un œil d’entomologiste
avisé…
A Aline Chauvel pour m’avoir encadrée à mes tout débuts dans le laboratoire et
enseignée la culture, les techniques de biologie moléculaire et de biochimie, avec patience,
mais surtout pour ne pas avoir tenu un cahier de mes meilleures boulettes, comme
ensemencer des plaques de culture sans cellules, ou envoyer, dans un mouvement ample et
gracieux, l’eppendorf contenant le contrôle de la manip après l’avoir malencontreusement
accroché à mes gants en le refermant, dans une faille spatio-temporelle. Je dois avouer que
secrètement, j’espérais que l’une d’elles serait à l’origine d’une découverte fortuite et
extraordinaire, à l’instar de la découverte de la pénicilline…Mes rêves de prix Nobel n’ont
malheureusement pas été exhaucés, mais je la remercie d’avoir continué à croire en moi et
tenter la fabuleuse aventure de la progastrine et des ovaires de souris, en m’aidant à réaliser
l’extraction des ovocytes, à l’aide de la très poétique technique du « toothpaste tube ».Un
grand merci aussi pour son aide de dernière minute sur le mise en page de ce manuscrit, un
peu irrégulière avant son passage.
A Nicolas Fénié qui n’a ni joué le jeu de la hiérarchie ni montré le respect qu’il devait à
son ainée en refusant de me faire mes cafés, de me les apporter et de les touiller, j’adresse
des non remerciements. D’ailleurs, tous les bons points qu’il a reçus mais qui, soit dit en
passant, pour la plupart, n’étaient pas mérités, seront confisqués jusqu’à ce qu’il change son
attidude irrévérencieuse.
A tous les membres de l’ex-unité INSERM U858, pour m’avoir accueillie, il y a 5 ans
déjà…
Aux membres des autres unités du 4
èmeétage de l’ex-I2MR, et tout particulièrement, à
Bernard Masri pour son humour et ses conseils de « motard ».
A toute l’équipe d’Oncomip, qui a récupéré un zombie tous les matins, après mes nuits
blanches passées à écrire cette thèse, pour ses encouragements et pour m’avoir changé les
idées à travers des discussions enflammées
métaphysico-philosophico-politico-DSKo-sociologiques (qui ne nous ont pas empêchés de travailler, pour rassurer Eric Bauvin).
Je
remercie également tout particulièrement Attika, qui s’est presque « vendue » pour m’obtenir
une piste de post-internat.
A tous mes amis, pour leur soutien, en particuliers à Sandrine, pour m’avoir inventé
cette histoire héroïque de baleineau et m’avoir supporté en colocation toutes ces années, à
Kim qui a essayé de me faire dormir la tête en position Nord-Est et les pieds non orientés
vers la porte, c’est-à-dire en travers du lit et toute tordue, pour attirer les bonnes ondes Feng
Shui, mais qui a été une oreille attentive et toujours de bon conseil pour mes choix de
carrière, de vie et de techniques de maturation de jambon italien, à Florence, encore une
ana-path (au passage, félicatations pour le petit Raphael), pour m’avoir toujours gardé une
place à l’internat de Rangueil à midi quand je montais au laboratoire, mais surtout pour les
discussions et les moments agréables partagés pendant ces déjeûners, à Raphael et Sylvain,
(et oui,vous êtes en couple même pour les remerciements) pour avoir bien voulu descendre du
bus une station trop tôt et finir le chemin à pied par -20°C car mon russe était trop rouillé
pour expliquer à la gentille dame que notre arrêt était le suivant, à Téma, avec qui j’ai pu
partager ma passion de la littérature et dont l’enthousiasme m’a donné l’envie de lire
Faulkner, à Benoît, qui est malheureusement reparti au Cambodge, pour ces soirées autour
d’une, voire plusieurs « femmes fatales » à l’Elephant Bar de Phnom Penh, pour avoir assisté
à mon concert d’Ehru désastreux, et m’avoir appris tout ce que je sais (c’est-à-dire pas
grand-chose) sur la culture du Plasmodium en condition extrême (sans hôte, sans gant, sans
pipette),et à Dushyant, pour être venu assister à la soutenance, même si je ne suis pas sûre
qu’il arrive à l’heure, surtout avec Kim qui est toujours en retard.
A ma famille, pour leur soutien et leur amour.
« …la rencontre fortuite sur une table de dissection
d’une machine à coudre et d’un parapluie. »
RESUME
Les polypes hyperplasiques (PH) constituent les lésions colorectales les plus fréquentes. Ces lésions ont pendant longtemps été considérées comme des lésions sans potentiel malin et le suivi coloscopique des patients ayant développé des PH n’est pas recommandé. Récemment, plusieurs études suggèrent qu’une partie de ces polypes pourrait constituer des précurseurs de certains cancers colorectaux sporadiques. Cependant, aucun biomarqueur permettant de détecter les PH à risque d’évolution maligne n’a pu être identifié. La progastrine est une prohormone impliquée dans la carcinogénèse colique. Dans un premier temps, nous avons étudié l’expression de la progastrine dans des PH provenant de 74 patients sans antécédent de pathologie colorectale et avons mis en évidence une surexpression de la prohormone dans 40% des cas. Dans un sous-échantillon composé de 39 patients pour lesquels un suivi coloscopique avait été réalisé, 41% des patients ayant présenté un PH ont développé des polypes adénomateux métachrones. Nous avons pu montrer une association significative entre l’expression de la progastrine et la survie sans néoplasme (p=0.001). En effet, la survie sans néoplasme à 5 ans était de 38% chez les patients présentant une forte surexpression de progastrine alors qu’elle était de 100% chez les patients avec une faible expression. Par ailleurs, nous avons démontré qu’un test prédictif composite basé sur le marquage de la progastrine et l’âge des patients permettraient de prédire la survenue d’un événement néoplasique métachrone chez les patients ayant développé un PH avec une sensibilité de 100% (Intervalle de confiance à 95% : 79%-100%) et une spécificité de 74% (51%-90%). La progastrine peut activer plusieurs voies pro-oncogéniques dans les cancers colorectaux. Certaines d'entre elles, en particulier les voies JAK/STAT ou ERK n’ont pas encore été explorées dans les polypes hyperplasiques. Afin de mieux caractériser les polypes exprimant la progastrine, nous avons, dans un second temps, étudié l’activation de STAT3 et ERK dans un deuxième échantillon de 49 polypes hyperplasiques. Le pourcentage de cellules marquées par des anticorps spécifiques anti-progastrine, anti-phospho-STAT3 et anti-phospho-ERK étaient en moyenne de 31% (écart-type : 35), 10% (23) et 34% (30%), respectivement et étaient significativement augmentée dans les PH par rapport aux tissus normaux contrôles pour la progastrine et P-ERK (p=0,0008 et 0.0003). De plus, l’augmentation progressive de l’expression de ces deux marqueurs entre le tissu normal, les PH, les adénomes de dysplasie légère, modérée et sévère étaient significative (p<0.0001 pour la progastrine et p=0.001 pour P-ERK) et suggèrent une activation de ces deux facteurs dans les PH intermédiaire entre le colon normal et les adénomes. Nous avons également montré, dans ces lésions, que l’expression de la progastrine était corrélée à celle de P-ERK (p=0,0184). L’ensemble de ces résultats suggèrent que la progastrine pourrait être associée à un sous-type de PH à risque d’évolution maligne et permettre de l’identifier.
ABREVIATIONS
ADN
Acide désoxyribonucléique
APC
Adenomatous polyposis coli
ARN
Acide ribonucléique
AT
Adénomes tubuleux
ATV
Adénomes tubulo-villeux
AV
Adénomes villeux
BAX
BCL-2-associated X protein
Bcl-2
B-cell lymphoma 2
bcl-xL
B-cell lymphoma-extra large
BER
Base excision repair
BRCA1
Breast cancer-associated gene 1
CCK2
Cholecystokinin 2
CCK2R
Cholecystokinin 2 receptor
CCR
Cancer colorectal
CD44
Cluster of Differentiation 44
cdc42
Cell division control protein 42
CDK4
Cyclin-dependent kinase 4
CDKN2A
Cyclin-dependent kinase inhibitor 2A
CDX2
Caudal-related homeobox
c-fos
Cellular fos
CHK1
Checkpoint kinase 1
CIMP
Cpg island methylator phenotype
CIN
Chromosomal instability
c-myc
Cellular myc
COX-2
Cycloexygenase 2
CTNNB1
Catenin beta1
CYP1a
Cytochrome P4501A
CYP1A2
Cytochrome P-450
DCC
Deleted in colorectal carcinoma
EB1
End binding 1
EGF
Epidermal growth factor
EGFR
Epidermal growth factor receptor
ERK
Extracellular signal-regulated kinases
ES
Embryonic stem,
FAK
Focal adhesion kinase
FCA
Foyer de cryptes aberrantes
G17-NH2
Gastrine amidée 17
G34-NH2
Gastrine amidée 34
GTPase
Guanosine triphosphatase
HAS
Haute Autorité de Santé
HE
Hématoxiline-éosine
hMLH1
Human Mutl-homolog 1
ICAT
Inhibitor of beta-catenin
IFNβ
Interferon beta
IκB
Inhibitor of nfκb
JAK
Janus kinase
LEF
Lymphoid enhancer-binding factor 1
LGR5
Leucine-rich repeat-containing G-protein coupled receptor 5
mcl-1
Myeloid cell leukemia 1
MCR
Mutation cluster region
MEK
Mitogen-activated protein kinase/extracellular signal-regulated kinase
kinaseMGMT
O-6-methylguanine-DNA- Methyltransferase
MINT1
Munc18-1-interacting protein 1
MINT2
Munc18-1-interacting protein 2
MLH3
Mutl-homolog 3
MMP2
Matrix metalloproteinase-2
MMR
Mismatch repair
MPHP
Mucin poor hyperplastic polyps
MSH3
Muts-homolog 6
MSH6
Muts-homolog 6
MSI
Microsatellite instability
MSI-H
Microsatellite instable high
MSI-L
Microsatellite instable low
MST1
Mammalian sterile 20-like 1
MTHFR
Méthylène-tétra-hydro-folate-réductase
MUTYH
Muty homolog
MVHP
Microvesicular hyperplastic polyps
NAT
N-acétyl-transférases
NFκB
Nuclear factor kappa-light-chain-enhancer of activated B cells
OMS
Organisation mondiale de la santé
PAF
Polypose adénomateuse familiale
PAM
Polypose associée à la mutation MUTYH
PH
Polype hyperplasique
PIP2
Phosphoinositides
PTEN
Phosphatase and tensin homolog
RASSF1
Ras association domain family protein 1
RASSF2
Ras association domain family protein 2
RASSF5
Ras association domain family protein 5
RCPG
Récepteurs couplés aux protéines G
ROS
Reactive oxygen species
SH2
Src homology 2
siARN
Small interfering ARN
SOS
Son of sevenless
SSA
Sessile serrated adenoma
STAT
Signal Transducers and Activators of Transcription
TCF
Transcription factor 4
TCF-4
Transcription factor 4
TGFβ
Transforming growth factor β
TGFβR2
Transforming growth factor β receptor 2
TNM
Tumor node metastasis
TSA
Traditional serrated adenoma
TYK
Tyrosine kinase
VEGF
Vascular endothelial growth factor
ZO-1
Zonula occludens-1
Sommaire
INTRODUCTION BIBLIOGRAPHIQUE ... 4
I-Epidémiologie du Cancer colorectal ... 5
II-Etiologie du Cancer colorectal ... 5
II-1-Régime et mode de vie ... 5
II-2-Inflammation chronique ... 5
II-3-Prédisposition génétique ... 6
II-3-1-Syndromes héréditaires ... 6
II-3-2-Les polymorphismes à forte pénétrance et polymorphismes nucléotidiques ... 8
III-Les différentes tumeurs épithéliales colorectales: la classification histo-pathologique ... 8
III -1- Les lésions prénéoplasiques: les foyers de cryptes aberrantes... 9
III -1-1-Les foyers de crypte aberrantes dysplasiques ... 9
III -1-2-Les foyers de cryptes aberrantes hyperplasiques ... 10
III-2-Les lésions précurseurs... 10
III-2-1-Les adénomes ... 11
III-2-2-Les tumeurs festonnées ... 12
III-3-Les adénocarcinomes colorectaux ... 14
IV-Les différentes voies de carcinogénèse colorectale : Vers une classification moléculaire ... 16
IV -1-Le modèle multi-étape de la carcinogénèse colique ... 16
IV -2-Une classification moléculaire basée sur les mécanismes moléculaires permettant l’accumulation des mutations génétiques ... 17
IV -2-1-L’instabilité chromosomique :... 18
IV -2-2-L’instabilité microsatellitaire ... 24
IV -2-3-l’Hyperméthylation des îlots CpG, CIMP (CpG island methylator phenotype) ... 28
V- Corrélation entre les différents types histologiques d’adénocarcinomes et les mécanismes moléculaires initiateurs ... 29
V-1-La voie classique ... 30
V-2-Les voies des tumeurs festonnées ... 30
V-2-1-La voie des tumeurs festonnées sessiles associées à une hyperméthylation forte de l’ADN et une instabilité microsatellitaire forte : groupe 1 ... 31
V-2-2-La voie des tumeurs festonnées sessiles associées à une hyperméthylation forte de l’ADN et microsatellite stable (ou à faible instabilité microsatellitaire) : groupe 2 ... 31
V-3-Le syndrome de Lynch : groupe 5 ... 32
VI -Les polypes hyperplasiques ... 32
VI -1-Epidémiologie et facteurs de risque des polypes hyperplasiques ... 32
VI -2-Caractéristiques histologiques et sous-types de polypes hyperplasiques ... 33
VI -3-Caractéristiques moléculaires des polypes hyperplasiques ... 34
VI -4-Syndromes associés à la présence de multiples PH ... 36
VI -5-Place des polypes hyperplasiques dans la carcinogénèse colique ... 37
VI -6- Valeur pronostic des PH ... 38
VI -7- Surveillance après exérèse d’un polype hyperplasique ... 39
RESULTATS EXPERIMENTAUX ... 41
ARTICLE 1 ET BREVET ... 42
INTRODUCTION : La progastrine ... 43
I-Synthèse et sécrétion de la progastrine dans les conditions physiologiques ... 43
II- Synthèse et sécrétion de la progastrine dans les tumeurs colorectales ... 44
III- La progastrine, un facteur de croissance des cellules épithéliales coliques ... 45
IV- Effets anti-apoptotiques de la progastrine sur les cellules épithéliales coliques ... 46
V- Effet de la progastrine sur l’adhésion et la migration des cellules épithéliales coliques ... 46
VI- Les récepteurs /protéines de liaison de la progastrine ... 47
OBJECTIFS ... 49
PATIENTS ET METHODES ... 50
I-Schéma de l’étude ... 50
II-Taille de l’échantillon, patients et recueil de données ... 50
III-Immunohistochimie ... 52
IV-Analyses statistiques ... 52
ARTICLE………54
ARTICLE 2... 77
INTRODUCTION ... 78
I-La voie WNT/β-caténine /TCF4 ... 78
I-1-L’activation de la voie WNT/β-caténine /TCF4 et ces cibles ... 78
I-2-La progastrine active la voie WNT/β-caténine /TCF4 ... 79
I--3-La voie WNT/β-caténine /TCF4 dans les tumeurs colorectales ... 79
II-La voie RAS/RAF/MEK/ERK ... 80
II-1-L’activation et les cibles de ERK... 80
II-2-La progastrine active la voie ERK ... 80
II-3-La voie ERK dans les cancers colorectaux ... 81
III-La voie JAK/STAT ... 81
III-1-Activation et cibles de la voie JAK/STAT ... 81
III-2-La progastrine active la voie JAK2/STAT3 ... 82
III-4-la voie Ras/Raf/MEK/ERK permettrait une activation maximale de la voie JAK/STAT ... 84
IV-1-Activation et cibles de la voie PI3K/AKT. ... 84
IV-2-La progastrine active la voie PI3K/AKT ... 85
IV-3-PI3K/AKT dans les cancers colorectaux ... 85
OBJECTIFS ... 87
PATIENTS ET METHODES ... 88
I-Schéma de l’étude ... 88
II-Taille de l’échantillon, patients et recueil de données ... 88
III-Immunohistochimie ... 88
IV-Analyses statistiques ... 89
ARTICLE………90
CONCLUSION ET DISCUSSION ... 113
ANNEXES : ... 119
A gastrin precursor, gastrin-gly, upregulates VEGF expression in colonic epithelial cells through an HIF-1-independent mechanism. ………..………. 120
A 'DNA replication' signature of progression and negative outcome in colorectal cancer……… ……….….121
Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: a rare clinicopathologic entity with poor prognosis………..122
Distribution, function and prognostic value of cytotoxic T lymphocytes in follicular lymphoma: a 3-D tissue imaging study………..…..123
REFERENCES BIBLIOGRAPHIQUES ... 125
INTRODUCTION
BIBLIOGRAPHIQUE
LES TUMEURS
COLORECTALES
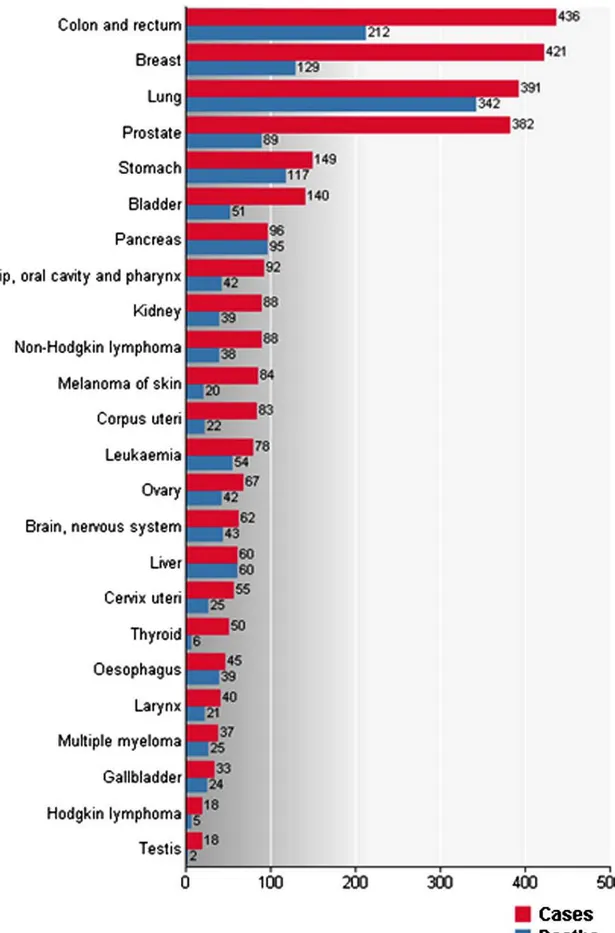
Figure 1 : Estimation de l’incidence (en milliers d’individus) des cancers et de leur mortalité en
Europe, en 2008 (Ferlay, Parkin et al. 2010)
I-‐Epidémiologie du Cancer colorectal
Le cancer colorectal (CCR) constitue le 3
èmecancer le plus fréquent chez l’homme et le
second chez la femme dans le monde en 2008. En termes de mortalité, il représente la 4
èmecause de décès par cancer (Ferlay, Shin et al. 2010). Cependant, près de 60% des cas sont
détectés dans les pays développés. Ainsi, en Europe, le cancer colorectal est le cancer le plus
fréquent et la 2
èmecause de mortalité par cancer (Ferlay, Parkin et al. 2010) (figure 1).
II-‐Etiologie du Cancer colorectal
Le cancer colorectal résulte de l’interaction complexe entre susceptibilité héréditaire et
facteurs environnementaux (Potter 1999).
II-‐1-‐Régime et mode de vie
La plus forte incidence du CCR dans les pays occidentaux pourrait s’expliquer en partie
par le mode de vie sédentaire et l’alimentation occidentale, calorique et riche en graisse
animale (Potter 1999). Plusieurs études épidémiologiques ont montré une association positive
avec l’abus d’alcool (Cho, Smith-Warner et al. 2004), le tabagisme (Botteri, Iodice et al.
2008) et l’obésité (Calle, Rodriguez et al. 2003). En revanche, l’activité physique (Wolin, Yan
et al. 2009) et la prise d’aspirine au long cours seraient des facteurs protecteurs. L’action
protectrice de l’aspirine serait expliquée par l’inhibition de COX-2, impliquée dans les
phénomènes inflammatoires (voir, ci-dessous, le chapitre inflammation chronique) et
surexprimé dans les cancers colorectaux (Rothwell, Wilson et al. 2010). Les régimes riches en
fibres ou pauvres en graisse pourraient aussi avoir un rôle protecteur (Potter 1999), bien que
certaines études d’intervention récentes n’aient pu mettre en évidence une réduction du risque
de CCR associée à ces régimes (Park, Hunter et al. 2005; Beresford, Johnson et al. 2006).
II-‐2-‐Inflammation chronique
Les maladies inflammatoires chroniques de l’intestin sont associées à un risque accru de
CCR, en particulier après 8 à 10 ans d’évolution. Parmi elles, la colite ulcéreuse est
considérée comme un état pré-néoplasique de CCR et la maladie de Crohn présente un risque
augmenté à la fois de cancer du grêle et du gros intestin (Gillen, Walmsley et al. 1994).
II-‐3-‐Prédisposition génétique
II-‐3-‐1-‐Syndromes héréditaires
La polypose adénomateuse familiale
La polypose adénomateuse familiale (PAF) est un syndrome autosomique dominant rare,
causé par une mutation germinale du gène suppresseur de tumeur, APC (adenomatous
polyposis coli), sur le bras long du chromosome 5q (5q21-22) (Groden, Thliveris et al. 1991).
Cette mutation conduit à la formation d’une protéine tronquée. La PAF est caractérisée par le
développement à un âge précoce, entre 10 et 20 ans, de multiples adénomes colorectaux,
allant de quelques polypes à plusieurs milliers. Ces adénomes non traités évoluent
naturellement vers des adénocarcinomes avec un âge moyen d’apparition de 40 ans (Jarvinen
1985).
Le syndrome de Lynch
Le syndrome de Lynch est un syndrome autosomique dominant. Il se caractérise par une
histoire familiale ou personnelle de CCR avant 50 ans, mais également de cancer de
l’endomètre, des voies urinaires, de l’estomac et des voies biliaires (Lynch, Lynch et al.
2009). Les adénocarcinomes colorectaux présentent une instabilité microsatellitaire, due à une
mutation germinale dans la famille de gènes impliqués dans le système de réparation des
mésappariements de l’ADN (Mismatch repair, MMR), principalement hMLH1 et hMSH2
(Leach, Nicolaides et al. 1993; Papadopoulos, Nicolaides et al. 1994). Le système MMR
permet de réparer les erreurs d’appariement produites par la DNA polymérase au niveau des
répétitions nucléotidiques (de 1 à 5 nucléotides) lors de la réplication (Kolodner 1995). La
perte du système MMR entraine donc une accumulation de petites délétions et insertions qui
peuvent survenir au niveau de gènes pro-oncogéniques ou suppresseurs de tumeur (Boland
and Goel 2010). Les cibles du système MMR sont détaillées dans le chapitre « gènes associés
à l’instabilité microsatellitaire ».
Le syndrome de Li-‐Fraumeni
Il s’agit d’un syndrome autosomique dominant qui se traduit par la présence de multiples
cancers chez l’enfant et le jeune adulte, avec une prédominance de sarcomes des tissus mous,
ostéosarcomes et cancer du sein, ainsi qu’une augmentation de l’incidence des tumeurs
cérébrales, leucémies et carcinomes adrénocorticaux. Les tumeurs du tractus digestif,
principalement CCR, représentent environ 10% des tumeurs observées dans les familles
Li-Fraumeni (Nichols, Malkin et al. 2001). L’âge moyen de survenue des CCR est de 33 ans et le
risque relatif est d’environ 2 par rapport à la population générale (Ruijs, Verhoef et al. 2010).
Ce syndrome est causé par une mutation germinale sur le gène suppresseur de tumeur TP53
dans 70% des cas (Li, Fraumeni et al. 1988)
La polypose associée aux mutations de MUTYH (PAM)
Il s’agit d’un syndrome autosomique récessif caractérisé par la présence d’un nombre
variable de polypes colorectaux (>10) de phénotypes histologiques différents (principalement
adénomes classiques mais également festonnés et polypes hyperplasiques), ayant tendance à
évoluer vers des carcinomes. Ces polypes sont localisés dans tout le colon mais également
dans l’intestin grêle (Lubbe, Di Bernardo et al. 2009). Ce syndrome est causé par une
mutation germinale biallélique au niveau du gène MUTYH. La protéine MUTYH est une
ADN
glycosylase du système de réparation par excision de bases (BER, Base Excision
Repair) qui joue un rôle majeur dans la réparation des lésions oxydatives de l’ADN. (Cleary,
Cotterchio et al. 2009). Une perte de fonction de MUTYH entraine donc un défaut de
réparation des lésions oxydatives de l’ADN, et une accumulation de transversions G : C vers
T : A dans les gènes suppresseurs de tumeurs et oncogènes. Ce type de transversions au
niveau des sites GAA du gène APC conduisent, notamment, à une sélection du codon stop
TAA. De même, dans l’étude de Boparai et al, 70% des polypes hyperplasiques (PH) et des
adénomes festonnés sessiles (sessile serrated adenoma, SSA) des patients avec une polypose
associée aux mutations de MUTYH présentaient des transversions au niveau du codon 12 de
KRAS comparés à 17% pour les PH et SSA sporadiques (Boparai, Dekker et al. 2008). Deux
transversions sont possibles au niveau de ce colon (GGTà TGT ou GTT) et conduisent à
l’inactivation de l’activité GTPase de Ras. Le risque associé aux mutations MUTYH et la
prévalence des porteurs bialléliques sont encore inconnus.
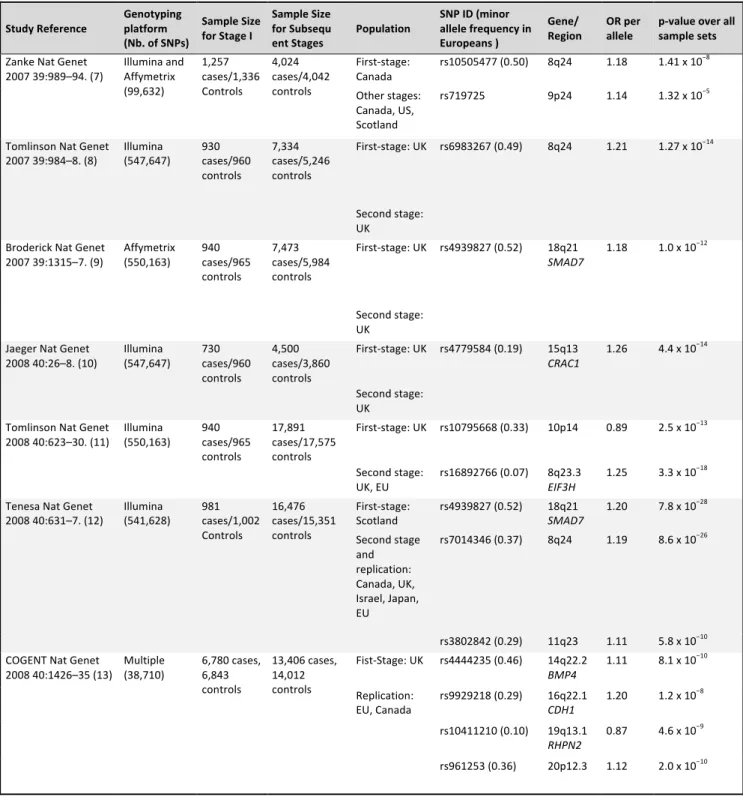
Study Reference Genotyping platform (Nb. of SNPs)
Sample Size for Stage I
Sample Size for Subsequ
ent Stages Population
SNP ID (minor allele frequency in Europeans )
Gene/
Region OR per allele p-‐value over all sample sets Zanke Nat Genet
2007 39:989–94. (7) Illumina and Affymetrix (99,632) 1,257 cases/1,336 Controls 4,024 cases/4,042 controls First-‐stage: Canada rs10505477 (0.50) 8q24 1.18 1.41 x 10 −8 Other stages: Canada, US, Scotland rs719725 9p24 1.14 1.32 x 10−5
Tomlinson Nat Genet 2007 39:984–8. (8) Illumina (547,647) 930 cases/960 controls 7,334 cases/5,246 controls First-‐stage: UK rs6983267 (0.49) 8q24 1.21 1.27 x 10−14 Second stage: UK
Broderick Nat Genet 2007 39:1315–7. (9) Affymetrix (550,163) 940 cases/965 controls 7,473 cases/5,984 controls First-‐stage: UK rs4939827 (0.52) 18q21 SMAD7 1.18 1.0 x 10 −12 Second stage: UK
Jaeger Nat Genet 2008 40:26–8. (10) Illumina (547,647) 730 cases/960 controls 4,500 cases/3,860 controls First-‐stage: UK rs4779584 (0.19) 15q13 CRAC1 1.26 4.4 x 10 −14 Second stage: UK
Tomlinson Nat Genet 2008 40:623–30. (11) Illumina (550,163) 940 cases/965 controls 17,891 cases/17,575 controls First-‐stage: UK rs10795668 (0.33) 10p14 0.89 2.5 x 10−13 Second stage:
UK, EU rs16892766 (0.07) 8q23.3 EIF3H 1.25 3.3 x 10
−18
Tenesa Nat Genet 2008 40:631–7. (12) Illumina (541,628) 981 cases/1,002 Controls 16,476 cases/15,351 controls First-‐stage: Scotland rs4939827 (0.52) 18q21 SMAD7 1.20 7.8 x 10 −28 Second stage and replication: Canada, UK, Israel, Japan, EU rs7014346 (0.37) 8q24 1.19 8.6 x 10−26 rs3802842 (0.29) 11q23 1.11 5.8 x 10−10 COGENT Nat Genet
2008 40:1426–35 (13) Multiple (38,710) 6,780 cases, 6,843 controls 13,406 cases, 14,012 controls Fist-‐Stage: UK rs4444235 (0.46) 14q22.2 BMP4 1.11 8.1 x 10 −10 Replication: EU, Canada rs9929218 (0.29) 16q22.1 CDH1 1.20 1.2 x 10 −8 rs10411210 (0.10) 19q13.1 RHPN2 0.87 4.6 x 10 −9 rs961253 (0.36) 20p12.3 1.12 2.0 x 10−10
Tableau 1. Etudes de GWAS publiées au 1er
janvier 2009 et loci de susceptibilité au cancer colorectal identifiés (Le Marchand 2009)
BRCA1
Les mutations germinales du gène BRCA1 situé sur le chromosome 17q sont responsables
d’une prédisposition héréditaire aux cancers du sein et de l’ovaire. Les familles BRCA1
présenteraient également un risque significativement augmenté de CCR, avec un risque relatif
d’environ 4 (Ford, Easton et al. 1994).
II-‐3-‐2-‐Les polymorphismes à forte pénétrance et polymorphismes
nucléotidiques
Les amines aromatiques hétérocycliques (contenues notamment dans la viande cuite à
haute température) pourraient constituer des carcinogènes colorectaux. Des polymorphismes
d’enzymes impliquées dans les voies métaboliques de ces amines, à savoir, la
méthylène-tétra-hydro-folate-réductase (MTHFR), les N-acétyl-transférases (NAT1, NAT2), les
glutathione-S-transférases, l’aldéhyde déshydrogénase et les enzymes du cytochrome P-450
(CYP1A2), pourraient donc expliquer les susceptibilités et prédispositions individuelles parmi
des populations soumises aux mêmes expositions (Rafter and Glinghammar 1998; Houlston
and Tomlinson 2001).
De même certains polymorphismes nucléotidiques (single nucleotide polymorphism) à
faible pénétrance ont été identifiés plus récemment par les études génomiques (Genome Wide
Association Studies) et restent à être confirmés comme facteurs de prédisposition (Le
Marchand 2009) (Tableau 1).
III-‐Les différentes tumeurs épithéliales colorectales: la classification histo-‐
pathologique
La classification de l’OMS distingue plusieurs types histologiques de tumeurs épithéliales
colorectales (Bosman, F. et al. 2010) :
Les lésions prénéoplasiques comprennent:
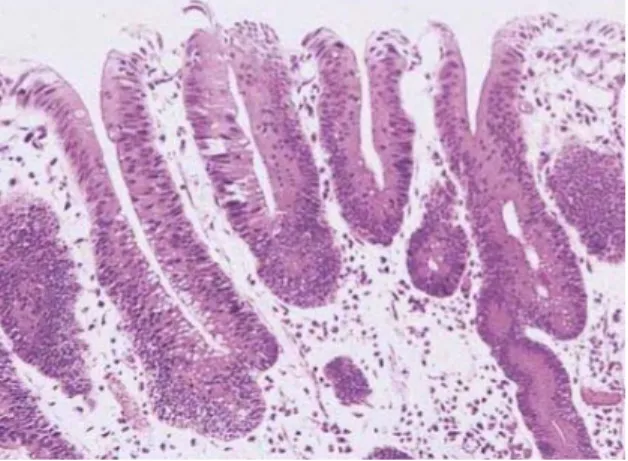
- Les foyers de cryptes aberrantes dysplasiques (Figure 2) et hyperplasiques (Figure 3)
Les lésions précurseurs incluent
-Les tumeurs festonnées : polypes hyperplasiques, adénomes festonnés sessiles (Figure 5),
adénomes festonnés traditionnels (Figure 7).
Les adénocarcinomes (Figure 8)
Parmi les adénocarcinomes, plusieurs variants plus rares sont distingués
Adénocarcinomes mucineux (Figure 9)
Carcinomes à cellules en bague à chaton (Figure 10)
Carcinomes médullaires (Figure 11)
Carcinomes festonnés (Figure 12)
Le diagnostic et la distinction entre ces tumeurs, au pronostic différent, reposent, dans la
plupart des cas, uniquement sur des critères histopathologiques.
III -‐1-‐ Les lésions prénéoplasiques: les foyers de cryptes aberrantes
Ils constituent les précurseurs morphologiques les plus précoces des néoplasies
épithéliales. Les foyers de cryptes aberrantes (FCA) sont composés de larges cryptes
recouvertes d’un épithélium épaissi pauvre en mucine. Il existe 2 principaux types de foyers
de cryptes aberrantes (Cheng and Lai 2003; Suehiro and Hinoda 2008) .
III -‐1-‐1-‐Les foyers de crypte aberrantes dysplasiques
Les foyers de cryptes aberrantes dysplasiques sont fréquents chez les patients présentant
une polypose adénomateuse familiale. Chez ces patients, ils sont également caractérisés par
une hyperprolifération épithéliale de la partie supérieure des cryptes, l’absence de méthylation
et de mutation KRAS, et la présence de mutations APC. L’hypothèse d’une séquence -foyers
de cryptes aberrantes dysplasiques / adénomes / carcinomes- a ainsi été proposée pour
caractériser la voie de carcinogénèse classique des CCR (Nascimbeni, Villanacci et al. 1999).
Cependant, alors que les foyers de cryptes aberrantes dysplasiques sont très fréquents chez les
patients atteints de polypose familiale adénomateuse, ils sont plus rares dans les formes
sporadiques, les foyers de cryptes aberrantes dysplasiques représentant environ 5% des foyers
de cryptes aberrantes sporadiques (Alrawi, Schiff et al. 2006). Par ailleurs, ces lésions
dysplasiques sporadiques présentent, moins souvent des mutations APC et plus fréquemment
des mutations KRAS et une hyperméthylation, comparées aux foyers de cryptes aberrantes
dysplasiques des polyposes adénomateuses familiales (Les mutations APC restent, cependant
plus fréquentes que dans les FCA hyperplasiques). Ces résultats suggèrent que la grande
majorité des foyers de cryptes aberrantes sporadiques (notamment hyperplasiques et une
fraction des dysplasiques) n’évolue pas en lésions néoplasiques.
(Takayama, Katsuki et al. 1998)
III -‐1-‐2-‐Les foyers de cryptes aberrantes hyperplasiques
Les foyers de cryptes aberrantes hyperplasiques possèdent certaines caractéristiques
morphologiques des polypes hyperplasiques et présentent fréquemment des mutations du
proto-oncogène KRAS. Ils pourraient constituer des précurseurs précoces d’une
sous-population de cancers colorectaux dans laquelle la mutation KRAS précéderait la mutation
APC (Takayama, Ohi et al. 2001). Par ailleurs, certains FCA hyperplasiques, festonnés,
présentent plus fréquemment des mutations BRAF et moins souvent des mutations KRAS que
les FCA hyperplasiques non festonnés (Rosenberg, Yang et al. 2007). Ils ont également été
associés à des méthylations des îlots CpG et pourraient donc être des précurseurs des
carcinomes festonnées à travers une séquence FCA hyperplasique festonnée/adénome
festonné/adénocarcinome (Chan, Broaddus et al. 2002).
(Takayama, Katsuki et al. 1998)
Figure 2. Foyer de cryptes aberrantes
dysplasiques, présentant une perte de polarité, un hyperchromatisme des noyaux et une stratification nucléaire. Grossissement x20. Coloration HE.
Figure 3. Foyer de cryptes
aberrantes hyperplasiques, présentant une lumière festonnée. Grossissement x20. Coloration HE.
III-‐2-‐Les lésions précurseurs
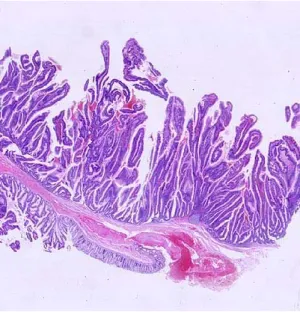
III-‐2-‐1-‐Les adénomes
Ces lésions sont définies par la présence d’une néoplasie intra-épithéliale et sont
caractérisées par une hypercellularité avec des noyaux volumineux et hyperchromatiques, une
stratification nucléaire et une perte de polarité. L’inactivation de la voie APC/β-caténine est
typique (Jen, Powell et al. 1994). Les adénomes sont classiquement considérés comme les
précurseurs des adénocarcinomes à microsatellite stable.
Les adénomes tubuleux (AT) peuvent être pédiculés ou plats. Au moins 80% des glandes
sont dysplasiques.
Les adénomes villeux (AV) sont typiquement sessiles (non pédiculés). Les projections
villeuses sont tapissées par des glandes dysplasiques sur plus de 80% du revêtement.
Figure 4. Petit adénome tubulaire
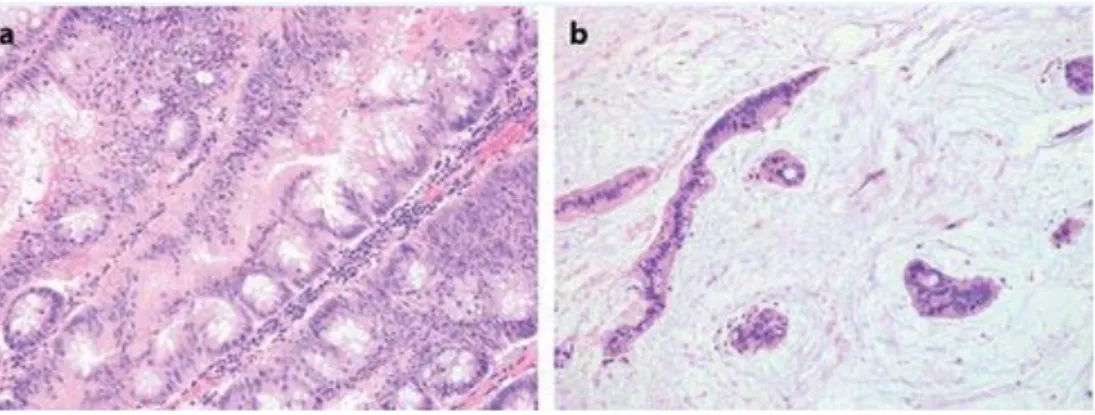
pédiculé du colon, présentant une prolifération et désorganisation glan-dulaire. Les cellules caliciformes sont moins nombreuses et les noyaux hyperchromatiques. Grossissement x4, coloration à l’hématoxyline-éosine (HE). (Source: Pathologie-Bilddatenbank)
Figure 5. Adénome villeux sessile du
colon, présentant de longues franges papillaires avec un axe conjonctif recouvert de glandes dysplasiques. Grossissement x4, coloration HE (Source: Pathologie-Bilddatenbank).
Les adénomes tubulo-villeux (ATV) présentent une architecture mixte, à la fois
tubuleuse et villeuse.
III-‐2-‐2-‐Les tumeurs festonnées
Elles font partie d’un groupe hétérogène de tumeurs dont les caractéristiques
histo-morphologiques ont été récemment précisées par Torlakovic (Torlakovic and Snover 1996;
Torlakovic, Skovlund et al. 2003).
Les polypes hyperplasiques sont des lésions prénéoplasiques. Ils font l’objet de cette
thèse et seront détaillés dans le chapitre 2,
Les adénomes festonnés sont caractérisés par une architecture en dent de scie (festonnée)
sur la totalité de la longueur des glandes. Celles-ci sont dysplasiques au niveau de leur partie
supérieure. On distingue les trois types histologiques décrits ci-dessous:
Les adénomes festonnées sessiles (SSA, sessile serrated adenoma) : Il s’agit de
lésions larges généralement supérieures à 5 mm, de localisation proximale qui seraient
présentes chez près de 9% des patients ayant subi une coloscopie (Bosman, F. et al. 2010).
Ces lésions sont caractérisées par une élongation et dilatation des glandes avec d’importants
festons, présents plutôt à la base des glandes qu’en superficie, comme pour les polypes
hyperplasiques. Les SSA non compliqués ne présentent pas de dysplasie, mais lors de la
progression carcinomateuse acquièrent des lésions de dysplasie semblables aux adénomes
classiques.
Figure 6. Adénome tubulo-villeux du
colon, présentant une architecture mixte avec des embranchements tubulaires (à gauche) et des franges villeuses (à droite). Grossissement x20. Coloration HE. (Source: John Hopkins medical school).
Figure 7. Adénome sessile festonné présentant typiquement (a) des anomalies architecturales avec
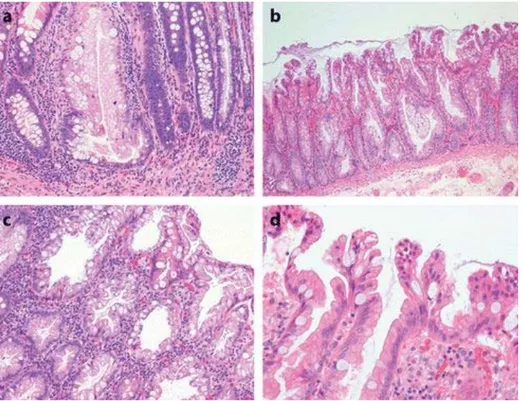
des cryptes en T ou L, (b) (c) des dilatations importantes des cryptes avec un aspect festonné marqué présent jusqu’à la base et (d) des cellules dans la partie supérieure des cryptes contenant des noyaux larges et désorganisés. Grossissement x20 (a et b) et x40 (c et d). Coloration HE. (Noffsinger 2009)
Les adénomes festonnés traditionnels (TSA, traditional serrated adenoma) : Ce sont
des lésions rares (<1% de tous les polypes) caractérisées par une architecture villeuse et
présentant des lésions cytologiques de type dysplasique. Ces anomalies sont à différencier des
dysplasies observées dans les adénomes classiques ou les SSA avec dysplasie.
Figure 6. Adénome festonné traditionnel présentant (a) des cryptes festonnées recouvertes de
cellules cylindriques élargies et nombreuses au cytoplasme très éosinophile avec (b) parfois une dysplasie de haut grade (Noffsinger 2009). Grossissement x40. Coloration HE.
III-‐3-‐Les adénocarcinomes colorectaux
La plupart des adénocarcinomes colorectaux sont d’architecture glandulaire. La taille et la
configuration des structures glandulaires sont variables et les cellules épithéliales sont souvent
larges et hautes dans les adénocarcinomes bien et moyennement différenciés. On distingue
plusieurs variants. Certains sont associés à des caractéristiques moléculaires spécifiques.
Les Adénocarcinomes mucineux
Ces tumeurs constituent une variante des adénocarcinomes, dans laquelle plus de 50% de
la lésion est constituée de mucine extracellulaire. Les nappes de mucines contiennent de
l’épithélium malin se présentant sous formes de structures glandulaires, de lambeaux ou de
cellules épithéliales isolées. Ce type histopathologique caractérise de nombreux carcinomes à
instabilité microsatellitaire forte (microsatellite instable high, MSI-H).
(Egashira, Yoshida et al. 2004)
Figure 8. Adénocarcinome moyennement
différencié du colon, présentant une prolifération glandulaire infiltrante.
Grossissement x4. Coloration HE.
Figure 9. Adénocarcinome mucineux bien
différencié (tubulaire), présentant de larges nappes de mucines. Grossissement x20. Coloration HE.
Carcinomes à cellules en bague à chaton
Les carcinomes à cellules en bague à chaton sont définis par la présence de plus de 50%
de cellules tumorales contenant de la mucine intra-cytoplasmique. Les cellules en bague à
chaton se caractérisent par une large vacuole de mucine repoussant le noyau en périphérie.
Certains carcinomes MSI-H sont de ce type.
(Egashira, Yoshida et al. 2004)
Carcinomes médullaires
Il s’agit d’un variant histopathologique rare caractérisé par des cellules contenant un
noyau vésiculaire avec un nucléole proéminent et un cytoplasme abondant. L’infiltration
lymphocytaire intra-épithéliale est importante. Les carcinomes médullaires sont associés de
manière invariable aux carcinomes MSI-H.
(Wick, Vitsky et al. 2005)
Figure 10. Carcinome à cellules en
bague à chaton. Grossissement x20. Coloration HE.
Figure 11. Carcinome médullaire.
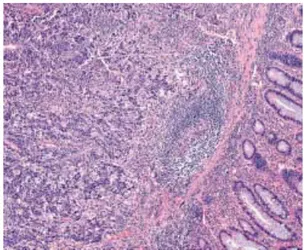
Les cellules tumorales de grande taille s’organisent en cordons et nids. Grossissement x20. Coloration HE.
Adénocarcinomes festonnés
Ces adénocarcinomes sont d’architecture festonnée, similaire à celle des adénomes
festonnés sessiles. Ils peuvent être à instabilité microsatellitaire forte ou faible (MSI low ou
MSI-L), BRAF muté et avec hyperméthylation des îlots CpG (segment d’ADN riche en paires
de bases GC).
Figure 12. Adénocarcinome festonné, présentant (a) des cryptes festonnées et des cellules au
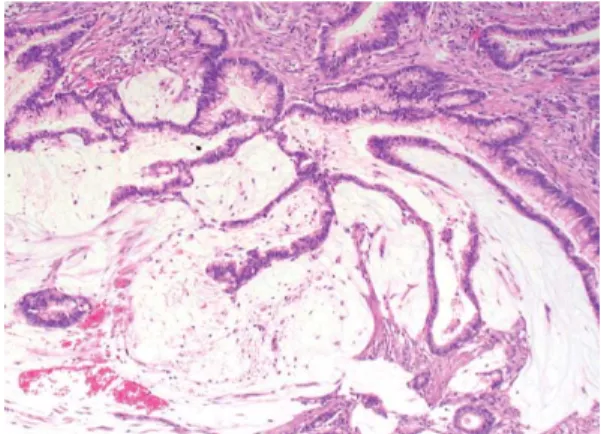
cytoplasme éosinophile semblables aux adénomes festonnés traditionnels. La nécrose sale (nécrose tumorale avec débris de cellules et nucléaires) est typiquement absente. (b) Ces tumeurs sont fréquemment mucineuses, les flaques de mucine contenant des amas arrondis ou cordons de cellules tumorales. Grossissement x40. Coloration HE. (Noffsinger 2009)
IV-‐Les différentes voies de carcinogénèse colorectale : Vers une classification
moléculaire
Les cancers colorectaux ne constituent donc pas un groupe homogène. Ces différents
types histologiques traduisent d’une part des étapes successives de la transformation tumorale
(modèle multi-étape de la carcinogénèse colique) et d’autre part, l’existence de différentes
voies de carcinogénèse
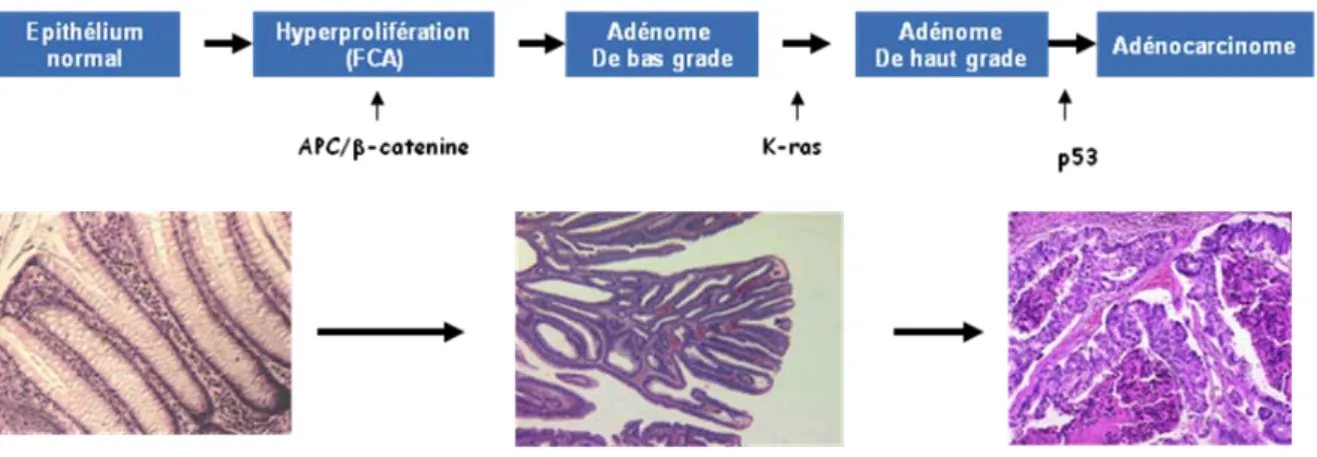
IV -‐1-‐Le modèle multi-‐étape de la carcinogénèse colique
Le concept de la progression adénome/carcinome a été proposé dans les années 1970 à la
suite d’une étude portant sur plus de 1000 échantillons histopathologiques par Morson et al
(Muto, Bussey et al. 1975). Dans cette étude, les auteurs ont observé la présence de foyers
malins au sein de larges adénomes et la présence d’adénomes bénins près des carcinomes,
ainsi qu’une similarité histologique entre ces lésions adjacentes.
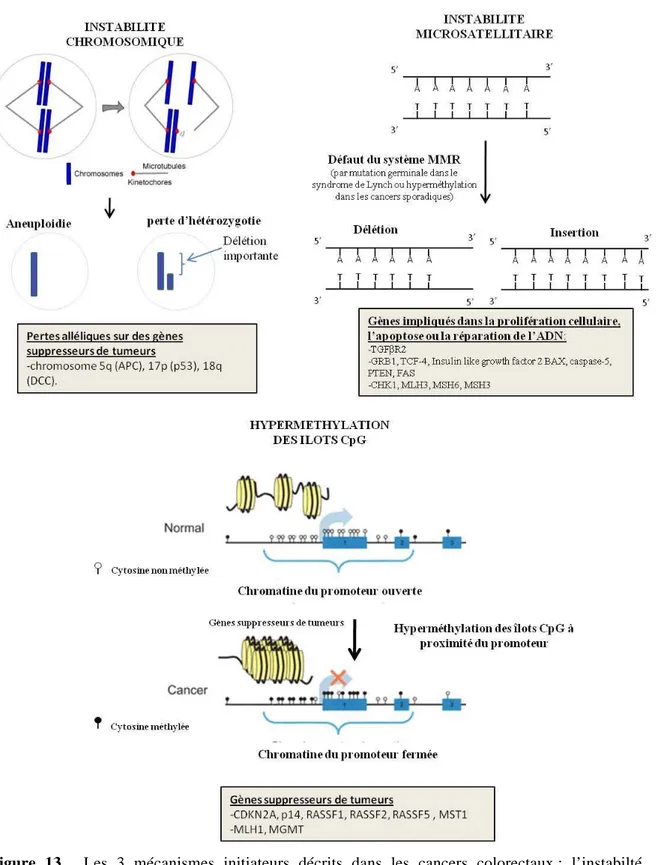
Figure 13. Les 3 mécanismes initiateurs décrits dans les cancers colorectaux : l’instabilté
Les différentes étapes morphologiques de la progression néoplasique colorectale, foyer de
cryptes aberrantes (FCA), adénomes et carcinomes, ont, par la suite, été mises en parallèle
avec l’acquisition progressive de mutations à chaque étape de la transformation
adénome-adénocarcinome, chacune d’elles étant associées à un nombre croissant de mutations
génétiques (Vogelstein, Fearon et al. 1988; Fearon and Vogelstein 1990).
Figure 17. Séquence adénome-carcinome classique. Les mutations APC initient le processus
néoplasique et la séquence histopathologique de transition adénome-carcinome reflète l’accumulation des mutations génétiques (Seules les mutations les plus fréquentes sont indiquées sur ce schéma) .
IV -‐2-‐Une classification moléculaire basée sur les mécanismes moléculaires permettant
l’accumulation des mutations génétiques
Trois mécanismes permettraient d’expliquer l’accumulation de mutations sur les gènes
contrôlant la prolifération et la mort cellulaire : l’instabilité chromosomique, l’instabilité
microsatellitaire et l’hyperméthylation de l’ADN (figure 13). Ils sont associés à des types
histologiques d’adénocarcinomes du colon et à des caractéristiques moléculaires distinctes.
Bien que certaines voies puissent s’entrecroiser et être communes à plusieurs groupes, les
cibles moléculaires de ces 3 mécanismes initiateurs sont différentes et expliquent l’existence
de plusieurs voies de carcinogénèse plus ou moins parallèles (Kruhoffer, Jensen et al. 2005).
La classification moléculaire des CRC, proposée en 2007 par Jass (Jass 2007), est basée à
la fois sur le type d’instabilité génétique (chromosomique, CIN ou microsatellitaire MSI) et la
présence de méthylation des îlots CpG (CIMP). .
De façon schématique et simplifiée, 5 groupes de CCR peuvent être distingués, en
fonction du ou des mécanisme(s) initiateur(s) impliqué(s) :
Les groupes 2, 4 et 5 correspondent à des tumeurs avec un mécanisme unique ou
prédominant, respectivement hyperméthylation, instabilité chromosomique et instabilité
microsatellitaire. Le groupe 1 comporte des tumeurs à la fois à instabilité microsatellitaire et
hyperméthylées alors que le groupe 2 est constitué des tumeurs à instabilité chromosomique et
hyperméthylées (figure 14).
Figure 14. Représentation graphique de la classification des cancers colorectaux, selon Jass
(Jass 2007).
MSI: microsatellite instability
CIMP: CpG island methylator phenotype CIN: chromosomal instability
IV -‐2-‐1-‐L’instabilité chromosomique :
L’instabilité chromosomique est le mécanisme initiateur caractérisant les cancers
colorectaux du groupe 4. Ils représentent près de 57 % des cancers colorectaux (Jass 2007). Il
s’agit d’un des premiers mécanismes décrit pour expliquer l’accumulation des mutations
génétiques au cours de la séquence adénome-carcinome du modèle multi-étape de Vogelstein
(Kinzler and Vogelstein 1996) mais également dans d’autres types de cancers (Lengauer,
Kinzler et al. 1998). Ainsi, la voie de carcinogénèse impliquée est également appelée voie de
l’instabilité chromosomique ou voie classique. L’instabilité chromosomique entraine une
aneuploidie ou des pertes d’hétérozygotie (Lengauer, Kinzler et al. 1998). La plupart des CCR
présentent une perte de 4 ou 5 allèles. La majorité de ces pertes alléliques portent sur les
chromosomes 5q (APC), 17p (p53) et 18q (DCC, deleted in colorectal carcinoma) (Gryfe,
Swallow et al. 1997).
Les mutations associées à l’instabilité chromosomique
Les cancers colorectaux associés à l’instabilité chromosomique présentent différentes
délétions ou mutations d’oncogènes ou suppresseurs de tumeur qui sont détaillées ci-dessous.
L’inactivation du gène APC
L’inactivation du gène APC serait l’événement le plus précoce et initiateur de la
carcinogenèse dans les tumeurs du groupe 4 (à instabilité chromosomique) (Kinzler and
Vogelstein 1996). La protéine APC agirait comme un gardien moléculaire contre le
développement des adénomes et son inactivation conduirait à un déséquilibre entre
prolifération et mort cellulaire (Morin, Vogelstein et al. 1996). En effet, l’activation de la voie
APC est impliquée dans de nombreux processus cellulaires, incluant la migration, l’adhésion,
la prolifération et la stabilité chromosomique.
Les fonctions suppresseur de tumeur d’APC s’expliquent en partie par son rôle dans la
régulation de la voie WNT/caténine, par différents mécanismes. L’interaction entre
β-caténine et APC dans un complexe comprenant la sérine/thréonine kinase GSK3β et l’axine
permet de réguler le niveau cytoplasmique de β-caténine. Dans ce complexe, la β-caténine est
phosphorylée puis ubiquitinylée et dégradée par le protéasome (Rubinfeld, Albert et al. 1996).
En l’absence de dégradation, la β-caténine se transloque dans le noyau où, après formation
d’un complexe avec les facteurs de transcription TCF/LEF, elle initie la transcription de
plusieurs gènes pro-oncogéniques dont c-myc et la cycline D1 (He, Sparks et al. 1998;
Shtutman, Zhurinsky et al. 1999). La protéine APC peut également favoriser l’exportation
nucléaire de β-caténine et ainsi empêcher la transcription de ses gènes cibles (Neufeld, Zhang
et al. 2000). Par ailleurs, il a été montré que le complexe APC/C-terminal binding protein
/β-TrCP (Sierra, Yoshida et al. 2006) peut inhiber la transcription des gènes cibles de la voie
WNT/β-caténine en entrant en compétition avec le complexe β-caténine activateur (figure 15).
La protéine APC serait également impliquée dans la régulation de la migration cellulaire par
différents mécanismes. D’une part, sa partie C-terminale peut interagir avec la proteine de
liaison au microtubule, EB1 et ainsi stabiliser les microtubules (Morrison, Wardleworth et al.
1998). D’autre part, APC peut également activer cdc42, impliquée dans la réorganisation du
cytosquelette d’actine et la formation de lamellipodes et filopodes (Kawasaki, Senda et al.
2000). Il a été, de même, montré qu’APC était capable d’interagir avec la protéine de soutien
IQGAP1, un des effecteurs de cdc42 (Watanabe, Wang et al. 2004).
La perte d’APC pourrait également induire une instabilité chromosomique (CIN) (Fodde,
Kuipers et al. 2001). En effet, EB1 est impliqué dans l’orientation des microtubules au cours
de la mitose. Fodde et al ont montré que des cellules souches ES (embryonic stem) exprimant
la protéine APC tronquée présentaient une instabilité chromosomique liée à la perte
d’accumulation de EB1 au niveau du kinétochore des cellules en mitose.
Les mutations bialléliques du gène APC surviennent à la fois dans les polyposes
adénomateuses familiales et les cancers colorectaux sporadiques. Environ 95% de ces
mutations conduisent à la formation d’une protéine APC tronquée et près de 60% des
mutations somatiques surviennent dans la région MCR (mutation cluster region) du gène
(Fearnhead, Wilding et al. 2002). Ces mutations confèrent à la protéine tronquée un fort effet
dominant négatif par absence des domaines nécessaires à la liaison à la β-caténine. Ainsi l’un
des principaux avantages sélectifs apporté par la protéine APC tronquée réside dans la perte
de régulation de la β-caténine. Cette hypothèse est confirmée par l’observation de la mutation
gain de fonction dans l’exon 3 du gène de la β-caténine, CTNNB1, principalement dans les
CCR sans mutations APC (Sparks, Morin et al. 1998).
Les dernières données de la littérature (Snover 2011) montrent que 60% des CCR
sporadiques présentent une activation constitutive de la voie Wnt/ β-caténine, que ce soit par
des mutations sur le gène APC ou CTNNB1.
Plusieurs modèles animaux transgéniques ont été développés pour confirmer le rôle de
l’inactivation d’APC dans la carcinogénèse colique. L’un des mieux caractérisés est le modèle
de souris APC
min/+(Min, multiple intestinal neoplasia). Ces souris présentent une mutation
d’APC au niveau du codon 850, transformant une leucine (TTG) en codon stop (TAG). La
délétion homozygote d’APC est létale. En revanche, les souris hétérozygotes développent des
centaines de polypes adénomateux au niveau de l’intestin grêle. Chez les animaux plus âgés,
des tumeurs localement invasives ont été détectées, ainsi que de petites zones
d’adénocarcinomes in situ (Moser, Pitot et al. 1990; Su, Kinzler et al. 1992). Ces lésions
présentent un niveau de β-caténine augmenté, à la fois, au niveau du cytoplasme et du noyau.
Cette surexpression est majeure aux stades précoces (foyers de cryptes aberrantes et petits
adénomes), les lésions plus avancées (adénomes de grande taille et carcinomes) présentant de
larges plages sans surexpression (Kongkanuntn, Bubb et al. 1999). Par la suite, d’autres
modèles de souris transgéniques APC muté, dont APC
Δ474, APC
Δ716et APC
Δ14ont été
développés par mutagénèse dirigée et confirment ce phénotype (Oshima, Oshima et al. 1995;
Sasai, Masaki et al. 2000). De plus, dans tous les adénomes des souris APC
Δ716, une perte
d’hétérozygotie a été détectée (l’allèle mutant APC restant inchangée). Ces résultats
confirment la théorie de « second événement » de Knudson (Knudson 1971).
Figure 16: Différents modèles de mutation APC et localisations principales des tumeurs induites,
modifié d’après Phelps et al (Phelps, Broadbent et al. 2009). Dans les cancers colorectaux humains, près de 60% des mutations somatiques surviennent dans la région MCR (mutation cluster region).