Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Pôle poitevin de recherche pour l'ingénieur en mécanique, matériaux et énergétique - PPRIMME (Poitiers)
(Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences et ingénierie en matériaux, mécanique, énergétique et aéronautique -SIMMEA (Poitiers)
Secteur de recherche : Milieux denses, matériaux et composants
Présentée par :
Kévin Alix
Développement d'une approche basée sur la microscopie électronique en transmission filtrée en énergie pour la détermination
des propriétés physiques de bulles d'hélium dans le silicium
Directeur(s) de Thèse :
Laurent Pizzagalli, Marie-Laure David Soutenue le 12 mai 2016 devant le jury Jury :
Président Jean-François Barbot Professeur des Universités, Université de Poitiers Rapporteur Steve Donnelly Professor, University of Huddersfield, UK
Rapporteur Cécile Hebert Professeur, École polytechnique fédérale de Lausanne (EPFL), Suisse Membre Laurent Pizzagalli Directeur de recherche CNRS, Université de Poitiers
Membre Marie-Laure David Maître de conférences, Université de Poitiers Membre Michael Walls Chargé de recherche CNRS, Université Paris Sud
Pour citer cette thèse :
Kévin Alix. Développement d'une approche basée sur la microscopie électronique en transmission filtrée en énergie
pour la détermination des propriétés physiques de bulles d'hélium dans le silicium [En ligne]. Thèse Milieux denses,
matériaux et composants. Poitiers : Université de Poitiers, 2016. Disponible sur Internet <http://theses.univ-poitiers.fr>
Pour l’obtention du grade de
DOCTEUR DE L’UNIVERSITÉ DE POITIERS
Faculté des Sciences Fondamentales et Appliquées Diplôme National - Arrêté du 7 août 2006
Ecole Doctorale Sciences et Ingénierie en Matériaux, Mécanique, Energétique et Aéronautique DOMAINE DE RECHERCHE : MILIEUX DENSES, MATERIAUX ET COMPOSANTS
Présentée par
Kévin Alix
Développement d’une approche basée sur la microscopie
électronique en transmission filtrée en énergie pour la
détermination des propriétés physiques de bulles
d’hélium dans le silicium
Directeur de thèse : Mr Laurent PIZZAGALLI Co-direction : Mme Marie-Laure DAVID
Soutenue le 12 mai 2016 Devant la Commission d’Examen
JURY
Président : Mr Jean-François Barbot Professeur des Universités - Institut P’, Poitiers Rapporteurs : Mr Steve Donnelly Professeur - Université d’Huddersfield, Angleterre
Mme Cécile Hébert Professeure - CIMé - EPFL, Lausanne, Suisse Examinateurs : Mr Michael Walls Chargé de Recherche CNRS - LPS, Orsay
Mme Marie-Laure David Maître de Conférence - Institut P’, Poitiers
Par Ordre d’Apparition :
En premier lieu, je tiens bien évidemment à remercier mes parents, sans qui mon nom ne pourrait logiquement pas être présent en première page de cette thèse. Même si leur implication dans sa rédaction a été limitée, leur soutien lors de ces trois années (et leur insistance lors de la quatrième...) a contribué à ma persistance, et à son achèvement.
Je remercie aussi ma soeur, pour ses perspectives sur mon travail, et son enthousiasme sur le sien.
Malgré leur abscence (notée) à ma soutenance, je remercie mes deux ex-collègues de master, puis de bureau, pour leur dynamisme, leur humour, leur présence, leurs colliers de coquillages, et leurs 653 types d’infusions différentes.
S’il y a peu de chances qu’ils lisent ceci, je dois néanmoins aussi mentionner les anciens thésards, motivation importante pour mon inscription en doctorat il y a bien longtemps.
Je remercie mes encadrants, obviously, parce que sans leur soutien tenant à l’acharnement thérapeutique tout ceci ne serait ni fait ni à faire, et je tiens à témoigner de la non-utilisation caractérisée du dispositif de motivation physique, pourtant offert par moi-même dans ce but. C’était bien la peine. J’ai bon espoir pour mon successeur, ceci dit.
Je remercie aussi les secrétaires (UFR, CNRS... UP... SIMMEA... IUT GTE aussi... il en manque peut-être), qui ont réussi à faire leur travail malgré un interlocuteur parfois pas bien locace.
Merci à l’ingénieur et aux techniciens (et affiliés, ma connaissance de la structure de Pprime demeurant à jamais incomplète et floue) qui ont fait leur possible pour faire fonctionner ce microscope tremblotant (et le reste), même si, au final...
... il aura fallu faire appel à l’alternative Suisse, naturellement, que je remercie pour leur accueil, leur aide, et pour la mise à disposition du microscope sur une bien longue période. Le titre de cette thèse aurait sans doute été bien différent sans eux.
Je souhaite beaucoup de courage aux thésards actuels, ceux qui finissent d’écrire, ceux qui vont soutenir, ceux qui pensent qu’ils ont encore le temps, ceux qui pensent qu’ils sont déjà en retard, ceux qui se demandent si on n’est pas en train de se moquer d’eux avec ces corrections désordonnées, et aussi ceux qui ne comprennent rien à leurs données, ceux qui n’en ont pas, ceux qui en ont trop, et sans oublier ceux qui sont de l’autre côté de la rue (si, si), qui ne mordent pas tant que ça. Aimez vous les uns les autres, bon sang.
Encore plus de courage aux presque-thésards, sans qui mes milieux de journée auraient été bien mornes en cette quatrième année. Ca passe plus vite qu’on s’y attend, ça finit un peu plus lentement...
Introduction générale 1 I Détermination de la densité d’hélium dans des bulles de taille nanométrique :
état de l’art 5
1 Mesures spectroscopiques . . . 5
1.1 Historique . . . 5
1.2 Mesures destructives . . . 10
2 Autres méthodes de mesure de la densité d’hélium . . . 11
3 Calculs atomistiques . . . 12
II Techniques Expérimentales 15 1 Synthèse des nanobulles . . . 15
2 Préparation des lames minces pour la microscopie électronique en transmission . 17 3 Imagerie spectrale pour les bulles . . . 19
3.1 Modes d’acquisition . . . 19
3.2 Limites du STEM-EELS pour l’étude des bulles d’hélium dans le silicium et le germanium . . . 23
3.3 Limite d’épaisseur pour la distinction des bulles . . . 25
III Méthode d’Acquisition et Traitement EFTEM 29 1 Paramètres d’acquisition . . . 29
1.1 Paramètres du microscope . . . 29
1.3 Paramètres de la camera CCD et dimension des spectres . . . 32
2 Non-IsoChromaticité . . . 33
2.1 Effets en STEM-EELS, EFTEM . . . 33
2.2 Méthode de mesure, exemples . . . 35
2.3 Correction . . . 37
3 Dérive spatiale . . . 37
3.1 Lien avec la Non-Isochromaticité . . . 38
3.2 Correction . . . 38
3.2.1 Filtrage du bruit . . . 38
3.2.2 Contraste, détection de contours et formes . . . 40
3.2.3 Corrélation et moyennage . . . 42
3.2.4 Combinaison avec la Non-IsoChromaticité . . . 47
4 Dérive de la haute tension . . . 48
4.1 Comparaison des effets en STEM-EELS, et en EFTEM . . . 49
4.2 Mesure et erreur induite . . . 51
4.3 Précautions expérimentales . . . 53
4.3.1 Standby du canon . . . 53
4.3.2 Influences des réglages . . . 54
4.3.3 Critères de stabilité . . . 54
5 Traitement des spectres . . . 55
5.1 Réduction du bruit . . . 56
5.1.1 Analyse Statistique Multivariée . . . 56
5.1.2 Médiane . . . 57
5.2 Pertes multiples . . . 57
5.3 Ajustement des plasmons . . . 57
5.4 Extraction du seuil K de l’hélium . . . 61
6 Erreur globale . . . 61
7 Discussion . . . 62
IV Caractérisation de bulles individuelles 65 1 Caractéristiques physiques d’une bulle . . . 65
1.1 Morphologie . . . 65
1.2 Densité d’hélium . . . 67
1.2.2 Validation de l’utilisation de la méthode du décalage du seuil K
de l’hélium . . . 68
1.3 Pression . . . 72
2 Nanobulles d’hélium dans le silicium . . . 74
2.1 Recuits ex situ à 700°C . . . . 74
2.2 Recuits ex situ à 500°C . . . . 79
2.3 Recuits in situ . . . . 83
2.3.1 Mesures préliminaires . . . 83
2.3.2 Évolution de la morphologie . . . 88
2.3.3 Cartes de densité d’hélium . . . 89
2.3.4 Évolution de la densité d’hélium . . . 95
3 Applications à d’autres matériaux . . . 97
3.1 Germanium . . . 97
3.2 Carbure de silicium . . . 99
3.3 Euxénite . . . 99
4 Discussion . . . 103
4.1 Effet de la surface des bulles sur la mesure . . . 103
4.2 Pression et équation de Laplace-Young . . . 104
Conclusion générale et perspectives 107
L’hélium est un élément chimique du début du tableau périodique, faisant partie de la famille des gaz nobles comme le néon, l’argon, etc... Ces derniers sont tous caractérisés par une structure électronique constituée de couches électroniques complètes (1s2 pour He), ce qui
explique leur très faible réactivité chimique. L’hélium, en particulier, ne peut former de liaisons chimiques stables avec d’autres éléments, et est considéré comme parfaitement inerte. C’est cette particularité qui le rend également insoluble de manière générale.
L’hélium peut toutefois se retrouver piégé dans les matériaux dans de multiples situations. Par exemple, il peut être produit naturellement par radiogénèse dans les minéraux contenant de l’uranium ou du thorium. L’estimation de la quantité présente permet ensuite d’effectuer une datation. De manière similaire, l’hélium est un produit final de réactions de fission et de fusion dans le domaine nucléaire. Enfin, les propriétés spécifiques de l’hélium font qu’il est également utilisé dans de nombreuses applications pour le traitement ou l’analyse des matériaux, au cours desquelles il peut effectivement se retrouver piégé. On peut citer par exemple le microscope à ions d’hélium qui peut être utilisé pour imager et structurer la matière à l’échelle nanométrique [1].
Du fait de l’absence d’affinité électronique, les seules interactions entre un atome d’hélium et les éléments chimiques constituant les matériaux sont de type électrostatique, avec une com-posante répulsive (à courte portée), et une autre faiblement attractive (à longue portée). La seconde, de type van der Waals, est généralement négligeable devant la première, ce qui ex-plique le caractère insoluble de l’hélium dans les matériaux. Afin de minimiser les interactions répulsives à courte portée, les atomes d’hélium vont donc avoir tendance à s’agréger dans des régions comportant une plus faible proportion d’atomes hôtes, comme les défauts lacunaires, joints de grain, interfaces etc... Selon les conditions, ces agrégats d’atomes d’hélium peuvent
évoluer jusqu’à atteindre une taille nanométrique voire sub-micrométrique. Ils sont générale-ment de forme sphérique, auquel cas on parle de bulles, mais ils peuvent aussi être de forme ellipsoïdale. Dans ce dernier cas, on parle de platelets.
Bulles et platelets sont bien documentés dans la littérature, et ont été observés dans de nombreux types de matériaux, que ce soit des métaux simples ou des matériaux plus com-plexes [2]. Toutefois, les divers mécanismes conduisant à leur formation et leur évolution ne sont pas encore totalement élucidés. Approfondir nos connaissances a bien évidemment un in-térêt en science fondamentale mais pas uniquement. En effet, ces bulles et platelets ont deux particularités importantes : d’une part ils se forment généralement dans des zones proches des surfaces, et d’autre part, ils peuvent contenir une forte densité d’hélium, se traduisant par une pression élevée exercée sur le matériau. Ceci conduit dans certains cas au gonflement et au clo-quage de ces surfaces [3], et une déterioration irréversible de la tenue mécanique des matériaux. Par ailleurs, ces contraintes locales élevées sont également mises à profit au cours de procédés employés dans la préparation de wafers en micro-électronique [4].
Afin d’améliorer nos connaissances dans ce domaine, des cadres théoriques ont déjà été proposés [5, 6]. Toutefois, pour vérifier et améliorer ces modèles, il est nécessaire de dispo-ser de données expérimentales et théoriques sur ces défauts. Une propriété essentielle est la densité d’atomes d’hélium contenus dans les bulles et platelets. Jusqu’à récemment, seules des estimations effectuées sur un ensemble de bulles étaient disponibles. L’avènement de nouvelles générations de microscopes électroniques et l’utilisation d’approches comme la Spectroscopie de Perte d’Énergie des Électrons (EELS) dans un microscope électronique en transmission à balayage (STEM-EELS) ont récemment permis de sonder des valeurs de densité au sein d’une bulle unique à l’échelle nanométrique [7, 8]. Ces expériences restent toutefois difficiles à mettre en oeuvre. Par exemple, il a été montré que l’hélium contenu dans des bulles dans du silicium était expulsé en raison du courant élevé associé au faisceau électronique focalisé [9]. Une alter-native moins destructrice consisterait à utiliser une approche de type microscopie électronique en transmission filtrée en énergie (EFTEM), basée sur l’acquisition en série d’images filtrées en énergie pour acquérir un spectre image. En effet, dans le cas de l’EFTEM, les conditions d’illu-mination sont totalement différentes de celles utilisées en STEM-EELS, notamment en terme de densité de courant puisque l’illumination est parallèle et non convergente. Il est alors envisa-geable que les effets d’endommagement dus au faisceau d’électrons soient moindres comme cela a été montré récemment dans des matériaux tels que LiFePO4 [10] et dans des polymères [11]. Une telle approche devrait donc permettre des mesures repétées sur de mêmes bulles, permet-tant ainsi un suivi de la densité d’hélium dans les bulles lors de recuit in situ dans le microscope électronique en transmission (MET) par exemple. Enfin, cette approche devrait permettre de
faire des mesures sur un grand nombre de bulles avec des temps d’acquisition plus courts qu’en STEM-EELS en utilisant un TEM/STEM conventionnel (canon Schottky-FEG classique).
Cette thèse, réalisée dans le département de physique et mécanique des matériaux de l’Ins-titut Pprime, avait pour objectif de développer des procédures d’acquisition et de traitement de données EFTEM, afin de déterminer la densité d’hélium dans des bulles. De telles procédures étaient inexistantes dans la littérature au commencement de ce travail. L’objectif était égale-ment d’optimiser notre approche afin d’obtenir des mesures pour des bulles de taille inférieures à 10 nm, ceci afin de permettre une convergence avec les résultats de simulations atomistiques disponibles. Nos travaux ont essentiellement été réalisés sur le silicium, un matériau modèle pour lequel il existe une forte expertise au sein du laboratoire.
Le plan de ce mémoire s’articule comme suit. Le premier chapitre dresse l’état de l’art dans le domaine, et le second présente en détail les conditions expérimentales de synthèse et préparation des échantillons, et des observations en microscopie. Les procédures d’acquisition et de traitement des données sont ensuite analysées et décrites dans le troisième chapitre. Le quatrième chapitre rapporte les résultats obtenus pour le silicium, et pour d’autres matériaux.
Détermination de la densité d’hélium dans des
bulles de taille nanométrique : état de l’art
Ce chapitre constitue un récapitulatif des méthodes expérimentales et de simulations utili-sées pour déterminer la densité d’hélium et la pression dans les bulles d’hélium.
1 Mesures spectroscopiques
1.1 Historique
Dans le milieu des années 60, la connaissance des propriétés physiques des bulles d’hélium dans les métaux est apparue comme essentielle à la compréhension des mécanismes physiques conduisant au gonflement des matériaux sous irradiation. La pression dans les bulles, supposées à l’équilibre grâce à la sursaturation de lacunes, est déterminée par la loi de Laplace-Young
P = 2γ/R où P est la pression dans la bulle, γ l’énergie de surface et R le rayon de la bulle. Le nombre d’atomes d’hélium dans les bulles est alors calculé en utilisant la loi des gaz parfaits [12]. Des modifications à cette première approche sont déjà discutées, en particulier l’influence d’une pression extérieure1 [12], la valeur de l’énergie de surface [13] et l’utilisation de lois telles que
l’équation de Van der Waals ou l’équation du viriel à la place de la loi des gaz parfaits [13].
En 1978, Ohtaka et Lucas [14] proposent de vérifier que l’équation de Laplace est en ef-fet applicable et donc de déterminer la pression dans les bulles en mesurant une propriété spectroscopique de l’hélium piégé qui dépende de la pression de façon non ambigüe. Ils pro-posent alors d’utiliser la position, la largeur et/ou la forme du seuil d’absorption optique (UV) (11S
0−n 1P1, n = 2) de l’hélium situé à 21,2 eV pour l’hélium atomique (584,3 Å [15]) comme 1. La loi de Laplace exprime en fait une différence de pression ∆P entre la pression dans et à l’exterieur de la bulle mais cela est très (trop ?) rarement considéré.
signature pour déterminer la densité d’hélium dans les bulles.
Une dizaine d’année auparavant, Surko et al. avaient montré que la transition 1s-2p de l’hé-lium est décalée de 1% lorsque l’on s’intéresse à de l’hél’hé-lium liquide [16] par rapport à la position de la même transition pour l’hélium atomique. Cet effet est alors attribué au recouvrement de la fonction d’onde de l’atome excité, peu mobile dans le liquide comparé au gaz, et de la fonction d’onde d’un atome proche voisin, conduisant à une interaction répulsive qui tend à décaler la transition atomique vers les hautes énergies.
Les premières expériences d’absorption UV puis de EELS sur des bulles d’hélium dans de l’aluminium sont reportées en 1980 (UV) et 1981 (EELS) par le groupe de Namur [17, 18]. Le seuil K de l’hélium est mesuré à 22,6 eV, un décalage de 1,4 eV et un élargissement de 1,2 eV sont alors mesurés pour des bulles qui font en moyenne 5 nm de diamètre. À l’aide d’une première approche théorique, les auteurs montrent que le décalage de ce seuil par rapport à sa position pour de l’hélium atomique, ∆E, est directement proportionnel à la densité d’hélium dans les bulles, nHe, selon :
∆E = CnHe (I.1)
Ceci leur permet d’estimer la densité d’hélium entre 70 et 140 atomes.nm−3.
Le groupe de Jülich va ensuite réaliser plusieurs autres expériences d’EELS sur des bulles d’hélium dans de l’aluminium et du nickel [19–22] mettant aussi en évidence le décalage du seuil K de l’hélium. Enfin, la relation donnée par l’équationI.1est confirmée par une approche théorique plus fine en 1983 [23], utilisant l’interaction dipolaire des atomes d’hélium excités et la répulsion de Pauli.
La détermination du paramètre C devient alors essentielle pour déterminer sans ambiguïté la densité d’hélium à partir de la position du seuil K de l’hélium lors d’expériences EELS. Le groupe de Jülich propose de déterminer ce paramètre en employant la méthode suivante [21]. Le diamètre moyen des bulles est tout d’abord déterminé à partir des micrographies obtenues en MET. La pression dans les bulles est ensuite calculée à partir d’une relation théorique liant pression et rayon. La pression dans la bulle est en effet supposée égale à une pression seuil permettant la mise en oeuvre du mécanisme de "loop punching"2 : P
L = (2γ + µb)/r où µ 2. On suppose que les bulles peuvent croitre par émission d’atomes interstitels ou d’amas d’interstiels sous la forme de boucles de dislocation, la concentration en lacunes et leur mobilité étant trop faible à la température ambiante pour jouer un rôle significatif dans la croissance des bulles.
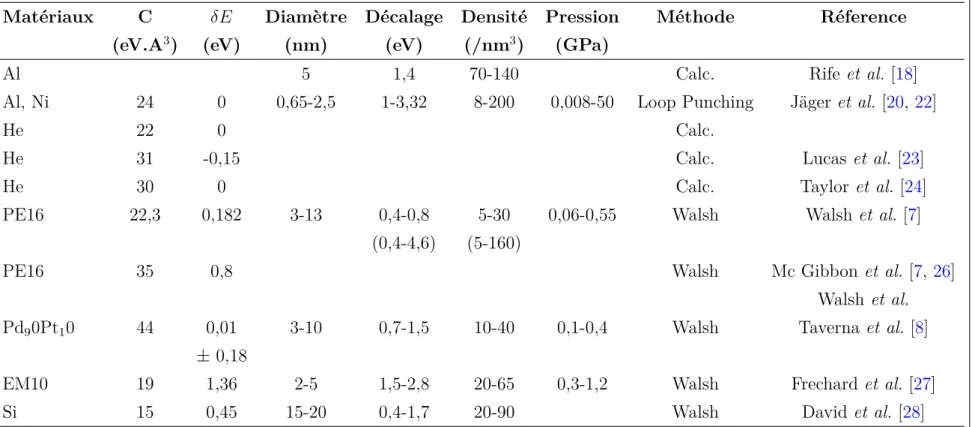
est le module de cisaillement du matériau, b est le vecteur de Burgers de la boucle et γ est l’énergie de surface. La densité d’hélium est déterminée via l’équation d’état appropriée et est ensuite corrélée à la valeur du décalage du seuil K de l’hélium mesurée sur les spectres EEL, permettant ainsi de déterminer le paramètre C. L’approche complémentaire à cette approche expérimentale est une approche théorique de calcul de structure électronique menée par diffé-rents groupes [21–24]. Comme le montre la TableI.1, dans laquelle sont consignés les principaux résultats émanants de ces différentes approches, les valeurs déterminées pour le paramètre C varient alors entre 22 et 30 eV.Å3.
Le développement de la microscopie électronique en transmission à balayage, avec la pos-sibilité de réaliser des expériences d’EELS résolues spatialement, conduit à l’émergence d’une autre méthode pour déterminer la densité d’hélium dans les bulles, basée cette fois sur la section efficace de diffusion inélastique des électrons et l’analyse de bulles uniques [25, 26]. En 2000, Walsh et al. publient un article très complet détaillant cette nouvelle procédure [7] et montrent que la densité d’hélium dans la bulle nHe peut s’écrire :
nHe = IHe IZL 1 σHe h (I.2)
avec IHe et IZL respectivement les intensités intégrées du seuil K de l’hélium et du pic
élastique, σHe la section efficace intégrée de diffusion inélastique sur la transition 1s → 2p et h
l’épaisseur de bulle traversée par le faisceau d’électrons.
Depuis, cette méthode a été utilisée à plusieurs reprises pour déterminer les densités d’hélium dans des bulles non seulement dans des alliages [8, 27], mais aussi dans des semi-conducteurs tels que le silicium et le germanium [28,29]. Au-delà de la détermination de la densité d’hélium dans des bulles individuelles, la résolution spatiale apportée par le STEM-EELS a permis de mettre en évidence des effets de surface lors des mesures [8].
D’autre part, cette méthode a récemment été adaptée pour estimer, à partir du seuil K de l’azote, la densité d’azote dans le coeur de nanosphères métalliques (CoPt) [30] ainsi que dans les pores de films minces de type SiOxNy [31].
Enfin, le fait de disposer d’une méthode de mesure de la densité d’hélium indépendante du décalage du seuil K permet a priori la détermination du paramètre C ; en traçant la densité d’hélium estimée par la méthode des intensités intégrées en fonction du décalage du seuil K
Figure I.1–Décalage du seuil K de l’hélium par rapport à la position Eabs
0 en fonction de la densité
d’hé-lium mesurée par la méthode des intensités intégrées (méthode A). Données reportées dans la littérature, d’après les références [7,8,21,26–28].
de l’hélium mesuré lors des expériences de STEM-EELS. Nous avons rassemblé les données expérimentales publiées dans la littérature sur la figure I.1 en traçant le décalage du seuil K de l’hélium en fonction de la densité d’hélium déterminée en STEM-EELS par la méthode des intensités intégrées. Nous avons aussi ajouté les plus anciennes mesures, pour lesquelles la pression seuil pour le déclenchement du mécanisme de "loop punching" était utilisée. Sur ce graphique, on constate que les données sont dispersées, d’autre part la tableI.1 montre que le paramètre C, évalué indépendamment pour chaque série de mesures, varie entre 15 et 44 eV.Å3.
Pourtant, comme nous l’avons déjà évoqué, la détermination de ce paramètre de façon suf-fisamment précise permettrait de déterminer la densité d’hélium en utilisant la méthode, plus directe, du décalage du seuil K de l’hélium. L’étude menée à l’Institut Pprime [28] un peu avant le début de cette thèse est encourageante en ce sens. En effet, en mettant à profit le fait que les bulles d’hélium dans le silicium se vident sous le faisceau d’électrons, il a été possible de faire varier la densité d’hélium dans une seule et même bulle et d’ainsi éliminer un certain nombre de causes d’incertitude sur la mesure de C, notamment sur la mesure du diamètre des bulles, et d’effectuer ces mesures sur une étendue de densités allant de 20 à 100 He.nm−3. Nous
I. 1 M es ur es sp ect ro sco pi q
Al 5 1,4 70-140 Calc. Rife et al. [18]
Al, Ni 24 0 0,65-2,5 1-3,32 8-200 0,008-50 Loop Punching Jäger et al. [20, 22]
He 22 0 Calc.
He 31 -0,15 Calc. Lucas et al. [23]
He 30 0 Calc. Taylor et al. [24]
PE16 22,3 0,182 3-13 0,4-0,8 5-30 0,06-0,55 Walsh Walsh et al. [7] (0,4-4,6) (5-160)
PE16 35 0,8 Walsh Mc Gibbon et al. [7, 26]
Walsh et al. Pd90Pt10 44 0,01 3-10 0,7-1,5 10-40 0,1-0,4 Walsh Taverna et al. [8]
± 0,18
EM10 19 1,36 2-5 1,5-2,8 20-65 0,3-1,2 Walsh Frechard et al. [27]
Si 15 0,45 15-20 0,4-1,7 20-90 Walsh David et al. [28]
Tableau I.1 – Valeurs du paramètre C liant la densité d’hélium au décalage du seuil K de l’hélium pour différents matériaux selon
∆E = C × nHe dans la littérature. Le décalage ∆E est déterminé par rapport à la valeur de 21,218 eV. Les gammes de valeurs de décalage
Figure I.2 – Bulles d’hélium dans le germanium [28]. a) Carte chimique de l’hélium, filtrée à 22 eV (±0,5 eV). Trois bulles sont visibles b) Seuil K de l’hélium extrait aux différentes positions, t1, t2, t3, t4,
dans la plus grosse des bulles. c) Densité d’hélium relative dans les trois bulles, la densité d’hélium est normalisée à la densité d’hélium maximale mesurée sur la carte. L’encart permet de s’affranchir des effets de saturation du contraste dans la plus petite bulle ; il représente la carte de densité d’hélium normalisée au maximum d’hélium estimé dans la plus petite bulle.
1.2 Mesures destructives
Dans le cas de bulles d’hélium dans le silicium et dans le germanium, le faisceau d’électrons très focalisé utilisé en STEM-EELS peut conduire au dépiégeage de l’hélium pendant la mesure, rendant ce type de mesures destructives [9, 28]. La figure I.2 montre par exemple trois bulles d’hélium dans du germanium étudiées en STEM-EELS. Comme on peut le voir sur la figureI.2b, le seuil K de l’hélium extrait à différentes positions du spectre image, de t1 à t4, sur la plus grosse
des bulles (27 nm de diamètre), est très clairement décalé vers les basses énergies au cours de l’expérience, de 22,3 à 21,5 eV. De plus, l’intensité même du pic diminue significativement. Cette variation n’est bien entendue pas une réelle variation spatiale, mais au contraire une variation temporelle ; elle est liée au temps durant lequel le faisceau d’électrons reste sur la bulle, de plus en plus important lorsque l’on parcourt l’image de gauche à droite et de haut en bas, ce qui correspond au balayage de la sonde. Dans cette bulle, la densité d’hélium est initialement de
60 He/nm3 et elle n’est plus que de 7 He/nm3 à la fin de l’acquisition du spectre image. Cet
effet a aussi été récemment observé dans des films minces de silicium poreux dont les nanopores contiennent de l’hélium [29]. Cet effet peut être mis à profit pour manipuler la pression dans les bulles, et il peut être contrôlé dans une certaine mesure en faisant varier l’énergie des électrons, le courant dans la sonde, et surtout le temps durant lequel le faisceau d’électrons interagit avec les bulles étudiées. Cependant, l’épaisseur de l’échantillon et la position de la bulle dans la lame mince sont autant de paramètres qu’il est plus difficile de contrôler et qui rendent le vidage des bulles aléatoires en fonction des paramètres contrôlables. Ainsi, l’observation répétée d’une même bulle peut être soumise à caution, et ce phénomène perturbe en outre l’étude des bulles lors de recuits in situ successifs, tels que ceux présentés au chapitre IV. Dans ce cas, la variation de la densité d’hélium dans une même bulle dépendrait en effet du temps de présence du faisceau et du temps de recuit.
2 Autres méthodes de mesure de la densité d’hélium
Une synthèse des méthodes utilisées pour déterminer la densité d’hélium et la pression dans les bulles a été effectuée par S. Donnelly en 1985 [32]. Une méthode souvent utilisée [32–34] consiste à déterminer la densité d’hélium en faisant le rapport entre le nombre total d’atomes d’hélium présents dans l’échantillon, NHe, et le volume total des bulles, Vtot : nHe = NHe/Vtot.
La première quantité peut être déterminée expérimentalement par des mesures de désorp-tion (Thermal Desorpdésorp-tion Spectroscopy), par des analyses de ERDA (Elastic Recoil Detecdésorp-tion Analysis), par Elastic Resonant Proton Backscattering, ou bien par des simulations Monte Carlo en utilisant le code SRIM [35] par exemple. Quant au volume total occupé par les bulles, il peut être estimé soit en mesurant le gonflement de l’échantillon après implantation (mesure d’une marche en microscopie à force atomique par exemple), soit en déterminant le volume total des bulles lors d’une analyse en MET en mesurant le diamètre des bulles et en déterminant la densité volumique de bulles. Notons que la morphologie des bulles peut aussi être déterminée par GISAXS (Grazing Incidence Small Angle X-ray Scattering) combiné ou non à une analyse en MET [36, 37] ou par holographie électronique [38].
Cette approche est basée sur plusieurs hypothèses :
— Seuls les défauts contenant de l’hélium contribuent au gonflement ; si l’échantillon com-prend de nombreuses bulles vides, la densité d’hélium sera sous-estimée.
— Tout l’hélium implanté est contenu dans les bulles
— Toutes les bulles sont visibles en MET ; si de nombreuses bulles ne sont pas visibles en MET, la densité d’hélium sera surestimée.
Outre les hypothèses très fortes qui sont faites ici, il faut remarquer que cette méthode comporte de nombreuses sources d’incertitudes, telles que la mesure du diamètre des bulles ainsi que les problèmes de recouvrement de bulles et de mesure d’épaisseur d’échantillon qui sont souvent peu pris en compte. D’autre part, ce type de mesure n’est pas local ; la valeur de densité d’hélium estimée est le résultat d’une moyenne sur l’échantillon. Or comme nous le montrerons par la suite, la densité d’hélium dans les bulles peut varier significativement d’une bulle à l’autre. Les observations au MET étant limitées en aire observable, la question de la représentativité des mesures de densité de bulles ou de morphologie peut aussi se poser.
Lorsque les bulles présentent une géométrie fortement ellipsoïdale, la pression peut aussi être déterminée en se basant sur un critère de fracture tel que le critère de Griffith [39–41]. En particulier, Tillmann et al. ont montré que le contraste de diffraction obtenu en MET en conditions dynamiques pouvait être exploité pour imager le champ de déformation élastique associé au cristal déformé à proximité des bulles. En comparant à des simulations d’images, basées sur l’expression du champ de déformation élastique induit par une fissure de Griffith, il est ensuite possible de déterminer le rapport P/µ de la pression dans la bulle sur le module de cisaillement de la matrice. Une telle étude a été effectuée sur des platelets d’hélium dans le silicium. Des pression de 5 à 15 GPa ont ainsi été déterminées dans des platelets de 17 à 65 nm de diamètre [40]. Il faut cependant noter que ces mesures ne sont rendues possibles que par la forme ellipsoïdale des bulles étudiées et qu’elle ne sera pas réalisable dans le cas de bulles sphériques pour lesquelles le critère de fracture ne peut s’appliquer.
3 Calculs atomistiques
Ces dernières années de nombreuses études de dynamique moléculaire ont été menées pour notamment déterminer l’état de l’hélium dans des bulles de taille nanométrique principalement dans le fer et le tungstène [42–47], mais aussi dernièrement dans des alliages fer-chrome [48] et dans le silicium [49] ; pour ce dernier matériau, notons qu’une thèse vient de débuter à l’Institut Pprime sur cette thématique. Dans la plupart des études menées jusqu’à présent, les bulles ont un diamètre inférieur à 5 nm. Les pressions déterminées sont souvent très grandes, de l’ordre de la dizaine de GPa ou plus dépendant du diamètre de la bulle, suggérant que l’hélium peut
Figure I.3 – a) Vue en coupe d’une boîte de simulation avec NHe= 8000. La couleur rouge correspond
aux atomes de tungstène ayant une structure locale bcc, le jaune aux atomes de tungstène localement désordonnés, et le noir aux atomes d’hélium localement désordonnés. b) Nombre moyen d’atomes par maille par couches sphériques concentriques, centrées au milieu de la boîte de simulation, pour NHe =
8000, avec couleurs correspondant à a). D’après Cui et al. [47].
être confiné à l’état solide dans les bulles [42]. Un autre résultat important qui ressort de ces études est le fait qu’une bulle d’hélium semble être constituée d’un coeur et d’une interface d’épaisseur finie et de dimension non négligeable par rapport au rayon de la bulle (environ 1/3). Comme le montre la figure I.3, le coeur de la bulle ne contient que des atomes d’hélium distribués uniformément, alors que l’interface contient à la fois des atomes d’hélium et de la matrice, et la densité d’hélium diminue progressivement lorsque l’on traverse l’interface [43,47].
D’autre part, en se basant sur des calculs atomistiques pour des bulles de différents dia-mètres, à différentes températures et densités d’hélium, Caro et al. ont récemment proposé une équation d’état pour l’hélium confiné dans des bulles [46].
Techniques Expérimentales
Ce chapitre a pour but de présenter les outils expérimentaux que nous avons utilisés dans le cadre de cette étude. La première partie est consacrée à la méthode de synthèse des nanobulles, tandis que la deuxième partie décrit la méthode de préparation des lames minces pour le MET. La troisième partie est quant à elle consacrée à l’imagerie spectrale en MET pour déterminer les propriétés physiques des nanobulles. En particulier, les approches STEM-EELS et EFTEM y sont comparées.
1 Synthèse des nanobulles
Dans le cadre de cette étude, les nanobulles sont formées par implantation ionique à forte fluence dans le matériau massif considéré, suivie d’un recuit thermique. Pour les échantillons de silicium, qui représentent la majorité des échantillons étudiés, l’implantation d’hélium est effectuée à une fluence de 7.1016 at.cm−2, une énergie de 50 keV et un courant de 1 µA.cm−2
dans du silicium de type P.
Dans ces conditions, le parcours projeté calculé par SRIM (Stopping and Range of Ions in Matter [35]) est de 420 nm et la déviation standard ∆Rp est de 110 nm. Comme le montre la figure II.1, ces conditions d’implantation conduisent à la formation d’un système condensé de bulles de 2 à 3 nm de diamètre réparties de façon homogène. La bande de bulles est située entre 250 et 630 nm de la surface, avec une plus forte densité de bulles entre 300 et 600 nm. La bande de bulles apparaît centrée sur le profil d’implantation de l’hélium [6]. Il est à noter que de nombreux amas d’interstitiels, mis en évidence par les contrastes sombres sur la figure II.1, sont aussi présents dans la zone endommagée.
Figure II.1 – a) Micrographie d’un échantillon de silicium implanté hélium et recuit à 300°C 30’. En superposition, les profils de dommage et profondeur d’implantation de l’hélium calculés par SRIM. b) Micrographie à plus fort grandissement de la partie centrale de la bande de bulles.
700°C, dans un four à canne sous vide secondaire (2,10−6 mbar). Ces paramètres d’implantation
et de recuit ont été choisis dans le but d’obtenir un nombre suffisant de bulles par aire observée, soit plusieurs dizaines pour une surface de 200×200 nm2. Ils permettent de plus d’observer des
bulles de tailles différentes, permettant de mettre en rapport leurs dimensions et la densité ou la pression de l’hélium qu’elles contiennent, individuellement.
À titre d’exemple, la figure II.2montre la microstructure observée en MET en champ clair après un recuit à 500°C pendant 30 min. La bande de bulles est localisée entre 300 et 570 nm de la surface. La plupart des bulles, d’environ 5 nm de diamètre, sont distribuées de façon homogène dans la bande de bulles. Cependant, en arrière de la bande, des bulles de diamètre plus important (environ 10 nm) sont regroupées sous la forme d’amas planaires de diverses tailles, parallèles à la surface (001) de l’échantillon. Ceci, vu en projection sur l’image de MET, donne lieu à une ligne discontinue de grosses bulles. On note aussi de nombreux contrastes sombres dans la zone endommagée, ces contrastes sont attribués à la présence d’amas d’interstitiels. Dans le cadre de cette thèse, nous nous sommes principalement intéressés aux bulles d’hélium et les amas d’interstitiels n’ont pas été caractérisés plus finement. On peut cependant supposer que certains de ces amas sont organisés sous la forme de défauts de type {113} ou sous la forme de boucles de dislocations comme cela est souvent observé pour des implantations à forte fluence
Figure II.2 – a) Micrographie en fond clair, sous-focalisé, d’un échantillon de silicium implanté hélium à température ambiante, et recuit à 500°C 30’. En superposition, les profils de dommage et profondeur d’implantation de l’hélium calculés par SRIM. b) Micrographie à plus fort grandissement de la partie arrière de la bande de bulles. Ici, l’orientation de l’échantillon a été choisie pour minimiser les contrastes de diffraction.
dans le silicium [50,51]. Enfin, les contours de diffraction observés notamment à l’arrière de la bande de bulles montrent que l’échantillon est fortement déformé.
2 Préparation des lames minces pour la microscopie
élec-tronique en transmission
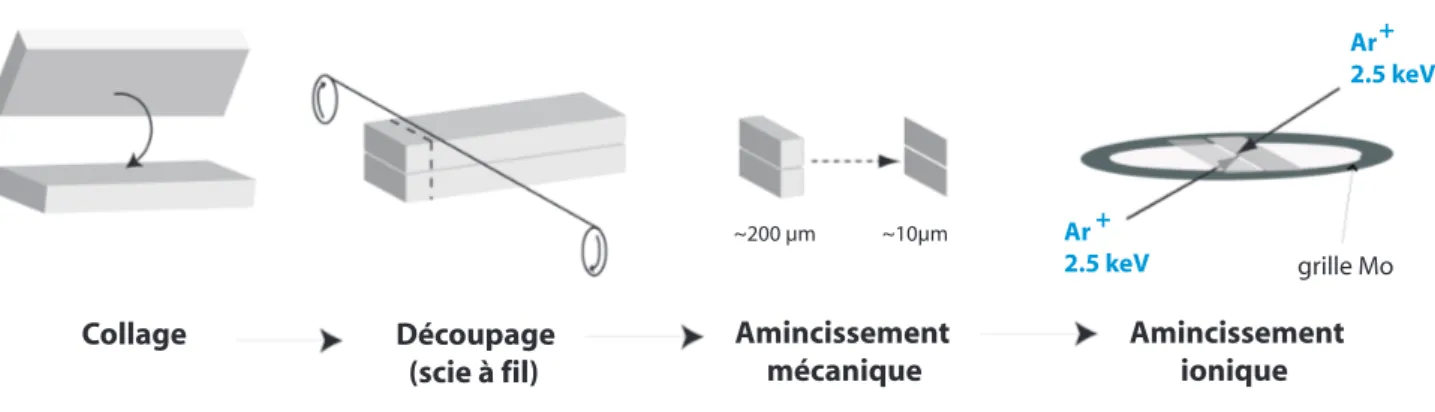
Les lames minces de MET sont préparées en plusieurs étapes, représentées en figure II.3 et détaillées ici.
Après recuit, les échantillons de silicium implantés sont coupés en lamelles de 1,5 mm de largeur. Ces lamelles sont collées face-à-face avec une colle polymère, afin d’obtenir des coupes transverses en fin de préparation. Ces "sandwichs" sont ensuite recuits à une température de l’ordre de 120°C pendant 30 minutes pour garantir l’adhésion des morceaux de silicium, puis découpés en lames de 200 µm d’épaisseur environ. L’influence de ce recuit est supposée négli-geable, notamment lorsque le recuit précédent est effectué à 500°C ou plus.
Figure II.3–Procédure de préparation des échantillons pour la microscopie électronique en transmission.
Ces lames sont ensuite amincies et polies par tripode sur disques diamantés. Les lames, collées sur un support en pyrex plan, sont orientées pour que l’interface Si/colle/Si soit per-pendiculaire à la direction de polissage des disques. Le polissage est fait sur les deux faces successivement, sur des disques de grain allant de 15 µm à 0,5 µm. L’épaisseur finale est de l’ordre de 10 à 20 µm.
Après ce polissage mécanique, les lames sont collées sur une grille en molybdène, permet-tant d’effectuer des recuits in situ dans le MET à haute température. Elles sont placées dans un PIPS (Precision Ion Polishing System) GATAN™ pour une étape d’amincissement ionique permettant de rendre les lames transparentes aux électrons. Pour cette étape, l’énergie des ions argon est fixée à 2,5 keV et l’angle d’incidence est fixé à 8° puis 4° pendant 2 min en fin d’amin-cissement. Ces conditions ont été choisies pour minimiser l’endommagement de l’échantillon. Cependant, il a été montré que l’étape d’amincissement ionique pouvait modifier certains sys-tèmes de nanocavités [52]. En particulier, les défauts ponctuels injectés lors du bombardement ionique peuvent interagir avec les cavités, et la migration de petites cavités (d<5 nm) situées à proximité (30-40 nm) de la surface d’implantation de l’échantillon a été observée dans le cas d’échantillons de silicium implantés à 10 keV. Cette migration n’a pas été observée pour des bulles plus éloignées de la surface d’implantation et plus grosses. Les bulles étudiées ici sont situées à plus de 200 nm de la surface, et la plupart des études sont menées sur des bulles de plus de 10 nm de diamètre, les effets de l’amincissement ionique sur les microstructures ob-servées sont donc supposés négligeables. Enfin, la direction des traces du polissage mécanique peut influencer la qualité du résultat de cette étape, ce qui explique l’importance donnée à l’orientation de l’interface par rapport à la rotation des disques.
Figure II.4– Photographie d’une lame mince de silicium, installée sur un porte-objet double tilt.
et d’autre de l’interface et selon l’amincissement le long du trou que cette étape peut créer. Ces zones peuvent s’étendre sur plusieurs dizaines de micromètres le long de l’interface. Un exemple d’échantillon dans un porte-objet est visible en figure II.4. On peut y voir l’interface au centre, horizontale. Ici, l’amincissement a provoqué la casse d’une partie de la lame, mais à droite du trou, l’échantillon est resté intact. Cette zone est a priori transparente aux électrons. La teinte rouge est caractéristique du silicium lorsqu’il atteint une épaisseur de l’ordre de 10 µm.
3 Imagerie spectrale pour les bulles
3.1 Modes d’acquisition
Pour obtenir les spectres de perte d’énergie des électrons dans des bulles individuelles, on utilise l’imagerie spectrale (Spectrum Imaging, SI) en microscopie électronique en transmission, qui permet d’obtenir des données spectrales résolues spatialement sur une zone. On peut avec cette méthode acquérir des spectres issus de nombreuses bulles, avec plusieurs spectres par bulle.
L’imagerie spectrale peut être employée de deux manières en microscopie électronique en transmission : en balayant la surface de l’échantillon, et en acquérant ainsi une grille de spectres
Figure II.5 – a) Schéma de structure général des données dans un datacube d’imagerie spectrale. En rouge/jaune, les plans constituent des images filtrées. En violet/bleu, les colonnes constituent des spectres. Les données peuvent être extraites d’une façon ou de l’autre : images filtrées en b), ou spectres en c). Il est aussi possible d’extraire des données en plans verticaux, non représentés.
(STEM-EELS), ou en acquérant des séries d’images filtrées en énergie (Energy-Filtered TEM). Dans les deux cas, on fait référence aux données acquises en tant que datacubes, puisque les données comportent trois dimensions : deux dans l’espace (x,y), et une dans le domaine des pertes d’énergies. En STEM-EELS, la dimension E est acquise en une fois et x et y en sé-quence, alors que l’EFTEM-SI fait l’inverse. La figure II.5 présente visuellement la structure de ces datacubes, et leur rapport aux deux méthodes d’acquisition. Ce schéma montre que les deux méthodes d’acquisition aboutissent à la même structure de données, et peut laisser penser que les données obtenues par l’une ou l’autre sont identiques. En pratique, il existe des diffé-rences dépendant notamment des capacités du microscope utilisé, selon le mode d’acquisition. Densité de courant et dose totale, étendue des spectres, et résolution en énergie sont parmi les distinctions à considérer lors du choix.
La spectroscopie de perte d’énergie des électrons par microscopie électronique en transmis-sion à balayage (STEM-EELS) utilise une sonde électronique focalisée subnanométrique, ici de 2 Å, qui balaye l’échantillon observé par intervalles réguliers. Pour chaque position de la sonde, un spectre, dont la dimension dépend de la dispersion utilisée, est acquis en une seule fois. Typiquement, dans les conditions utilisées pour l’étude des bulles sur un 2200FS, on acquière des spectres d’un peu plus d’une centaine d’eV. D’autre part, les électrons diffusés élastique-ment à grand angle sont recueillis sur un détecteur annulaire (High Angle Annular Dark Field). Ce signal donne lieu à un contraste masse-épaisseur, qui lorsque l’épaisseur est constante est directement proportionnel à Z1,7 [53]. C’est ce signal qui est utilisé pour obtenir l’information
spatiale relative à l’échantillon. Ainsi, dans le cas du STEM-EELS, l’information bidimension-nelle relative à l’échantillon ainsi que les spectres sont acquis simultanément.
Dans le cas de l’EFTEM-SI, l’échantillon est observé avec un faisceau large et parallèle. Chaque datacube est constitué d’images filtrées, acquises séquentiellement au même endroit, sur un domaine d’énergie donné. Ce filtrage est effectué par une fente située dans le plan de dispersion du filtre. On obtient les spectres en extrayant les données du datacube dans le sens des énergies [54–56].
Les deux méthodes sont schématisées en figureII.6, qui montre les différences techniques entre elles. En STEM-EELS, le faisceau convergent interagit avec l’échantillon. Les électrons diffusés à grand angle sont recueillis sur le détecteur HAADF. Les électrons diffusés selon un angle inférieur sont dispersés par le filtre, et l’image du plan de dispersion est projetée sur la caméra CCD. Cette image est sommée dans la direction perpendiculaire à l’axe de dispersion, formant ainsi le spectre final. En EFTEM, le faisceau parallèle interagit avec l’échantillon, traverse le
Figure II.6 – Schéma de fonctionnement de l’acquisition des spectres en STEM-EELS et des images filtrées en EFTEM.
filtre et est aussi dispersé. La fente placée dans le plan de dispersion sélectionne l’énergie des électrons dans ce plan. Le système de projection forme ensuite l’image de l’échantillon à partir des électrons filtrés, qui est captée par la caméra CCD.
Deux microscopes JEOL™ 2200FS ont été utilisés dans le cadre de cette étude. Le premier, appartenant à l’institut Pprime et situé à Poitiers (France), a été utilisé initialement pour les essais et l’établissement de la procédure d’acquisition, ainsi que pour le développement des pro-cédures de traitement et d’analyse des données acquises, qui sont détaillées au chapitre suivant. Le deuxième, appartenant au Centre Interdisciplinaire de Microscopie Électronique (CIMe−)
et situé à Lausanne (Suisse), a été utilisé pour une grande partie des acquisitions faites sur les échantillons de silicium implanté hélium. En effet, la dégradation de l’environnement du microscope à Poitiers, et en particulier la présence d’un champ magnétique non négligeable généré par le bâtiment, a été détectée lors des premières expériences réalisées dans le cadre de ce travail. L’amplitude de la dérive de la haute tension du microscope ne permettant pas de réa-liser des analyses quantitatives suffisamment précises, l’utilisation d’un microscope situé dans un environnement plus stable a été nécessaire. Au delà des différences d’infrastructure entre ces deux équipements, la seule différence fonctionnelle est le type de lentille objectif (configuration dite Cryo pour le microscope du CIMe−, High Resolution pour celui de Pprime), qui n’est pas
censé avoir d’influence en spectroscopie en opération normale. De même, les caractéristiques proches du canon, des différents diaphragmes et des systèmes d’acquisition permettent d’utiliser une méthode commune aux deux appareils.
3.2 Limites du STEM-EELS pour l’étude des bulles d’hélium dans
le silicium et le germanium
Comme nous l’avons rappelé dans le chapitre précédent, après les premières expériences en faisceau parallèle des années 80, le STEM-EELS s’est imposé comme une technique de choix pour l’étude des propriétés physiques des bulles d’hélium car la résolution spatiale de ce mode de mesure permet de déterminer la densité d’hélium en même temps que la morphologie et la taille des bulles à l’échelle locale, c’est-à-dire pour des bulles individuelles.
L’objectif de cette thèse était notamment d’étudier l’évolution d’un système de bulles dans le silicium pendant un recuit thermique dans le MET. Or il a été montré, dans le cas de bulles d’hélium dans du silicum ou du germanium, que la sonde électronique très focalisée utilisée
pour l’acquisition de datacubes en STEM-EELS pouvait conduire au dépiégeage de l’hélium et donc au vidage progressif des bulles [9,28].
Ceci rend l’étude répétée de bulles spécifiques après sollicitation (recuit thermique, irra-diation) difficile sans forte réduction du rapport signal/bruit ou de la résolution spatiale. De plus, la sonde peut provoquer localement une contamination au carbone, qui ajoute un signal spectral évoluant avec le temps et perturbant la mesure de la position du seuil K de l’hélium.
D’autre part, nous souhaitions pouvoir étudier l’évolution de plus d’une dizaine de bulles pour des raisons statistiques. Sur les échantillons considérés ici et en gardant une bonne réso-lution spatiale, ceci aurait demandé un temps d’expériences d’une dizaine de minutes et donc une correction de dérive spatiale automatique durant l’expérience, qui est rendue difficile par les contrastes présents sur les échantillons.
L’EFTEM-SI s’est donc imposée comme une technique alternative possible pour l’étude des bulles. Tout d’abord, les conditions d’illumination sont très différentes de celles utilisées en STEM-EELS, en particulier en terme de densité de courant sur un même microscope : malgré l’absence de mesures spécifiques de celle-ci, la différence entre les tailles de sonde (<1 nm2
en STEM-EELS et approximativement 1 µm2 en EFTEM) permet de supposer un rapport de
densités en faveur de l’EFTEM. Il est donc envisageable que l’endommagement sous le fais-ceau électronique soit réduit, comme cela a été observé pour le LiFePO4 [10] et pour certains
polymères [11]. D’autre part, l’EFTEM-SI permet d’acquérir des images sur un large champ d’observation avec des temps d’exposition relativement courts, ce qui permet d’étudier de nom-breuses bulles avec un seul datacube. Enfin, chaque image étant acquise en une fois, la dérive spatiale peut être observée et compensée, pendant et après l’acquisition de chaque datacube. On peut donc acquérir directement plusieurs bulles sur un même datacube, et localiser de façon fiable les spectres extraits même en cas de dérive. La cartographie chimique permet de plus de déterminer au préalable si une zone contient des bulles pleines.
La résolution en énergie, limitée par la taille de la fente (ici 0,5 eV) est inférieure à celle possible en STEM-EELS, mais en contrepartie l’étude simultanée d’un nombre de bulles im-portant est possible. La figure II.7montre 2 spectres, acquis par les deux méthodes. Malgré la perte de résolution, 1,3 eV en EFTEM-SI pour 1,0 eV en STEM-EELS (largeur à mi-hauteur du pic élastique), et un bruit plus important en EFTEM-SI, le signal du seuil K de l’hélium est visible et distinct du plasmon du silicium.
Figure II.7 – Spectres acquis en EFTEM et STEM-EELS sur la même bulle. Les principaux signaux, pic élastique, plasmon du silicium (17 eV) et seuil K de l’hélium (22-24 eV) sont bien visibles dans les deux cas.
3.3 Limite d’épaisseur pour la distinction des bulles
Dû à la méthode de polissage et d’amincissement, l’épaisseur des échantillons n’est pas constante sur toute leur surface. Or, outre la restriction habituelle que cela apporte sur les zones dites transparentes, l’épaisseur limite aussi directement les zones observables en EFTEM comme en STEM-EELS, pour l’étude des bulles.
En effet, puisque l’on cherche à étudier chaque bulle individuellement, il est nécessaire de pouvoir acquérir des données qui ne proviennent que de la bulle considérée. Or, dû à la pro-jection en MET, un spectre acquis peut être une combinaison des spectres issus de plusieurs bulles traversées par le faisceau.
Les bulles sont a priori réparties de façon homogène parallèlement à la surface d’implan-tation. En coupe transverse, l’épaisseur de l’échantillon fait donc varier le nombre de bulles projetées. La probabilité d’avoir des bulles superposées augmente donc avec l’épaisseur de l’échantillon. Ceci impose une limitation de l’épaisseur maximale de l’échantillon sur les zones
observées, qui est estimée à 50 nm pour les échantillons de silicium recuits à 700°C. Cette valeur correspond à plus du double du diamètre des plus grosses bulles pour ce type d’échantillon, soit 20 nm, et limite donc la superposition de celles-ci seulement. Sur ces mêmes échantillons, les plus petites bulles (environ 10 nm) peuvent demander une épaisseur plus faible pour leur observation. En effet, leur superposition est moins probable du fait de leur surface projetée plus faible, mais elles sont plus nombreuses. Il en va de même pour les échantillons recuits aux températures plus basses.
Expérimentalement, les zones utilisables pour la spectroscopie sont déterminées visuelle-ment, en s’assurant que le moins possible de bulles sont superposées. Une superposition partielle est acceptable dès lors qu’il n’y a pas de recouvrement au centre des bulles observées, car c’est aux points centraux que la moyenne de densité est calculée.
Inversement, lors de l’amincissement, certaines bulles peuvent être vidées par intersection de leur surface avec celle de la surface amincie. Une zone trop fine peut comporter une majorité de bulles vides. On effectue une cartographie chimique autour du seuil K de l’hélium pour déterminer si c’est le cas. Par exemple, la figure II.8 montre une bulle vide mais néanmoins visible. La cavité originale est bien visible sur l’image filtrée sur le plasmon du silicium, ainsi que celle des bulles pleines, mais le contraste du signal du seuil K de l’hélium n’est pas présent dans la bulle vide.
Une zone utilisable doit donc être suffisamment fine pour limiter les superpositions, et suffisamment épaisse pour contenir majoritairement des bulles pleines. Selon la microstructure, la combinaison de ces deux critères peut ne pas être possible pour toutes les bulles d’une zone.
Figure II.8 – Images filtrées (fente 1 eV) sur a) le plasmon du silicium à 17 eV et b) le seuil K de l’hélium. L’absence de signal issu du seuil K de l’hélium sur une des trois bulles centrales (indiquée par la flèche sur chaque image) montre qu’elle est vide.
Méthode d’Acquisition et Traitement EFTEM
Dans ce chapitre, les étapes nécessaires à la détermination de la position du seuil K de l’hélium par voie EFTEM sont détaillées. L’acquisition du signal et la correction des artefacts sont spécifiques à l’EFTEM, alors que les étapes de traitement des spectres donnant les cartes de densité comportent des points communs avec les méthodes employées en STEM-EELS. Les différentes contraintes, menant aux paramètres et méthodes choisis, sont détaillées en parallèle.
Le choix de la plupart des paramètres a été fait au cours de séries d’expériences dont les résultats seront présentés dans le chapitre suivant. Par manque de temps, certains paramètres n’ont donc pu être optimisés pleinement, et d’autres n’ont jamais été modifiés suite à leur choix initial. La discussion en fin de ce chapitre donne les principales pistes à explorer dans le but d’aboutir à une optimisation plus poussée.
1 Paramètres d’acquisition
1.1 Paramètres du microscope
Le grandissement employé sur le JEOL 2200FS est, pour la majeure partie des images pré-sentées ici, de 100k. Cette valeur a été choisie afin d’obtenir une aire d’observation de l’ordre de 200 par 200 nanomètres carrés, permettant d’observer plusieurs dizaines de bulles simulta-nément selon l’échantillon. Dans le cas d’échantillons de silicium, tels que présentés au chapitre précédent, jusqu’à une dizaine des bulles les plus grosses peut être observée dans les zones jugées utilisables pour l’EFTEM.
Afin de réduire le contraste de diffraction, l’échantillon est orienté hors axe de zone. Un diaphragme objectif est inséré afin de réduire les aberrations d’une part, et d’autre part afin de définir l’angle de collecte des électrons. Cette donnée est en effet nécessaire si l’on veut utiliser
la méthode des intensités intégrées pour déterminer la densité d’hélium. Enfin, on se place dans des conditions d’éclairement parallèle.
1.2 Filtrage en énergie
Le filtrage en énergie est effectué par un filtre de type oméga intercalé dans le système de projection, divisé en deux parties pré- et post-filtre afin de le prendre en compte. La largeur de la fente de sélection est modifiable par pas de 0,5 eV, de 0,5 eV à plus de 200 eV. Pour l’obtention des datacubes, le 2200FS permet de faire varier la valeur de l’énergie filtrée de deux manières.
La première consiste à faire changer par pas constant l’excitation du filtre et donc la dé-viation subie par les électrons. Le spectre projeté dans le plan de dispersion est ainsi déplacé par le filtre le long de l’axe de dispersion, et la fente fixe balaye progressivement l’étendue en énergie voulue.
La deuxième possibilité consiste à faire varier la tension d’accélération des électrons, avant leur interaction avec l’échantillon. La fente et l’excitation du filtre étant cette fois fixés, l’énergie des électrons arrivant en un point donné du plan de dispersion ne change pas en cours d’acqui-sition. Le spectre est déplacé le long de l’axe de dispersion par la variation de l’énergie initiale des électrons, là aussi par pas constant.
Pour la première méthode, l’énergie des électrons avant interaction est donc fixe, et elle varie en sortie de fente. L’inverse se produit pour la deuxième méthode. Cette différence est une des raisons pour laquelle la variation par haute tension est utilisée ici, en EFTEM, puis-qu’elle permet d’éviter les problèmes liés à la variation d’énergie dans le système de projection du microscope (notamment l’aberration chromatique), alors que le microscope compense cette variation pour le système d’illumination.
De plus, bien que les deux méthodes permettent la même variation de position du spectre, de l’ordre de 3000 eV, seule la variation par haute tension permet d’atteindre un pas minimal de 0,2 eV, alors que la variation par le filtre est limitée à environ 0,6 eV. La variation par haute tension est donc la méthode utilisée ici.
Figure III.1 –Images filtrées obtenues dans les mêmes conditions, pour une fente de 1 eV. a) est filtrée
autour du pic élastique, b) autour du plasmon du silicium (17 eV).
relative du spectre et de la fente dans le plan de dispersion du filtre. Il suit que tout mouvement parasite de l’un ou de l’autre fausserait cette valeur. Afin d’éviter ce phénomène, l’excitation du filtre, et la position et la largeur de la fente ne sont modifiées si possible que pendant les réglages initiaux du microscope.
Le but premier de la méthode présentée ici étant la mesure de la position du seuil K de l’hélium, ce pas minimal de 0,2 eV entre chaque image filtrée en énergie est utilisé pour donner un maximum de points utilisables pour les étapes de traitement des spectres. La largeur de la fente est quant à elle de 1,0 eV. La valeur minimale de 0,5 eV, envisagée en premier lieu afin de minimiser la convolution en énergie de la forme de la fente avec les spectres, présente un contraste de stries important tel qu’observé sur la figure III.1. Cette figure montre que ce contraste domine celui de l’échantillon, mais n’est pas présent pour tous les plans filtrés en énergie. Il peut donc perturber à la fois les étapes de correction des données et l’analyse. Ce contraste est dû aux imperfections de la fente de sélection. Une fente neuve, dont les bords n’ont pas encore été endommagés par le faisceau d’électrons, présente un contraste moindre, alors simplement dû à une éventuelle contamination des bords. Ce contraste peut être atténué en augmentant la largeur de la fente, mais aussi en la déplaçant sur l’axe des énergies. Ce mouvement peut en effet provoquer le déplacement les stries sur la CCD, idéalement jusqu’à en faire sortir les plus prononcées pour obtenir une intensité plus uniforme. Ceci est fait au cours des réglages initiaux du microscope.
1.3 Paramètres de la camera CCD et dimension des spectres
Le temps de pose est optimisé pour obtenir un rapport signal/bruit suffisant pour l’exploi-tation des spectres, tout en n’amenant pas l’acquisition à des durées excessives et en restant suffisamment éloigné de la limite de saturation de la camera. Afin de réduire ce temps de pose, un compromis est fait sur la résolution des images en employant un binning 4×4, donnant des images de 512×512 pixels pour autant de spectres.
Pour les spectres de pertes faibles attendus, il est nécessaire de faire varier le temps de pose lors de l’acquisition des images filtrées. En effet, la faible épaisseur des zones observées mène à un rapport d’intensité entre le pic élastique et le reste du signal de 10 ou plus, et il n’est pas envisageable d’acquérir l’ensemble avec un temps de pose unique sans effets négatifs. L’acqui-sition des datacubes est donc faite en deux parties : une incluant le pic élastique et le début du spectre de pertes faibles, et une autre excluant le pic élastique. Ils utilisent respectivement des temps de pose de 0,5 et 5 s.
Le recouvrement entre ces datacubes, nécessaire à la reconstitution après acquisition, s’étend sur 1 eV. Puisque l’on cherche à éviter la saturation de la caméra CCD, la position de ce re-couvrement sur le spectre dépend de l’intensité des signaux. Le temps de pose du datacube encadrant le pic élastique est choisi en fonction de l’intensité de celui-ci, il n’y a donc pas de risque de saturer la CCD sur le reste du spectre, et il n’y a pas de limite à l’étendue de ce datacube autre que le temps de pose total. L’énergie minimale du datacube encadrant le plas-mon du silicium et le seuil K de l’hélium doit cependant être suffisament élevée pour éviter la saturation en tout début de datacube, à cause du temps de pose dix fois plus élevé et de la proximité du pic élastique. Dans les conditions d’acquisitions utilisées ici, un minimum de 3 eV au-delà du pic élastique est nécessaire, mais il est possible de limiter le temps d’acquisition total en plaçant le recouvrement plus loin, juste avant le début du signal qui sera effectivement analysé par la suite (voir la section5). Ici, le plasmon d’interface Si/SiO2, placé à environ 8 eV,
est donc la limite haute.
Au cours des expériences menant aux résultats présentés ici, plusieurs positions pour ce recouvrement ont été utilisées avant d’aboutir à cette limite. Ainsi, une partie des datacubes a été acquise avec un recouvrement à 12 eV. Des problèmes ont été rencontrés lors du traitement, dû au fait que le plasmon sus-nommé présentait un bruit important amené par le temps de pose plus court. Les datacubes suivants ont donc été acquis avec un recouvrement à 3 eV, présentant
parfois une saturation partielle des premières images. Finalement, un recouvrement entre 6 et 7 eV semble être un juste milieu.
Les datacubes s’étendent au total de -3 à 32 eV. On dépasse de cette façon la position maxi-male observée pour le seuil K de l’hélium (approx. 26 eV) de plusieurs eV. Cette dimension de 35 eV est considérée suffisante car, sur des spectres simulés correspondant à ceux obtenus ex-périmentalement, la dimension des spectres n’a d’influence notable sur la mesure de la position du seuil K de l’hélium que pour un signal sans aucun bruit. Si un bruit électronique approprié est ajouté sur les spectres simulés, la déviation des valeurs dûe au bruit occulte la différence dûe à la longueur des spectres.
Le microscope ne permettant pas une variation de haute tension négative, un décalage initial d’environ 3 eV est effectué préalablement à l’acquisition. Le pic élastique est ensuite recentré sur la fente via l’excitation du filtre. L’acquisition est donc effectuée de 0 à 35 eV, avec un décalage initial du couple fente-filtre de 3 eV.
En incluant le temps de transition de la haute tension et l’enregistrement de 175 images filtrées, une acquisition effectuée avec les paramètres indiqués ici prend entre 15 et 20 minutes.
2 Non-IsoChromaticité
2.1 Effets en STEM-EELS, EFTEM
Dans le cas idéal, les électrons traversant le filtre avec une même énergie doivent arriver dans le plan de dispersion en un seul point, et les électrons filtrés ont tous la même énergie, à la largeur de la fente près. Ce n’est pas le cas en pratique dû aux aberrations du filtre, il existe en effet un décalage en énergie entre les électrons [57]. Ce décalage est fonction de la position dans le plan objet et donc dans le plan image.
Les effets de la Non-IsoChromaticité (NIC) sont visibles à la fois en mode spectroscopie et en imagerie filtrée. Sur la figure III.2, on peut voir l’effet de la NIC sur un datacube EFTEM non corrigé. La différence entre les positions extrêmes du pic élastique est ici de l’ordre de 4 pixels, soit 0,8 eV. L’effet sur les images filtrées est évident près du pic élastique (Fig. III.2b).
Figure III.2–a) Coupe issue d’un datacube EFTEM brut, en conditions normales d’acquisition, autour
du pic élastique. b) Image issue du même datacube, filtrée par une fente de 1 eV autour du pic élastique. c) Orientation des images dans le datacube. La variation d’intensité globale de l’image filtrée b) correspond au changement de position du pic élastique visible en a) à travers l’image, équivalent à un déplacement de la fente de sélection en énergie. Ici, seuls les électrons au centre de l’image b) sont issus du maximum du pic élastique. Les taches claires sont en revanche dûes à une variation d’épaisseur, liée à la présence des bulles dans l’échantillon.
Figure III.3– Images filtrées sur le pic élastique avec une fente de 1 eV, et une défocalisation d’environ
700 nm, montrant l’effet visuel de la NIC pour a) un filtre de type oméga dans un JEOL 2200FS et b) un filtre GIF en extrémité de colonne, dans un TECNAI G2.
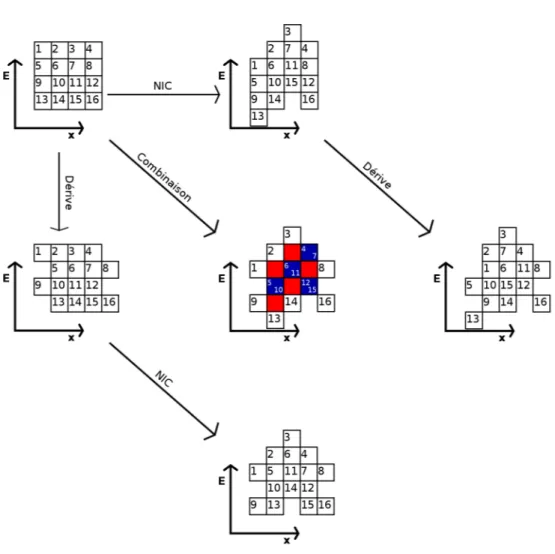
2.2 Méthode de mesure, exemples
La NIC est déterminée à partir des positions ponctuelles d’un élément commun fixe des spectres d’un datacube. Cette détermination est exécutée séparément pour chaque acquisition, afin de pallier à toute variation qui pourrait apparaître entre deux séries d’acquisition ou même au cours d’une série.
La méthode utilisée ici inclut l’acquisition d’un datacube séparé pour la partie élastique des spectres, le pic élastique est donc l’élément de comparaison entre les spectres. Il a été tenté d’utiliser la position du plasmon du silicium dans le deuxième datacube, mais les cartes de NIC obtenues se montraient bien plus sensibles à la position des bulles et à la dérive de l’échantillon, et n’ont donc pas été utilisées.
La position des pics élastiques est obtenue par le calcul du moment d’ordre 1 de chaque spectre, en ne considérant que les intensités supérieures à 20% du maximum afin de ne pas prendre en compte le plasmon du silicium et ses variations d’intensité. Un filtre médian de surface 3x3 est appliqué sur cette carte de positions pour éliminer partiellement le bruit, et la carte elle-même est ensuite ajustée par un polynôme d’ordre 2 ou plus. L’ordre du polynôme dépend du type de filtre en énergie et des corrections qu’il comprend : sur le filtre omega du
Figure III.4– a) et c) montrent les cartes de NIC correspondant respectivement aux figuresIII.3a) et
III.3b), pré-ajustement, faisant apparaître clairement la différence de forme de NIC entre les deux types de filtre. b) et d) montrent des profils pris dans les diagonales de chaque carte, et le résultat des ajustements des cartes pour plusieurs ordres.
![Figure I.2 – Bulles d’hélium dans le germanium [28]. a) Carte chimique de l’hélium, filtrée à 22 eV (±0,5 eV)](https://thumb-eu.123doks.com/thumbv2/123doknet/7996314.267924/21.892.156.738.200.634/figure-bulles-hélium-germanium-carte-chimique-hélium-filtrée.webp)