Mécanismes de calcification et de rigidité artérielle en
insuffisance rénale chronique
Mémoire
Alexandra Gauthier-Bastien
Maîtrise en médecine expérimentale
Maître ès sciences (M. Sc.)
Québec, Canada
iii
Résumé
En insuffisance rénale chronique (IRC), les désordres du métabolisme minéral osseux causent des pathologies osseuses et vasculaires. La calcification vasculaire s’accompagne de rigidité artérielle et de remodelage vasculaire. Nos objectifs sont de décrire le rôle du remodelage vasculaire dans le développement de la rigidité et de la calcification artérielle et de décrire les anomalies osseuses corticales en IRC. L’IRC et la calcification vasculaire sont induites par néphrectomie 5/6 et supplémentation en calcium, phosphore et vitamine D. La diminution de la détection en immunofluorescence d’α-actine du muscle lisse et d’élastine et l’augmentation d’ostéocalcine et d’endothéline-1 sont associées à l’augmentation de la calcification vasculaire et de la vélocité de l’onde de pouls. Ces atteintes vasculaires s’accompagnent d’une diminution du volume, de l’aire et de l’épaisseur de l’os cortical tibial. Ainsi, la calcification artérielle s’accompagne d’un remodelage vasculaire responsable, du moins partiellement, de la rigidité artérielle et s’accompagne aussi de perte osseuse corticale.
v
Abstract
In chronic kidney disease (CKD), mineral metabolism disorders cause vascular and bone disease. Vascular calcification is associated with vascular remodeling. Our objectives were to describe the role of vascular remodeling in arterial stiffness and calcification and to describe cortical bone abnormalities in CKD. CKD and vascular calcification were induced by 5/6 nephrectomy and calcium, phosphorus and vitamin D supplementation. The decrease in immunofluorescence detection of α-smooth muscle actin and elastin and the increase of osteocalcin and endothelin-1 were associated with increased arterial stiffness and calcification. These changes were accompanied by a decrease in volume, area and thickness of cortical bone of the tibia. Therefore, vascular calcification is associated with vascular remodeling which is responsible, at least in part, for arterial stiffness and is also accompanied by cortical bone loss.
vii
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des tableaux ... xi
Liste des figures ... xiii
Liste des abréviations... xv
Remerciements ... xvii
Avant-propos ... xix
1. Introduction ... 1
1.1. Physiologie générale du rein ... 1
1.2. Physiologie des hormones régulant le calcium et le phosphore ... 3
1.2.1. Calcitonine ... 4
1.2.2. Hormone parathyroïdienne ... 5
1.2.3. Vitamine D ... 6
1.2.4. Fibroblast growth factor-23 ... 7
1.3. Insuffisance rénale chronique ... 8
1.3.1. Causes ... 8
1.3.2. Conséquences ... 8
1.3.3. Impact sur la régulation du métabolisme minéral osseux ... 9
1.3.3.1. Fibroblast growth factor-23 ... 9
1.3.3.2. Vitamine D ... 9
1.3.3.3. Phosphatémie et calcémie ... 10
1.3.3.4. Hormone parathyroïdienne ... 10
1.3.4. Modalités thérapeutiques ... 12
1.3.5. Mortalité cardiovasculaire ... 12
1.4. L’insuffisance rénale chronique et l’os ... 13
1.4.1. Anatomie et physiologie de l’os ... 13
1.4.1.1. Remodelage osseux ... 15
1.4.2. Pathologies osseuses ... 17
1.4.2.1. Facteurs de risque ... 18
1.4.2.2. Types d’atteintes... 18
viii
1.4.2.2.2. Os adynamique ... 19
1.4.2.2.3. Trouble de la minéralisation ... 20
1.5. Conséquences extra-osseuses du désordre du métabolisme minéral osseux ... 21
1.5.1. Calcification des tissus mous ... 21
1.5.2. Calcification vasculaire ... 22
1.5.2.1. Mécanismes de calcification ... 23
1.5.2.1.1. Inhibiteurs de la calcification ... 24
1.5.2.1.1.1. Fétuine-a ... 26
1.5.2.1.1.2. Protéine matricielle Gla ... 26
1.5.2.1.1.3. Ostéoprotégérine ... 26
1.5.2.1.2. Promoteurs de la calcification ... 27
1.5.2.1.2.1. Phosphatémie et calcémie ... 28
1.5.2.1.2.2. Vitamine D ... 29
1.5.2.1.2.3. Hormone parathyroïdienne ... 30
1.5.2.1.2.4. Fibroblast growth factor-23 ... 30
1.5.2.1.2.5. Inflammation ... 31
1.5.2.1.2.6. Endothéline-1 ... 31
2. Hypothèses ... 33
3. Vascular remodeling and media calcification increases arterial stiffness in chronic kidney disease ... 35
3.1. Résumé ... 36
3.2. Abstract ... 37
3.3. Manuscript ... 38
3.3.1. Introduction ... 38
3.3.2. Materials and methods ... 39
3.3.2.1. Animal experiments ... 39
3.3.2.2. Methods ... 40
3.3.2.2.1. Biochemical parameters ... 40
3.3.2.2.2. In vivo Micro-CT imaging ... 40
3.3.2.2.3. von Kossa staining ... 40
3.3.2.2.4. Immunofluorescence analysis ... 40
3.3.2.2.5. Analysis of data... 41
3.3.3. Results ... 41
3.3.3.1. Biochemical parameters ... 41
3.3.3.2. Hemodynamic parameters and pulse wave velocity ... 41
ix 3.3.3.4. Vascular remodeling ... 42 3.3.4. Discussion ... 42 3.3.5. Acknowledgments... 44 3.3.6. Declaration of interest ... 44 3.3.7. References ... 45 3.3.8. Legends to figures ... 47 4. Expression d’endothéline-1 ... 55 4.1. Méthodes ... 55 4.1.1. Immunofluorescence ... 55
4.1.2. PCR quantitative par transcription inverse... 55
4.1.3. Analyse des données ... 56
4.2. Résultats ... 56
5. Anomalies osseuses... 59
5.1. Méthodes ... 59
5.1.1. Prélèvements ... 59
5.1.2. Tomographie par ordinateur ... 59
5.1.3. Analyse des données ... 59
5.2. Résultats ... 59
6. Discussion ... 61
7. Conclusion ... 65
xi
Liste des tableaux
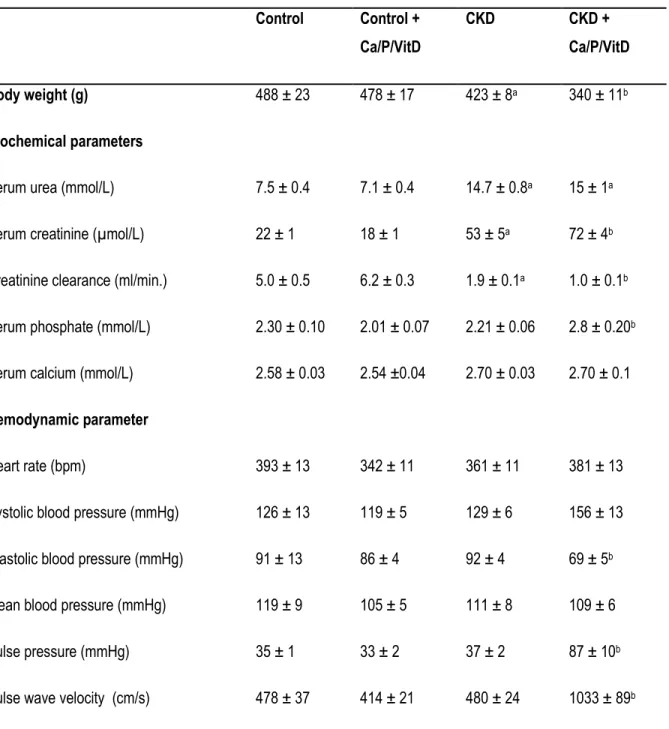
Tableau 3.1: Biochemical and hemodynamic parameters measured at the end of the study Tableau 4.1 : Séquences et température d’hybridation des amorces
xiii
Liste des figures
Figure 1.1 : Anatomie macroscopique du rein Figure 1.2 : Le néphron et ses capillaires
Figure 1.3 : Homéostasie du calcium et du phosphore Figure 1.4 : Synthèse et activation de la vitamine D
Figure 1.5 : Causes de mortalité chez les patients dialysés aux États-Unis entre 2008 et 2010 Figure 1.6 : Interaction entre les ostéoblastes et les ostéoclastes
Figure 1.7 : Rôle de BMP-2 et MSX2 dans la calcification vasculaire Figure 1.8 : Mécanismes impliqués dans la calcification artérielle en IRC Figure 1.9 : Remodelage vasculaire associé à l’IRC
Figure 3.1: 24-hour urinary calcium and phosphate excretion Figure 3.2: SBP, MBP, PP and PWV
Figure 3.3: Micro CT-Scan images
Figure 3.4: Von Kossa histology and detection of osteocalcin by immunofluorescence on thoracic aorta sections
Figure 3.5: Detection of α-SMA, elastin and collagen-1 by immunofluorescence on thoracic aorta sections
Figure 3.6: Correlation between PWV and either elastin or α-SMA Figure 4.1 : Détection d’endothéline-1 en immunofluorescence
Figure 4.2 : Expression d’endothéline-1 dans l’aorte thoracique par RT-qPCR Figure 4.3 : Concentration plasmatique d’ET-1
xv
Liste des abréviations
1,25(OH)2D : 1,25-dihydroxyvitamine D 1,25OHD : 1,25-dihydroxyvitamine D 25OHD : 25-hydroxyvitamine D α-SMA: α actine du muscle lisse ADMA : Diméthylarginine asymétrique ARNm : Acide ribonucléique messager Β2M : β-2-microglobuline
BMP-2 : « Bone morphogenetic protein-2 »
Ca/P/VitD : Diète enrichie en Ca (1,2 %) et en phosphore (1,2 %) et injections sous-cutanées de vitamine D (0,5μg/kg, 3x/semaine)
CathS : Cathepsine S
C-FMS: « Colony-stimulating factor 1 receptor »
CIHR: « Canadian Institutes of Health Research » CKD : « Chronic kidney disease »
CKD-MBD : Désordres du métabolisme minéral osseux en IRC CPP : « Calciprotein particles »
ET : Endothéline
ETA : Récepteur A de l’ET ETB : Récepteur B de l’ET
FGF-23 : « Fibroblast growth factor-23 » FGFR1: Récepteur du FGF-23
FRSQ: Le Fonds de la recherche en santé du Québec Gas 6 : « Growth arrest specific-6 »
HyAp : Hydroxyapatite IL: Interleukine
IRC : Insuffisance rénale chronique
M-CSF: « Macrophage colony-stimulating factor ». MGP : Protéine matricielle Gla
MMP : Métalloprotéinase matricielle MSX2 : « Msh homeobox 2 » OPG : Ostéoprotégérine
PBS : « Phosphate buffer saline »
PCR : Réaction en chaîne par polymérase PTH : Hormone parathyroïdienne
PTH1R: Récepteur-1 de la PTH PTHrP: « PTH-related protein » PWV : « Pulse wave velocity »
RANK: « Receptor activator of nuclear factor kappa B » RANKL: « RANK ligand »
SRA : Système rénine-angiotensine
RT-qPCR : PCR quantitative par transcription inverse RUNX2: « Runt related transcription factor-2 » SEM : Erreur standard de la moyenne
SM22: « Smooth muscle 22 »
xvi
TGF-α : « Transforming growth factor-α » TNF-α: « Tumor necrosis factor-α »
TRPV5/6 : « Transient receptor potential cation channel subfamily V 5 et 6 » UV : Ultraviolet
VDR : Récepteur de la vitamine D
VEGF: Facteur de croissance de l’endothélium vasculaire VOP : Vélocité de l’onde de pouls
xvii
Remerciements
Je ne peux que remercier tous ces gens, que ce soit au laboratoire ou dans la vie, qui m’ont aidée, conseillée ou encouragée durant ce périple.
Je remercie Dr Richard Larivière et Dr Mohsen Agharazii, vous sans qui rien n’aurait été possible, de m’avoir accordée votre confiance et d’avoir cru que je pouvais y arriver. Merci pour tout le temps, l’énergie et la passion que vous avez investis dans ma réussite. Pareils remerciements au Dr Fabrice Mac-Way, pour tous ces enseignements et ces précieux conseils.
Merci à mon équipe de travail débordante de personnalités extraordinaires. Remerciements au Dr Roth-Visal Ung pour ton aide offerte à tout moment et pour tes réponses patientes à mes milliards de questions farfelues journalières. Merci à Mélissa Legault, pour ton accueil sympathique, à Sylvain Picard, de démontrer une passion pour la science tellement rafraîchissante et à Éric Côté, Catherine Fortier, Dre Caroline Basoni, Dr Olivier Désy et Dre Karine Marquis pour vos précieux conseils, votre support et votre amitié.
Remerciements à Aude Bellavance-Laramée, Mylène Bilodeau-Mercure, Jean-Philippe Blais, Caroline Corbeil et Gabrielle Lalancette, parce que votre amitié, votre écoute et vos propositions de sorties extravagantes ont grandement contribué à ma réussite.
Je remercie aussi Cindy et Kévin Gauthier-Bastien, Sylvie Gauthier et Réginald Bastien de m’avoir soutenue sans relâche et de m’avoir inculqué dès le début de mon cheminement scolaire ce que je comprends aujourd’hui être la plus belle devise de travail qui soit : « Travaille fort, mais surtout….garde le sourire! ».
Je conclus en remerciant Philippe Deschênes d’avoir parcouru cette aventure avec moi si patiemment.
xix
Avant-propos
Le chapitre 3 présente l’article « Vascular remodeling and media calcification increases arterial stiffness in chronic kidney disease » accepté pour publication dans la revue « Clinical and Experimental Hypertension ». L’étude présentée a été entièrement réalisée dans le cadre de mes études à la maîtrise en médecine expérimentale à l’Université Laval et a été mise sur pied grâce à la collaboration entre les Dr Richard Larivière, Fabrice Mac-Way, Marcel Lebel et Mohsen Agharazii. J’ai réalisé les expérimentations animales et de laboratoire conjointement avec le Dr Roth-Visal Ung et la rédaction du manuscrit sous la supervision des Dr Richard Larivière et Mohsen Agharazii.
1
1. Introduction
1.1. Physiologie générale du rein
Les reins sont des organes rétropéritonéaux localisés dans l’abdomen supérieur de chaque côté de la colonne vertébrale. Ils ont une masse entre 120 et 150g chacun.Ils filtrent plus de 170L de plasma par jour pour former l’urine et ils assurent ainsi l’excrétion de plusieurs produits du métabolisme comme l’urée, la créatinine et l’acide urique. Ils ajustent aussi l’excrétion d’eau, de sodium, de calcium, de phosphate et de potassium. Les reins sont finalement impliqués dans la sécrétion d’hormones comme la rénine, les prostaglandines, le monoxyde d’azote (NO), l’endothéline (ET), l’érythropoïétine et la 1,25-dihydroxyvitamine D (1,25OHD).[1]
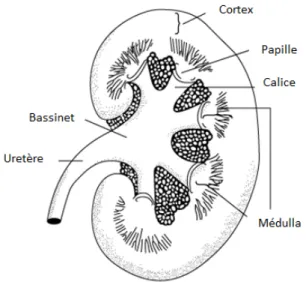
L’unité fonctionnelle du rein est le néphron (figure 1.1). Ils sont présents au nombre de 1 à 1,3 millions dans chaque rein. Chaque néphron est formé d’un glomérule et d’un tube divisé en plusieurs segments. Le glomérule est formé de capillaires entre une artériole afférente et une efférente et est entouré de la capsule de Bowman.Le tubule proximal origine dans la capsule et est composé du tubule contourné proximal suivi du tubule droit proximal. Il est suivi de l’anse de Henle, qui est divisée en trois segments: la branche descendante mince, la branche ascendante mince et la branche ascendante épaisse. Se trouvent ensuite le tubule contourné distal et le tubule collecteur dans lequel plusieurs néphrons vont se drainer. Le tubule collecteur médullaire se draine dans les calices, qui sont suivis du bassinet, de l’uretère et finalement de la vessie (figure 1.2).[1]
2
Figure 1.1 : Le néphron et ses capillaires
Tiré et traduit de : Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J : Harrisson’s Principles of
Internal Medicine, 18e édition : www.accessmedicine.com.
Figure 1.2 : Anatomie macroscopique du rein
3
Lors de la fonction de filtration du sang par les reins, il y a tout d’abord formation d’un ultrafiltrat avec passage d’une partie du plasma de l’artériole afférente et des capillaires glomérulaires dans la capsule de Bowman, puis dans les tubules. C’est durant son passage à travers les tubules que l’ultrafiltrat se transforme peu à peu en urine, suite à la sécrétion et à la réabsorption d’eau, d’ions et de produits du métabolisme. Ces modifications de la composition de l’urine sont effectuées par les cellules tubulaires qui, selon leur localisation, ont des fonctions différentes. En somme, il y a réabsorption de 99 % de l’ultrafiltrat et excrétion de 1 à 2L d’urine par jour.[1]
1.2. Physiologie des hormones régulant le calcium et le phosphore
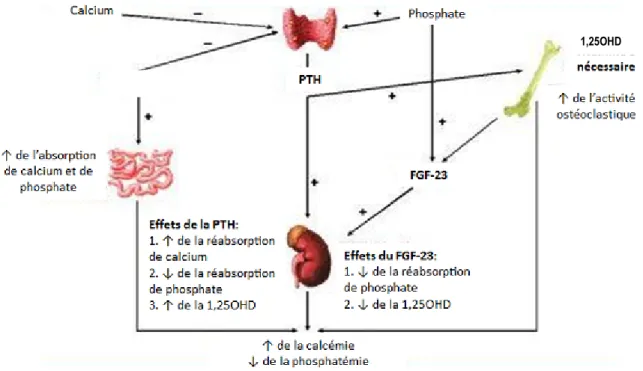
Le calcium et le phosphore sont des constituants essentiels de l’os. Le calcium est aussi impliqué, entre autres fonctions, dans la transmission neuronale et dans plusieurs cascades de signalisation cellulaire. Le métabolisme phosphocalcique permet donc le maintien de niveaux sériques de calcium ionisé et de phosphore adéquats, soit entre 1,1 et 1,3 mmol/L et 0,75 et 1,45 mmol/L respectivement.[2-3] La calcitonine, l’hormone parathyroïdienne (PTH), la 1,25OHD et le « fibroblast growth factor-23 » (FGF-23) sont toutes des hormones régulatrices du métabolisme du phosphore, du calcium et de l’os (figure 1.3).
4
1,25OHD
Figure 1.3 : Homéostasie du calcium et du phosphore
La sécrétion de PTH par les glandes parathyroïdiennes, principalement régulée par la calcémie, la phosphatémie et la concentration sanguine de 1,25OHD, a pour effet d’augmenter la calcémie et d’abaisser la phosphatémie. Ces effets sont médiés par l’augmentation de l’activité ostéoclastique de l’os et l’augmentation de la réabsorption de calcium, la diminution de la réabsorption de phosphate et l’augmentation de la production de 1,25OHD par le rein. La 1,25OHD augmente l’absorption intestinale de phosphate et de calcium. Le FGF-23, dont la production est principalement régulée par la phosphatémie, diminue la réabsorption rénale de phosphate et diminue la production rénale de 1,25OHD.
Tiré et traduit de: J Am Board Fam Med, 2009. PTH : hormone parathyroïdienne, FGF-23 : « fibroblast growth factor-23 » et 1,25OHD: 1,25-dihydroxyvitamine D.
1.2.1. Calcitonine
La calcitonine est principalement sécrétée par la glande thyroïde. Ses actions sont opposées à celles de la PTH.En agissant sur les récepteurs de la calcitonine présents sur les ostéoclastes, elle inhibe ces derniers et diminue ainsi la résorption osseuse. Elle diminue aussi la réabsorption tubulaire de calcium par action sur les récepteurs de la calcitonine des cellules tubulaires distales. Ces deux actions conjuguées diminuent les niveaux sériques de calcium.[4] Les concentrations normales sériques sont < 2,93 pmol/ml chez les hommes et <1,46 pmol/ml chez les femmes.[5] Le rôle physiologique semble de faible importance et, en dehors de situations de grande demande en
5
calcium, ni les niveaux trop élevés ni les niveaux trop bas ne semblent avoir de conséquences sur la masse osseuse et sur la régulation de la calcémie.[6]
1.2.2. Hormone parathyroïdienne
La PTH est sécrétée par les parathyroïdes, avec une concentration normale sérique de 1,0 à 6,5 pmol/L.[5]Elle agit principalement en se liant au récepteur PTHR1, mais deux autres récepteurs, dont les fonctions demeurent peu connues, ont été identifiés. La PTH augmente la réabsorption tubulaire rénale du calcium et la résorption osseuse (figure 1.3).[7] Elle stimule aussi l’activité de la 1-α-hydroxylase et donc la conversion de la 25-hydroxyvitamine D (25OHD) en 1,25OHD dans les cellules tubulaires rénales (figure 1.4). Toutes ces actions combinées augmentent la calcémie.La PTH diminue aussi la réabsorption rénale de phosphate.[8]
L’augmentation de la résorption osseuse par la PTH semble plutôt provenir d’une sécrétion continue. La sécrétion intermittente, quant à elle, a des effets anaboliques qui permettent de compenser la résorption osseuse résultant de l’élévation continue. La PTH diminue l’apoptose des ostéoblastes, stimule leur prolifération et leur maturation et favorise la différenciation des cellules ostéo-progénitrices de la moelle en ostéoblastes. Cette activité anabolique pourrait résulter de la modulation de la sécrétion de certains facteurs de croissance, comme la diminution de la sclérostine et la sécrétion de facteur de croissance insulinique. La sclérostine est une protéine qui inhibe la différenciation ostéoblastique en diminuant la stabilisation de la β-caténine par inhibition de la voie de signalisation Wnt.[9-11]
La concentration sérique de calcium, dont les changements sont détectés par les récepteurs du calcium dans la parathyroïde, est le principal régulateur de la sécrétion de PTH (figure 1.3).[8] La diminution du calcium ionisé sanguin augmente à la fois la sécrétion de PTH et la prolifération des cellules parathyroïdiennes.La phosphatémie est aussi régulatrice, indirectement, par diminution des niveaux de calcium ionisé et par diminution de l’activité de la 1-α-hydroxylase, mais aussi directement, par augmentation de la prolifération des cellules parathyroïdiennes, de l’expression de l’acide ribonucléique messager (ARNm) de la PTH, de l’expression de prepro-PTH et de la sécrétion de PTH indépendamment des niveaux de calcium et de vitamine D.[12]Aussi, l’augmentation de la concentration de phosphate intracellulaire dans la glande parathyroïde augmente la durée de vie de l’ARNm de la PTH (figure 1.3).[10]
6
7-déhydrocholestérol
Calciférol
Rayons UVB (peau)
Apports alimentaires
25-hydroxylase (foie) 1-α-hydroxylase (rein)
1,25OHD 25OHD
1.2.3. Vitamine D
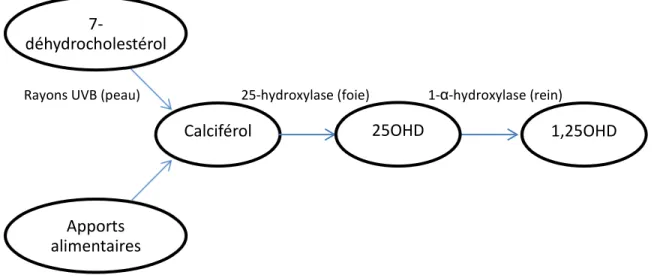
La vitamine D, ou calciférol, est une vitamine impliquée dans le métabolisme phosphocalcique dont l’apport peut être endogène ou exogène. Dans l’alimentation, elle se trouve sous forme d’ergocalciférol (vitamine D2) lorsqu’elle est d’origine végétale et de cholécalciférol (vitamine D3) lorsqu’elle est d’origine animale. Outre les apports alimentaires, la peau assure une production endogène de cholécalciférol par photoisomérisation de la provitamine D (7-déhydrocholestérol) grâce aux rayons ultraviolets B. L’activation du calciférol nécessite deux hydroxylations (figure 1.4). Dans le foie, il est hydroxylé par la 25-hydroxylase pour former de la 25OHD. Celle-ci représente la forme la plus abondante et circule majoritairement liée à la protéine liant la vitamine D. Une deuxième hydroxylation a ensuite lieu pour transformer la 25OHD en 1,25OHD, la forme active de la vitamine D.[3] 98 % de cette deuxième hydroxylation a lieu dans les mitochondries des tubules proximaux du rein.[12] L’augmentation des concentrations de phosphate et de calcium intracellulaires réduit l’efficacité de la 1-α-hydroxylase.[3, 13]
Figure 1.4 : Synthèse et activation de la vitamine D
Le calciférol provient de deux sources, soit les apports alimentaires en ergocalciférol et en cholécalciférol et la production par la peau de cholécalciférol par photoisomérisation du 7-déhydrocholéstérol par les rayons UVB. Le calciférol est ensuite hydroxylé en 25OHD par la 25-hydroxylase dans le foie, puis hydroxylé une deuxième fois en 1,25OHD par la 1-α-hydroxylase dans les reins. La 1,25OHD constitue la forme active de la vitamine D.
25OHD : 25-hydroxyvitamine D, 1,25OHD : 1,25-dihydroxyvitamine D et UV : ultraviolet.
La 1,25OHD agit en se liant à un récepteur nucléaire, le récepteur de la vitamine D (VDR). L’activation du VDR a pour effet d’augmenter l’absorption intestinale de phosphate et de calcium
7
notamment en augmentant l’expression de « transient receptor potential cation channel subfamily V 5 et 6» (TRPV5/6), des transporteurs du calcium, et de calbindines 9K, des protéines liant le calcium.[3] Des VDR sont aussi présents dans les cellules parathyroïdiennes et leur activation diminue la synthèse et la sécrétion de PTH.[10, 12] L’activation des VDR sur les ostéoblastes augmente leur expression de «receptor activator of nuclear factor kappa B ligand» (RANKL). Cela a pour effet d’augmenter la différenciation et l’activation des ostéoclastes qui participent à la résorption osseuse.[3] La 1,25OHD est aussi impliquée dans la différenciation des cellules ostéo-progénitrices de la moelle en ostéoblastes.[12]
1.2.4. Fibroblast growth factor-23
FGF-23 est une protéine produite par les ostéocytes et, dans une moindre mesure, par les ostéoblastes et les cellules parathyroïdiennes.[10, 14-15] Sa concentration normale sérique est d’environ 50 RU/ml.[10]Elle est principalement impliquée dans la régulation de la phosphatémie. En diminuant l’expression des co-transporteurs sodium-phosphore de type 2a et 2c à la surface apicale des cellules épithéliales du tubule proximal du rein, elle diminue la réabsorption rénale de phosphate.[15-16] Elle diminue aussi l’expression de ces mêmes co-transporteurs dans l’intestin, ce qui diminue l’absorption intestinale de phosphate.[15]Elle inhibe la synthèse de 1,25OHD dans les reins en diminuant l’expression de la 1-α-hydroxylase et augmente la dégradation de la 1,25OHD en augmentant l’expression de la 25-hydroxyvitamine D-24-hydroxylase.[16] Ces deux effets se conjuguent donc pour diminuer les niveaux de 1,25OHD et l’absorption intestinale de phosphate et de calcium. Le FGF-23 agit aussi dans la parathyroïde où elle inhibe la transcription et la sécrétion de PTH.[17] Les pathologies qui s’accompagnent d’une sécrétion inappropriée de FGF-23 causent donc des pertes importantes de phosphate qui résultent en de l’hypophosphatémie et de l’ostéomalacie ou du rachitisme.[15] La diminution de la phosphatémie et la hausse de 1,25OHD exercent une rétroaction positive sur la sécrétion de FGF-23.[15-16]
Les effets du FGF-23 sont médiés par l’activation de récepteurs du FGF (FGF-R). Toutefois, l’affinité du FGF-23 pour ses récepteurs est faible et cette activation dépend donc de klotho, son co-récepteur. Klotho est exprimé majoritairement dans les tubules rénaux.[14, 17] L’affinité du FGF-23 est plus grande pour le complexe klotho-FGF-R que pour le FGF-R seul.[18]
8
1.3. Insuffisance rénale chronique
L’insuffisance rénale chronique (IRC) se définit par une perte de fonction rénale lente et progressive, déterminée par la diminution du taux de filtration glomérulaire.[19] Les patients atteints présentent fréquemment de l’hypertension. Une protéinurie persistante, des petits reins échogènes dans les stades plus avancés et une variété d’autres signes peuvent aussi suggérer le diagnostic. En général, le taux de filtration glomérulaire s’abaisse sous 10-15 ml/min./1,73m2 avant l’apparition de symptômes. Ils sont donc rares dans les stades initiaux de la maladie. Ils sont non spécifiques et très variés et incluent, entre autres, de la fatigue, de la nycturie, du prurit et des troubles gastro-intestinaux.[20]
1.3.1. Causes
En Amérique du Nord et en Europe, le diabète et l’hypertension sont responsables de la vaste majorité des cas d’IRC.[19-20] Ces deux pathologies coexistent fréquemment; la majorité des patients atteints de diabète de type 2 présentant de l’hypertension.[21] Le risque d’IRC augmente avec la hausse de la pression artérielle (principalement systolique), dès qu’elle se situe au-dessus de la normale.[22-23] Dans ces cas de glomérulosclérose hypertensive, on retrouve à la fois des signes d’hyperperfusion et d’hypertrophie glomérulaire et de l’ischémie glomérulaire et post-glomérulaire, de l’inflammation et de l’atrophie tubulaire. Par ailleurs, un cercle vicieux se développe, la perte d’autorégulation du flot sanguin rénal et la diminution du taux de filtration glomérulaire accentuant le coefficient de relation entre la pression artérielle et les dommages rénaux.[23]
Le diabète est responsable de 44 % des nouveaux cas d’IRC terminale. Les lésions microvasculaires du diabète causent des atteintes glomérulaires avec épaississement de la membrane basale glomérulaire et perte de podocytes.[21]Les glomérulonéphrites, les maladies kystiques, les maladies tubulointerstitielles chroniques ainsi que d’autres atteintes urologiques peuvent aussi causer une IRC.[20]
1.3.2. Conséquences
Avec la perte de fonction rénale, on observe une rétention de sodium et d’eau et, conséquemment, une expansion du volume extracellulaire. Pour autant que les apports en eau ne dépassent pas la capacité de clairance par les reins, la natrémie et l’osmolalité sont maintenus. Toutefois, l’augmentation de volume peut causer des symptômes comme de l’œdème périphérique ou contribuer à l’hypertension. Les maladies rénales représentent d’ailleurs la principale cause
9
d’hypertension secondaire. Mis à part la rétention sodée, la sécrétion excessive de rénine et l’hyperactivité du système nerveux sympathique ont été incriminées.[19]
Par diminution de l’excrétion des acides endogènes, l’IRC peut aussi s’accompagner d’acidose métabolique, généralement lorsque le taux de filtration glomérulaire descend sous 20-25 ml/min.[24] L’anémie et l’hyperkaliémie sont aussi d’autres conséquences potentielles de l’IRC.[19]
1.3.3. Impact sur la régulation du métabolisme minéral osseux
En IRC, la réabsorption du calcium, l’excrétion de phosphate et la synthèse de la 1,25OHD sont affectées et on observe donc, tôt dans l’évolution de la maladie, des altérations du métabolisme minéral osseux. Ces désordres se présentent souvent par une hypocalcémie, une hyperphosphatémie, une déficience en 1,25OHD, une hyperparathyroïdie et une hausse du FGF-23.[1] Malgré que la chronologie selon laquelle ces désordres apparaissent en IRC suscite encore des questionnements, il semble que la hausse de FGF-23 soit la première manifestation, suivie de la déficience en 1,25OHD. L’hyperphosphatémie, l’hypocalcémie et l’hyperparathyroïdie surviennent dans les stades plus avancés d’IRC.[25]
1.3.3.1. Fibroblast growth factor-23
Avant même qu’il y ait augmentation de la phosphatémie, une hausse de FGF-23 survient en réponse à la diminution de l’excrétion de phosphate et par diminution de l’excrétion de FGF-23 par les reins. Cette hausse permet d’augmenter l’excrétion de phosphate par les néphrons fonctionnels restants et assure donc le maintien de la phosphatémie dans les stades initiaux de l’IRC.[25]Les concentrations sériques atteignent 500 RU/ml au début de la maladie rénale et plus de 10 000 RU/ml chez les patients dialysés chroniques.[10, 15] De plus, les niveaux de klotho sont abaissés en IRC.[26] Ce phénomène cause une résistance au FGF-23 et en augmente les concentrations. Les facteurs pouvant expliquer cette diminution de klotho incluent l’inflammation et le stress oxydatif.
1.3.3.2. Vitamine D
Puisque le FGF-23 inhibe la synthèse de 1,25OHD et augmente sa dégradation, la déficience en 1,25OHD survient aussi dans les stades initiaux de la maladie. Une étude regroupant des patients à différents stades d’IRC a reconnu le FGF-23 comme prédicteur de la déficience en 1,25OHD, indépendamment de la phosphatémie et du taux de filtration glomérulaire.[25] La perte de fonction rénale diminue la 1-α-hydroxylation de la 25OHD, causant ainsi une diminution de la concentration
10
de 1,25OHD. De plus, certains patients présentent des déficiences en 25OHD indépendantes de l’IRC, par diminution des apports alimentaires et/ou de l’exposition au soleil.[12, 14]
1.3.3.3. Phosphatémie et calcémie
La phosphatémie demeure normale au début de la maladie. Toutefois, elle augmente progressivement avec le déclin de la fonction rénale et la diminution de l’excrétion rénale de phosphate.[27] L’hyperparathyroïdie chez ces patients est aussi impliquée dans l’aggravation de l’hyperphosphatémie. En dehors de ce contexte pathologique, la PTH tend à diminuer la réabsorption de phosphate, et donc à diminuer la phosphatémie. Toutefois, lorsque la PTH est trop élevée et que la perte de fonction rénale s’aggrave, la phosphaturie ne peut plus s’élever davantage, mais les effets osseux de la PTH continuent de s’accentuer. Cela signifie qu’il y a une relâche osseuse de calcium et de phosphate de plus en plus importante. C’est alors que la PTH amplifie l’hyperphosphatémie.
L’hypocalcémie résulte d’un ensemble de facteurs dont la baisse des taux de 1,25OHD, la rétention de phosphate et la résistance périphérique à l’action de la PTH.[3, 27]
1.3.3.4. Hormone parathyroïdienne
La glande parathyroïdienne répond principalement aux variations de calcium et l’hypocalcémie en IRC est donc impliquée dans le développement d’hyperparathyroïdie. L’hyperphosphatémie et la déficience en 1,25OHD sont aussi impliquées. Dans la population atteinte d’IRC, on retrouve d’ailleurs une association entre les niveaux de PTH intacte et des niveaux abaissés de 1,25OHD.[27] Cela s’explique par l’inhibition physiologique de la 1,25OHD sur la sécrétion de PTH. L’accumulation de divers fragments de PTH qui ont parfois des effets biologiques différents ou même antagonistes (fragment 7-84) contribue à l’élévation de la PTH sans qu’il y ait nécessairement augmentation de l’activité de la PTH.[10, 28] On observe aussi une diminution de l'expression des récepteurs de la PTH dans ses tissus cibles, comme le rein. Cette baisse, qui est impliquée dans le développement de l’hyperparathyroïdie, serait indépendante des variations des niveaux de PTH et de phosphate et pourrait être expliquée par la baisse des niveaux de 1,25OHD.[12]
Dans la parathyroïde, en IRC, on observe aussi une diminution des récepteurs de la 1,25OHD et du calcium, du FGF-R1 et de klotho.[10]Puisque la 1,25OHD, le calcium et le FGF-23 diminuent la sécrétion de PTH, la diminution de leurs récepteurs et co-récepteurs dans la parathyroïde pourrait
11
expliquer des changements de la relation entre la calcémie et la PTH (résistance à la suppression de la sécrétion de PTH par le calcium) et causer de l’hyperparathyroïdie.[10, 12]
L’hyperparathyroïdie en IRC origine donc dans les stades initiaux de la maladie d’une augmentation de production et de sécrétion qui consiste en une adaptation aux autres désordres du métabolisme phosphocalcique en IRC. Plus tardivement toutefois, la stimulation prolongée par les taux élevés de phosphate et la baisse de calcium et de 1,25OHD causent l’hypertrophie et la prolifération cellulaire de la parathyroïde avec augmentation de la masse de la glande.[12, 29] En effet, des apports élevés en phosphate en IRC causent une augmentation de l’expression de « transforming growth factor-α » (TGF-α) dans la parathyroïde causant la prolifération des cellules parathyroïdiennes. De plus, à la fois la 1,25OHD et une diète riche en calcium préviennent cette prolifération en augmentant l’expression de p21 et en prévenant l’augmentation de production de TGF-α.[30-31]
Le volume de la glande a été associé avec les taux de PTH sériques et l’augmentation de volume pourrait donc causer de l’hyperparathyroïdie.[29] De plus, cette hyperplasie diffuse s’accompagne éventuellement d’hyperplasie nodulaire avec prolifération monoclonale et développement d’adénomes autonomes.[32] Dans les régions avec formation de nodules, on observe une diminution de l’expression des récepteurs du calcium et de la 1,25OHD plus grande que dans les régions avec hyperplasie diffuse. Ces patients présentent de l’hyperparathyroïdie tertiaire (réfractaire) qui proviendrait non seulement de l’augmentation du volume de la glande, mais aussi de la diminution de la réponse aux éléments inhibiteurs de la sécrétion et de la production autonome. Elle se manifeste cliniquement par de l’hypercalcémie et une hyperparathyroïdie sévère qui ne répond plus aux agents qui devraient réguler sa sécrétion.[29]
Ces atteintes du métabolisme minéral osseux se composent donc d’anomalies du calcium, du phosphate, de la vitamine D, du FGF-23 et de la PTH. Comme le métabolisme phosphocalcique est important pour la physiologie de l’os et de son remodelage, les désordres en IRC ne sont pas sans conséquences cliniques et causent des pathologies osseuses, des fractures, des douleurs osseuses, de la calcification vasculaire et de la calcification des tissus mous. Comme les atteintes biochimiques, osseuses et extra-osseuses sont intimement liées entre elles, l’appellation « chronic kidney disease mineral and bone disorder» (CKD-MBD) a été retenue pour regrouper les atteintes minérales et osseuses en IRC.
12
1.3.4. Modalités thérapeutiques
Les possibilités de traitement de l’IRC terminale incluent l’hémodialyse, la dialyse péritonéale ou la transplantation rénale. Cette dernière option constitue le traitement de choix et représente une augmentation de la durée et de la qualité de vie par rapport à la dialyse. [33-34] Les critères d’initiation de la dialyse chronique incluent la présence de symptômes urémiques, d’hyperkaliémie, d’augmentation de volume extracellulaire ou d’acidose métabolique qui ne répondent pas aux traitements, de diathèse hémorragique ou d’un taux de filtration glomérulaire sous 10ml/min./1,73m2. Aucune différence quant à la mortalité n’a été démontrée selon le type de dialyse choisie et les préférences du patient guident la décision.[34]
Toutefois, si la sévérité de l’IRC n’est pas suffisante pour justifier la dialyse ou la transplantation ou si ces traitements ne permettent pas d’éviter toutes les complications, celles-ci peuvent aussi être traitées individuellement. L’hypertension et l’augmentation de volume peuvent être traitées par restriction sodique et/ou diurétiques.[19] De plus, le traitement des dyslipidémies, de l’anémie et du diabète revêt une importance toute particulière dans cette population afin de réduire le risque de maladies cardiovasculaires.[35]
Le traitement des désordres du métabolisme phosphocalcique inclut le traitement de l’hyperphosphatémie avec des chélateurs du phosphore. Les chélateurs non-calciques sont privilégiés, surtout lorsque le produit phosphocalcique est élevé. Toutefois, ils sont plus chers et les chélateurs calciques sont donc parfois utilisés et sont à privilégier chez les patients avec une calcémie basse. La vitamine D est le principal traitement de l’hyperparathyroïdie. Dans les cas réfractaires, les calcimimétiques ou la parathyroïdectomie peuvent être utilisés. Toutefois, l’efficacité du traitement des désordres du métabolisme minéral osseux pour réduire la mortalité demeure controversée.[35]
1.3.5. Mortalité cardiovasculaire
L’IRC est un facteur de risque de maladies cardiovasculaires. En IRC terminale, elles représentent la principale cause de mortalité, avec environ 40 % des décès. Les deux principales causes de mortalité cardiovasculaire chez les dialysés sont les arythmies et l’insuffisance cardiaque (figure 1.5).[36] Plusieurs facteurs de risque traditionnels de maladies cardiovasculaires sont fréquents dans la population atteinte d’IRC dont le diabète, l’hypertension, l’âge, l’obésité et les dyslipidémies. [37-38] Toutefois, des facteurs de risque non-traditionnels typiques de cette population ont aussi été
13
associés avec le développement de maladies cardiovasculaires dont la diminution de la filtration glomérulaire, l’augmentation des apports en calcium, l’hyperphosphorémie, l’hyperparathyroïdie et l’augmentation des concentrations de FGF-23.[39-41] La calcification artérielle et la rigidité artérielle, complications fréquentes de l’IRC, sont aussi des facteurs de risques de maladies cardiovasculaires dans cette population.[40, 42]
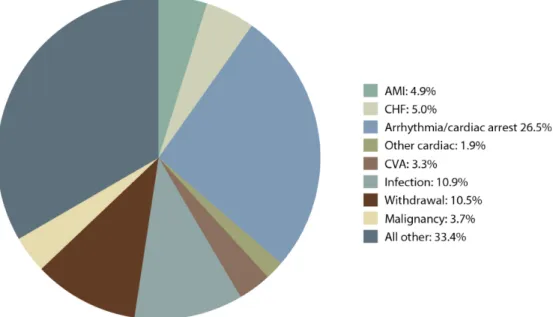
Figure 1.5 : Causes de mortalité chez les patients dialysés aux États-Unis entre 2008 et 2010
Graphique présentant les principales causes de mortalité chez les patients dialysés aux États-Unis entre 2008 et 2010. Les maladies cardiovasculaires représentent la principale cause de mortalité avec 41,6 %. La majorité de cette mortalité est attribuable aux arythmies et aux arrêts cardiaques qui représentent 26,5 % de la mortalité toutes causes confondues. Tiré intégralement de : Collins, A.J., et al., US Renal Data System 2012 Annual Data Report. Am J Kidney Dis, 2013.
61(1 Suppl 1): p. A7, e1-476. AMI : Infarctus du myocarde, CHF : Insuffisance cardiaque chronique, « arrhythmia/cardiac
arrest » : arythmies et arrêts cardiaques, « other cardiac » : autres causes cardiaques, CVA : accident vasculaire cérébral, « withdrawal » : arrêt du traitement, « malignancy » : néoplasie et « all other » : autres causes.
1.4. L’insuffisance rénale chronique et l’os
1.4.1. Anatomie et physiologie de l’os
Les os possèdent deux compartiments principaux, soit l’os cortical et l’os trabéculaire. L’os cortical, qui constitue 80 % de la masse osseuse, forme les parois extérieures de l’os, alors que l’os trabéculaire se trouve à l’intérieur des limites de l’os cortical.L’os cortical est formé d’ostéons qui sont constitués de plusieurs couches concentriques de tissu osseux comportant en leur centre un canal de Havers dans lequel voyage une veine, une artère et un nerf.[15] L’os trabéculaire est formé
14
de plaques entrecoupées d’espaces intertrabéculaires.[15, 43] Ces plaques sont formées de lamelles parallèles de tissu osseux entourées d’une mince couche d’ostéoblastes avec quelques ostéoclastes (endostéum).[15] Les espaces intertrabéculaires abritent la moelle osseuse. La moelle osseuse rouge (hematopoïétique active) est composée de cellules hématopoïétiques et de cellules lipidiques, ces dernières occupant 50 % de l’espace. Selon l’os dont il est question, l’âge du patient et la demande hématopoïétique, on retrouve plutôt de la moelle jaune, dont les cellules hématopoïétiques ont été entièrement remplacées par des cellules lipidiques.[44]
Le tissu osseux est formé de tissu ostéoïde (matrice organique) et de composés inorganiques. Le tissu ostéoïde constitue environ le 1/3 de la masse osseuse et il est principalement composé de collagène de type 1, mais contient aussi d’autres protéines, comme la fibronectine, l’ostéonectine, l’ostéopontine et l’ostéocalcine, des lipides et des protéoglycans.[12, 45] La matrice inorganique est constituée majoritairement de cristaux d’hydroxyapatite sous forme hexagonale (Ca10(PO4)6(OH)2), mais contient aussi de l’hydroxyapatite sous forme monoclinique et du phosphate de calcium amorphe, ce dernier étant surtout présent dans les zones de formation osseuse active. En effet, le phosphate de calcium amorphe est initialement déposé, puis au cours des semaines suivantes, les cristaux d’hydroxyapatite se développent.[12, 45-47]Cette combinaison de protéines et de minéraux confère à l’os sa rigidité et sa résistance, par combinaison des propriétés de chaque constituant. Grossièrement, la matrice organique possède une bonne résistance à la traction et la matrice inorganique une bonne résistance à la compression.[47]
Les principales cellules constituant l’os sont les ostéocytes (95 %), les ostéoblastes (5 %) et les ostéoclastes (1-2 %).[9] Les ostéoblastes sont responsables de la sécrétion de la matrice extracellulaire osseuse et de sa minéralisation. Ils proviennent de la différenciation de cellules souches mésenchymateuses qui deviennent pré-ostéoblastes, puis ostéoblastes et ne se localisent qu’en surface de l’os. Lors du processus de production de la matrice extracellulaire calcifiée, ils restent figés dans cette matrice et deviennent alors des ostéocytes. Ces derniers sont impliqués dans la signalisation régulant le processus de minéralisation et dans la régulation de l’activité ostéoblastique et ostéoclastique.[48] Les ostéoclastes, quant à eux, sont responsables de la résorption de l’os. L’équilibre entre les actions des ostéoclastes et des ostéoblastes est donc crucial et hautement régulé.[3]
15
1.4.1.1. Remodelage osseux
Le remodelage osseux touche environ 15 % de la masse osseuse totale chaque année chez l’adulte. Il permet la résolution de microfractures spontanées et l’ajustement des os aux changements du centre de gravité et de l’axe du corps.[12] Ce sont les ostéocytes qui perçoivent ces changements mécaniques et initient le processus de remodelage. Ils permettent, en modulant leur production de sclérostine, la différenciation des cellules souches mésenchymateuses en ostéoblastes.[10-11] Les ostéoblastes peuvent aussi être stimulés par l’interleukine-1 et le facteur de nécrose tumorale α (TNF-α). Comme ils présentent des PTH1R et des VDR, leur activité peut aussi être modulée par la 1,25OHD et par la PTH.[3]
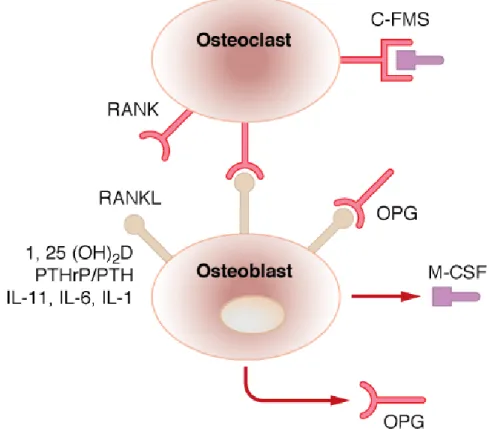
Les ostéoblastes activés stimulent la maturation et la différenciation des pré-ostéoclastes en ostéoclastes matures et activés par production d’interleukine-6 et 11, de « granulocyte macrophage colony-stimulating factor » (GM-CSF) et de « macrophage colony-stimulating factor » (M-CSF).[12, 43] Les ostéoblastes sont aussi des régulateurs de l’ostéoclastogénèse par un autre mécanisme, soit l’interaction entre RANKL, une protéine membranaire exprimée par les ostéoblastes et « receptor activator of nuclear factor kappa B » (RANK), une protéine exprimée sur les progéniteurs ostéoclastiques. L’interaction entre RANK et RANKL induit la différenciation et l’activation des ostéoclastes.[43]Les ostéocytes modulent aussi directement l’activité ostéoclastique en modifiant le ratio RANKL/ostéoprotégérine par la voie de signalisation Wnt/β-caténine.[11] L’ostéoprotégérine (OPG) est un récepteur soluble de RANKL qui empêche sa liaison à RANK (figure 1.6).[49]
16
Figure 1.6 : Interaction entre les ostéoblastes et les ostéoclastes
L’activation des ostéoclastes par les ostéoblastes est médiée par l’interaction entre le récepteur RANK des ostéoclastes et RANKL des ostéoblastes. L’OPG produite par les ostéoblastes bloque ce processus d’activation ostéoclastique en se liant à RANKL pour empêcher son interaction avec RANK, alors que le M-CSF produit par les ostéoblastes favorise cette activation des ostéoclastes par l’interaction RANK/RANKL en se liant au récepteur C-FMS présent sur les ostéoclastes. 1,25(OH)2D, PTH/PTHrP, IL-11, IL-6 et IL-1 stimulent la production de RANKL par les ostéoblastes.
C-FMS: « colony-stimulating factor 1 receptor », OPG: ostéoprotégérine, RANK: « receptor activator of nuclear factor
kappa B », RANKL: « RANK ligand », 1,25(OH)2D: 1,25-dihydroxyvitamine D, PTH: hormone parathyroïdienne, PTHrP:
« PTH-related protein », IL: interleukine, M-CSF: « macrophage colony-stimulating factor ».
Tous ces stimuli induisent ainsi la formation d’ostéoclastes activés par la fusion de progéniteurs ostéoclastiques mononucléés. Ces ostéoclastes activés s’arriment ensuite à la matrice extracellulaire par des intégrines αvβ3 et sécrètent, dans une région bien délimitée nommée lacune de Howship, des ions hydrogènes pour solubiliser les minéraux et des enzymes protéolytiques, comme l’acide phosphatase tartrate résistante (TRAP) et des cathepsines (B, D, L et K), pour dégrader la matrice organique.[3, 12]
17
La résorption est suivie par la production de matrice ostéoïde par les ostéoblastes recrutés. Celle-ci est ensuite minéralisée par déposition d’hydroxyapatite le long des fibres de collagène-1. Ce processus de minéralisation dépend du calcium et du phosphate disponible, mais aussi des ostéoblastes, dont l’activité alcaline phosphatase permet d’inactiver les inhibiteurs de la calcification.[3]
1.4.2. Pathologies osseuses
Considérant le rôle important du calcium, du phosphate et des hormones comme la PTH et la vitamine D dans le maintien de la physiologie de l’os, les désordres du métabolisme minéral en IRC ont des impacts sur le métabolisme osseux. L’incidence de fracture de hanche est d’ailleurs 14 et 17 fois plus élevée en IRC pour les hommes et pour les femmes respectivement que dans la population normale, témoignant d’un processus pathologique accentué dans cette population.[50]
Quatre types d’atteinte sont le plus fréquemment rencontrées, soit l’ostéite fibrosante, l’os adynamique, l’ostéomalacie et l’atteinte mixte. Dans la caractérisation de ces atteintes, quatre principaux éléments doivent être évalués soit le volume osseux, le taux de remodelage, la minéralisation et la quantification des dépôts métalliques (aluminium ou fer) sur la surface trabéculaire.[51] Les quatre atteintes se différencient par un taux de remodelage et une minéralisation différents, mais peuvent toutes se présenter avec un volume osseux normal, augmenté ou diminué.
La terminologie ostéodystrophie rénale regroupe toutes les atteintes osseuses en IRC et touche environ 60 % des patients en IRC terminale non-dialysés.[52] Toutefois, la prévalence d’ostéodystrophie rénale varie selon la population étudiée. Plus encore, la prévalence de chaque type d’atteinte varie selon les populations et selon les traitements reçus par les patients. Comme les traitements ne cessent d’évoluer, la prévalence de ces pathologies s’est aussi modifiée au cours des dernières décennies. La prévalence de l’ostéite fibrosante a diminué, mais il y a eu augmentation de la prévalence de l’os adynamique, qui est devenu l’atteinte la plus fréquente. Ce changement pourrait s’expliquer au moins en partie par le traitement plus agressif de l’hyperparathyroïdie. En effet, l’abaissement des niveaux de PTH permet de diminuer les atteintes de l’ostéite fibrosante, mais la suppression excessive cause un os adynamique.[53] Toutefois, avec les nouvelles recommandations internationales « kidney disease : improving global outcome » (KDIGO) qui recommandent de viser des valeurs de PTH sérique entre 2 et 9 fois les valeurs normales, la suppression excessive
18
deviendra peut-être moins fréquente et on pourrait observer une recrudescence d’ostéite fibrosante aux dépens de l’os adynamique.[51]
1.4.2.1. Facteurs de risque
Ainsi, les niveaux de PTH jouent un rôle important dans le développement de pathologies osseuses, que les valeurs soient trop basses (os adynamique) ou trop élevées (ostéite fibrosante). Cet impact provient du rôle important de la PTH dans l’initiation de la résorption osseuse par augmentation de l’expression de RANKL.[7]
L’acidose métabolique peut aussi causer des atteintes de la minéralisation.[24] Un processus actif de résorption se déroule en acidose métabolique avec activation des ostéoclastes et diminution de l’activité ostéoblastique.[53] Dans un modèle ex vivo chez la souris, l’acidose métabolique a diminué la synthèse ostéoblastique de collagène et l’activité alcaline phosphatase et stimulé la sécrétion ostéoclastique de β-glucuronidase, une enzyme lysosomale impliquée dans la résorption osseuse. Toutefois, même en présence d’un inhibiteur des ostéoclastes (calcitonine), une excrétion osseuse augmentée de calcium se produit en acidose métabolique, suggérant que le processus actif de dissolution s’additionne à une dissolution physicochimique passive.[54]
L’inflammation systémique chez les dialysés pourrait aussi s’ajouter aux éléments en cause dans les désordres osseux en IRC. Cette inflammation se caractérise par l’élévation chez les dialysés des niveaux sériques de protéine C réactive et de nombreuses cytokines pro-inflammatoires, comme les interleukines-1, 6 et 12 et le « Tumor necrosis factor-α » (TNF-α).[51, 55-56]Toutefois, le rôle de ce statut inflammatoire est controversé. Alors que dans plusieurs pathologies osseuses, l’inflammation augmente la résorption osseuse, en hémodialyse, certaines études démontrent l’effet inverse. Une étude clinique a d’ailleurs établi une association négative entre les niveaux sériques d’interleukine-6 et certains marqueurs du remodelage osseux (fragment N-terminal de l’ostéocalcine et « Betaisomerized C-terminal cross-linked peptide of collagen type I »).[56-57]
1.4.2.2. Types d’atteintes
1.4.2.2.1. Hyper-remodelage osseux
L’ostéite fibrosante est une atteinte qui se caractérise par un hyper-remodelage osseux. L’hyperparathyroïdie secondaire, complication fréquente de l’IRC, est la cause principale de cette augmentation du remodelage, car la PTH est un stimulateur important de l’activité ostéoblastique et
19
ostéoclastique.La sévérité de l’atteinte osseuse est d’ailleurs associée avec l’élévation de la PTH.À la biopsie, il y a une augmentation du nombre et de la taille des ostéoclastes. De plus, l’activité ostéoblastique augmente la quantité de tissu ostéoïde, mais de façon désorganisée avec modification de sa structure.[53] L’hyperparathyroïdie et l’augmentation du remodelage osseux ont en effet été associés avec la diminution de l’épaisseur et de la densité de l’os cortical et l’augmentation de la porosité de l’os cortical en IRC.[58] La diminution de l’épaisseur de l’os cortical a de plus été associée avec le développement de calcification de la média vasculaire dans un modèle animal d’insuffisance rénale induite par une diète riche en adénine.[59] Ces anomalies de l’os cortical s’accompagnent aussi d’un épaississement anormal de l’os trabéculaire.[58-59]
1.4.2.2.2. Os adynamique
L’os adynamique se caractérise par un remodelage diminué. En IRC, la prévalence de cette pathologie est encore plus élevée dans la population diabétique ou âgée.[52, 60] La PTH est encore une fois incriminée, car ce sont cette fois-ci des valeurs de PTH trop basses qui sont en cause. La baisse de la PTH intacte a d’ailleurs été associée avec une diminution de la prolifération ostéoblastique.[61] Comme l’IRC s’associe plutôt avec une hausse de PTH, les valeurs abaissées de PTH sont généralement de cause iatrogénique et sont le résultat d’une suppression excessive de la parathyroïde avec une parathyroïdectomie ou l’administration de dérivés de 1,25OHD ou de sels de calcium.[62]
La résistance osseuse à la PTH peut aussi être impliquée dans l’apparition de la pathologie malgré des valeurs normales ou légèrement élevées de PTH. L’accumulation de fragments de PTH qui inhibent la résorption osseuse cause aussi une élévation de la PTH qui ne s’associe pas nécessairement avec une augmentation du remodelage. Toutefois, la PTH n’est pas le seul facteur responsable de l’os adynamique. En effet, certains présentent un os adynamique même en présence d’hyperparathyroïdie secondaire. L’exposition à l’aluminium est une des causes d’os adynamique, mais est de moins en moins fréquente.[60] La dialyse a aussi été décrite comme facteur de risque, probablement à cause du calcium présent dans le dialysat. L’hémodialyse chronique avec des membranes en polyacrylonitrile est aussi associée avec une activité ostéoblastique et ostéoclastique plus basse que l’hémodialyse avec des membranes en cuprophane. Cette différence pourrait s’expliquer par un processus inflammatoire plus marqué avec les membranes en cuprophane.[63]
20
Le diagnostic repose sur les niveaux sériques de PTH et sur l’observation de la pathologie à la biopsie osseuse, ce dernier élément permettant un diagnostic définitif. La biopsie permet d’observer l’absence d’ostéoblastes activés et une diminution marquée de la résorption osseuse et de la production de collagène-1 par les ostéoblastes. Comme les autres pathologies osseuses, la plupart des patients atteints sont asymptomatiques. Toutefois, comme le remodelage est lent, le captage de calcium par les os est déficient et certains patients développent de l’hypercalcémie réfractaire et des calcifications vasculaires. L’association entre les apports en calcium et la calcification vasculaire est d’ailleurs plus forte chez les patients présentant un os adynamique que chez ceux présentant une ostéite fibrosante.[39] Les patients avec os adynamique sont aussi plus sujets aux fractures de hanche que les patients avec hyper-remodelage osseux.[50]
1.4.2.2.3. Trouble de la minéralisation
Le trouble de minéralisation, ou ostéomalacie, est l’atteinte la moins fréquente parmi les quatre atteintes mentionnées. Elle se caractérise par une vitesse de formation de la matrice ostéoïde plus grande que la vitesse de minéralisation, et donc, par une augmentation du tissu ostéoïde. Dans cette atteinte, le remodelage osseux est normal.[53]
Par le passé, l’ostéomalacie était principalement causée par l’accumulation d’aluminium, qui réduit la prolifération ostéoblastique, la production de collagène-1 et augmente l’activité ostéoclastique. Cet aluminium provenait de l’utilisation de sels d’aluminium comme chélateurs du phosphate et de la présence d’aluminium dans les solutions de dialyse. L’élimination de ces sources d’exposition a permis de diminuer la prévalence de l’ostéomalacie en IRC. La déficience en vitamine D a aussi été associée avec cette atteinte, mais elle est, tout comme l’accumulation d’aluminium, de plus en plus rare. [12, 53, 60]
Il y a donc trois atteintes pathologiques bien définies par des caractéristiques spécifiques en IRC, soit l’ostéite fibrosante, l’os adynamique et l’ostéomalacie. Toutefois, les pathologies ne se présentent pas toujours aussi distinctement et certains patients présentent parfois ce que l’on appelle une atteinte mixte. Ces patients présentent des caractéristiques d’atteinte causée par l’hyperparathyroïdie avec hyper-remodelage osseux et des troubles de la minéralisation tout à la fois. Ces patients présentent aussi une augmentation de volume du tissu ostéoïde et de la fibrose de la moelle.[53]
21
1.5. Conséquences extra-osseuses du désordre du métabolisme minéral osseux
Les désordres du métabolisme minéral osseux ont aussi des conséquences extra-osseuses avec un impact important sur la mortalité des patients en IRC. Il s’agit du développement de calcifications qui touchent les tissus mous comme les vaisseaux, les valves cardiaques, les viscères et les articulations.[60, 64]Ces calcifications peuvent être divisées selon leur localisation, soit valvulaire, vasculaire (intimale ou médiale) ou touchant les autres tissus mous. Selon le site de l’atteinte, les causes et les facteurs de risque varient. Les désordres du métabolisme du calcium et du phosphate constituent une cause importante de calcification extra-osseuse.[65] Le remodelage osseux étant intimement lié au métabolisme minéral, les atteintes osseuses discutées précédemment sont aussi associées aux complications extra-osseuses de l’IRC.[39]
1.5.1. Calcification des tissus mous
Les organes les plus souvent atteints par les calcifications viscérales sont les poumons, le cœur, les reins, les muscles squelettiques et l’estomac. Leur atteinte peut d’ailleurs s’associer à une perte de fonction de l’organe.[60, 64] Les calcifications péri-articulaires (aussi appelées calcinose tumorale), touchent plus souvent les hanches, les épaules, les coudes et les genoux et peuvent elles aussi s’associer à une perte de fonction.
Les sites des atteintes vont varier selon le profil biochimique du patient. Les calcifications qui surviennent dans un contexte d’hypercalcémie avec phosphatémie normale ou basse ont une prédilection pour les reins, les poumons et la muqueuse gastrique, alors que celles qui surviennent dans un contexte d’hyperphosphatémie avec calcémie normale ou diminuée ont une prédilection pour les reins et les vaisseaux.[66]
Les calcifications valvulaires peuvent être présentes dans la population générale et sont causées principalement par le stress mécanique et l’inflammation chronique. Toutefois, dans la population atteinte d’IRC, les désordres du métabolisme minéral osseux et le statut d’inflammation chronique constituent des facteurs de risque supplémentaires de calcification valvulaire.[67]Histologiquement, la pathologie se manifeste par la déposition de lipides et l’infiltration de macrophages, mastocytes et lymphocytes B et T, et possède donc plusieurs similitudes avec l’athérosclérose.[68-69] Un processus actif de formation osseuse a aussi été décrit dans les zones de calcification, avec présence d’ostéoblastes, d’ostéoclastes et de « bone morphogenetic protein-2 » (BMP-2), un
22
puissant inducteur de la calcification.[69]La calcification valvulaire, particulièrement la calcification de la valve mitrale, est associée avec une prévalence plus élevée de maladies cardiovasculaires et avec la mortalité à la fois dans la population générale et en IRC.[67, 69]
1.5.2. Calcification vasculaire
La calcification vasculaire est directement associée avec la diminution de la filtration glomérulaire et elle apparait rapidement dans l’évolution de la maladie, soit dès le stade 3.[49, 70] De plus, elle est associée avec une augmentation de la rigidité artérielle déterminée par la hausse de la vélocité de l’onde de pouls (VOP).[42, 71] La calcification artérielle et la rigidité artérielle sont toutes deux associées avec la mortalité cardiovasculaire et la mortalité toutes causes confondues en IRC.[42] La contribution de la calcification vasculaire et de la rigidité artérielle à cette surmortalité est reliée à la hausse de la pression artérielle et au développement d’hypertrophie ventriculaire gauche. Ces atteintes sont des éléments en cause dans le développement d’arythmies et d’insuffisance cardiaque.[40]
Chez les insuffisants rénaux, on retrouve deux types de calcifications vasculaires distinctes selon leur localisation dans le vaisseau, soit intimale et médiale. La calcification intimale, principalement localisée dans les artères de gros et de moyen calibre, se développe par calcification des plaques athérosclérotiques et survient donc dans la population générale (sans IRC), comme dans la population atteinte d'IRC. Elle s’associe avec des facteurs de risque traditionnels de maladies cardiovasculaires et cause des complications telles que l’ischémie et l’infarctus du myocarde.[37-38, 72]
Les calcifications médiales, aussi connues sous le nom de sclérose de Mönckeberg, affectent plus souvent les patients atteints d’IRC et de diabète et surtout les patients âgés. Elle est caractérisée par des dépôts diffus d’hydroxyapatite dans la média, une couche artérielle constituée en grande partie de cellules musculaires lisses vasculaires (VSMC), et ne s’associe pas nécessairement à un processus athérosclérotique. Les calcifications médiales sont associées à des facteurs de risque non-traditionnels de maladies cardiovasculaires et au développement de rigidité artérielle, d’hypertension, d’hypertrophie ventriculaire gauche, d’arythmies et d’insuffisance cardiaque.[39-40, 72]
La calciphylaxie, ou artériopathie urémique calcifiante, est une complication rare de l’IRC qui se caractérise par la calcification médiale et la thrombose des artères et des veines de petit et de moyen
23
calibre.[73]Il y a prolifération et épaississement de l’intima, infiltration de neutrophiles, lymphocytes et macrophages, développement de fibrose, occlusions thrombotiques, et conséquemment, ischémie et nécrose. Cette complication peut atteindre les organes internes et les muscles, mais touche plus fréquemment les tissus cutanés et sous-cutanés. Il y a cliniquement apparition de lésions douloureuses et nécrotiques, qui s’ulcèrent et peuvent s’infecter. La pathologie touche principalement les membres inférieurs et les extrémités et la mortalité est estimée à 80 %. Les facteurs de risque sont semblables à ceux de la calcification médiale, dont principalement les désordres du métabolisme phosphocalcique. Cela inclut l’hyperphosphatémie, l’hypercalcémie, l’augmentation du produit phosphocalcique et l’hyperparathyroïdie. Dans certains cas, cette complication peut être guérie avec la parathyroïdectomie, mais elle n’est pourtant pas spécifique à l’hyperparathyroïdie. Des patients avec des niveaux abaissés de PTH, des patients présentant un os adynamique ou même des patients ne souffrant pas d’IRC ont présenté de la calciphylaxie.[65, 74]
1.5.2.1. Mécanismes de calcification
La calcification vasculaire a longtemps été considérée comme un phénomène causé par le dépôt passif de phosphate de calcium. Elle apparaît maintenant comme un processus actif hautement régulé similaire à la formation osseuse.
Le processus débute dans des foyers de nucléation, soit des zones à risque pour la calcification vasculaire comme le sont les corps apoptotiques et les vésicules matricielles sécrétées par les VSMC. Les corps apoptotiques sont relâchés par les VSMC en apoptose alors que les VSMC viables sécrètent les vésicules matricielles. Ces dernières contiennent de l’hydroxyapatite, de l’alcaline phosphatase, des transporteurs de phosphate sodium dépendants, des annexines 2, 5 et 6 et une quantité réduite de protéines protectrices naturelles de la calcification. L’alcaline phosphatase est une enzyme qui dégrade le pyrophosphate, un inhibiteur naturel de la minéralisation. Lors de cette réaction de déphosphorylation, il y a libération de phosphate. Les annexines servent de canaux calciques et elles augmentent la quantité de calcium dans la vésicule.[15, 38, 75-76]
Aussi, les VSMC ont la capacité de se différencier en d’autres types cellulaires qui originent de cellules mésenchymateuses comme les ostéoblastes, les chondrocytes et les adipocytes. L’augmentation de l’expression de BMP-2 et de facteurs de transcription comme «runt related transcription factor-2 » (RUNX2), ostérix, « (sex determining region Y)-box 9 » (SOX9) et « msh homeobox 2 » (MSX2) induit la différenciation ostéoblastique ou chondrocytique des VSMC (figure
24
1.7 et 1.9). Les cellules différenciées sécrètent des protéines osseuses comme l’ostéocalcine, l’ostéopontine, l’ostéonectine et le collagène-1 et exercent une activité alcaline phosphatase.[38, 72] D’ailleurs, il y a détection de ces facteurs de transcription et de ces protéines caractéristiques du tissu osseux et diminution de la détection de protéines sécrétées par les VSMC dans les vaisseaux calcifiés des patients en IRC, témoignant de la différenciation cellulaire qui s’y produit.[77] Cette différenciation a été associée avec le développement de la calcification vasculaire.[65, 72]
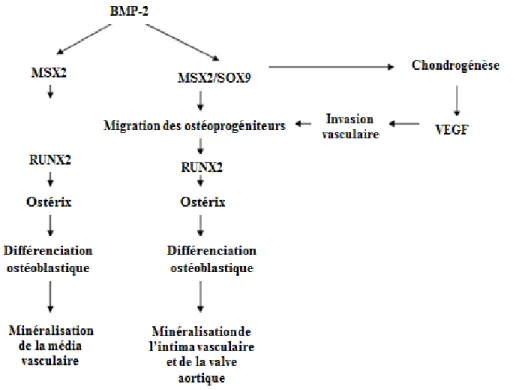
Figure 1.7 : Rôle de BMP-2 et MSX2 dans la calcification vasculaire
La production de BMP-2 favorise la production des facteurs de transcription MSX2, SOX9, RUNX2 et ostérix et induit ainsi la différenciation ostéoblastique contribuant à la calcification vasculaire et valvulaire.
Tiré et traduit de : Hruska, K.A., et al., Bone morphogenetic proteins in vascular calcification. Circ Res., 2005. 97(2): p. 105-14. BMP-2 : « Bone morphogenetic protein-2 », MSX2 : « msh homeobox 2 », SOX9: « (sex determining region Y)-box 9 », VEGF: facteur de croissance de l’endothélium vasculaire et RUNX2 : « Runt related transcription factor-2 ».
1.5.2.1.1. Inhibiteurs de la calcification
Le développement de la calcification vasculaire est prévenu dans les vaisseaux sains par des mécanismes de protection qui sont efficaces même en présence de facteurs de risque de calcification. En effet, lors d’une étude ex vivo, des artères ont été prélevées d’individus sains, de patients en IRC pré-dialysés et de patients en IRC dialysés. Les vaisseaux ont par la suite été
25
soumis à un milieu riche en calcium et en phosphate. Même après 21 jours dans ce milieu calcifiant, les vaisseaux sains n’ont pas développé de calcification, alors qu’une accumulation de calcium a été démontrée dans les vaisseaux provenant d’individus en pré-dialyse ou dialysés. Plus encore, l’accumulation de calcium était significativement plus importante dans les artères provenant d’individus dialysés que dans celles de patients pré-dialysés.[72] Cela démontre l’importance des mécanismes de protection physiologiques contre la calcification et leur perte d’efficacité en IRC. Les principaux protecteurs naturels sont la fétuine-a, la protéine matricielle Gla (MGP) et l’ostéoprotégérine (OPG) (figure 1.8).[65]
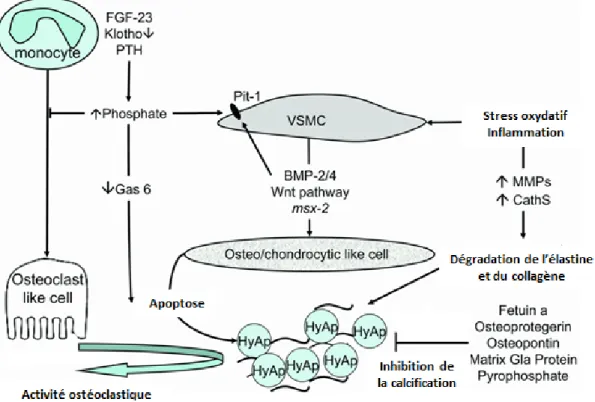
Figure 1.8 : Mécanismes impliqués dans la calcification artérielle en IRC
La calcification vasculaire en IRC est un processus multifactoriel. Tout d’abord, la perte d’inhibiteurs de la calcification comme la fétuine-a, l’ostéoprotégérine, l’ostéopontine, la protéine matricielle Gla et les pyrophosphates contribuent au développement de la calcification vasculaire, De plus, l’état inflammatoire chronique, le stress oxydatif et les désordres du métabolisme phosphocalcique contribuent à la calcification en induisant la différenciation des VSMC en cellules ressemblant aux ostéoblastes et la modification de la composition de la matrice extracellulaire vasculaire.
Tiré et traduit de : Briet, M., et Burns, K.D., Chronic kidney disease and vascular remodeling: molecular mechanisms and clinical implications. Clin Sci, 2012. 123(7): p. 399-416. FGF-23 : « fibroblast growth factor-23 », PTH : hormone parathyroïdienne, VSMC: cellule musculaire lisse vasculaire, BMP: « bone morphogenetic protein », msx-2 : « msh homeobox-2 », Gas 6 : « growth arrest specific-6 », HyAp : hydroxyapatite, MMP: métalloprotéinase matricielle et CathS : cathépsine S.
26
1.5.2.1.1.1. Fétuine-a
La fétuine-a est une glycoprotéine circulante qui forme des complexes de phosphate de calcium nommés « calciprotein particles » (CPP). La formation de tels complexes inhibe la précipitation du phosphate de calcium dans les tissus. De plus, la fétuine-a libre est captée par les cellules et incorporée dans les vésicules matricielles pour y prévenir la minéralisation. Elle inhibe aussi l’apoptose des VSMC et augmente la clairance des corps apoptotiques par phagocytose.[78] Les concentrations abaissées de fétuine-a en IRC ont été associées avec la présence de CPP, de calcification vasculaire et avec la mortalité.[55]
1.5.2.1.1.2. Protéine matricielle Gla
La MGP est une protéine vitamine K dépendante, c’est-à-dire qu’elle doit être glutamate-γ-carboxylée par la vitamine K pour produire son activité de protection contre la calcification. Elle peut aussi être sérine-phosphorylée, quoique le rôle de cette phosphorylation demeure à préciser.[79] Deux mécanismes permettent à la MGP de prévenir les calcifications extra-osseuses. Premièrement, elle forme des CPP avec la fétuine-a et le phosphate de calcium.[80] Deuxièmement, la liaison du calcium aux résidus Gla de la MGP favorise la liaison et l’inhibition de BMP-2 et 4 par la MGP. Ces protéines ne peuvent donc plus exercer leurs effets qui favorisent la différenciation ostéoblastique.[81] Les artères des patients dialysés présentent une diminution de la carboxylation de la MGP, et donc de son activité. La diminution des taux plasmatiques de MGP carboxylée déphosphorylée s’associe, du moins chez les hémodialysés, à la mortalité cardiovasculaire et la mortalité toutes causes confondues. De plus, les patients avec des niveaux plus bas de MGP carboxylée déphosphorylée ont davantage de calcification vasculaire.[79]
Ce rôle de la MGP pourrait expliquer l’association entre l’administration de warfarine et le développement de calcification artérielle. Pour effectuer son action, la warfarine bloque la gamma-carboxylation vitamine K dépendante de protéines impliquées dans la cascade de coagulation. Elle bloque donc aussi la gamma-carboxylation de la MGP, inhibant ainsi ses actions protectrices.[82]
1.5.2.1.1.3. Ostéoprotégérine
L’OPG est un récepteur soluble de RANKL. Elle empêche donc RANKL de se lier à RANK, exprimé par les ostéoclastes. Ainsi, l’effet de RANKL qui active les ostéoclastes et la résorption osseuse est inhibé (figure 1.6). Des niveaux sériques d’OPG >757,7 pg/ml sont prédicteurs de la présence de