Novel pharmacogenomic markers of
irinotecan-induced severe toxicity in
metastatic colorectal cancer patients
Thèse
Siwen Sylvialin Chen
Doctorat en sciences pharmaceutiques
Philosophiae doctor (Ph.D.)
Québec, Canada
Résumé
L’irinotécan est un agent de chimiothérapie largement utilisé pour le traitement de tumeurs solides, particulièrement pour le cancer colorectal métastatique (mCRC). Fréquemment, le traitement par l’irinotécan conduit à la neutropénie et la diarrhée, des effets secondaires sévères qui peuvent limiter la poursuite du traitement et la qualité de vie des patients. Plusieurs études pharmacogénomiques ont évalué les risques associés à la chimiothérapie à base d’irinotécan, en particulier en lien avec le gène UGT1A, alors que peu d’études ont examiné l’impact des gènes codant pour des transporteurs. Par exemple, le marqueur UGT1A1*28 a été associé à une augmentation de 2 fois du risque de neutropénie, mais ce marqueur ne permet pas de prédire la toxicité gastrointestinale ou l’issue clinique. L’objectif de cette étude était de découvrir de nouveaux marqueurs génétiques associés au risque de toxicité induite par l’irinotécan, en utilisant une stratégie d’haplotype/SNP-étiquette permettant de maximiser la couverture des loci génétiques ciblés. Nous avons analysé les associations génétiques des loci UGT1 et sept gènes codants pour des transporteurs ABC impliqués dans la pharmacocinétique de l’irinotécan, soient ABCB1, ABCC1, ABCC2, ABCC5, ABCG1, ABCG2 ainsi que SLCO1B1. Les profils de 167 patients canadiens atteints de mCRC sous traitement FOLFIRI (à base d’irinotécan) ont été examinés et les marqueurs significatifs ont par la suite été validés dans une cohorte indépendante de 250 patients italiens. Nous avons découvert dans la région intergénique en aval du gène UGT1, un nouveau marqueur (rs11563250G) associé à un moindre risque de neutropénie sévère (rapport des cotes (RC)=0.21; p=0.043 chez les canadiens, RC=0.27; p=0.036 chez les italiens, et RC=0.31 p=0.001 pour les deux cohortes combinées). De plus, le RC est demeuré significatif après correction pour multiples comparaisons (p=0.041). Par ailleurs, pour l’haplotype défini par les marqueurs rs11563250G et UGT1A1*1 (rs8175347 TA6), le RC était de 0.17 (p=0.0004). Un test génétique évaluant ces marqueurs permettrait d’identifier les patients susceptibles de bénéficier d’une augmentation de dose d’irinotécan. En revanche, une autre combinaison de marqueurs, ABCC5 rs3749438 et rs10937158 (T–C), a prédit un risque plus faible de diarrhée sévère dans les deux cohortes (RC = 0.43; p=0.001). La coexistence des marqueurs ABCG1 rs225440T et ABCC5 rs2292997A a prédit un risque accru de neutropénie (RC=5.93; p=0.0002), alors qu’une prédiction encore plus significative a été obtenue lorsque ces marqueurs sont combinés au marqueur de risque bien établi UGT1A1*28 rs8175347 (RC=7.68; p<0.0001). Enfin, les porteurs de l’allèle de protection
UGT1 rs11563250G en absence d’allèles de risque, ont montré une incidence réduite de neutropénie sévère (8.2% vs. 34.0%; p<0.0001). Nous concluons que ces nouveaux marqueurs génétiques prédictifs pourraient permettre d’améliorer l’évaluation du risque de toxicité et personnaliser le traitement à base d’irinotécan pour les patients atteints du cancer colorectal métastatique.
Abstract
Irinotecan is a cytotoxic agent widely used for the treatment of solid tumors, most particularly for metastatic colorectal cancers (mCRC). Treatment with this drug frequently results in severe neutropenia and diarrhea that can seriously impact the course of treatment and patients’ quality of life. Pharmacogenomic tailoring of irinotecan-based chemotherapy has been the subject of several investigations, especially for the UGT1A1 gene, but with limited data regarding transporter genes. In this study, we sought to discover toxicity-associated markers using a haplotype-tagging SNP (htSNP) strategy to maximize gene coverage. We examined the genetic association across the UGT1 locus, and in seven transporter genes participating in irinotecan pharmacokinetics involving the ABC transporter genes ABCB1, ABCC1, ABCC2, ABCC5, ABCG1, ABCG2 and the solute carrier organic anion transporter gene SLCO1B1. The profiles of 167 mCRC Canadian patients treated with FOLFIRI-based regimens were examined and findings were replicated in an independent cohort of 250 Italian patients. We found rs11563250G, located in the intergenic region downstream of UGT1, to be significantly associated with reduced risk of severe neutropenia (odds ratio (OR)=0.21; p=0.043 and OR=0.27; p=0.036, respectively, and OR=0.31 when combined; p=0.001), which remained significant upon correction for multiple testing in the combined cohort (p=0.041). For the two-marker haplotype rs11563250G and UGT1A1*1 (rs8175347 TA6), the OR was of 0.17 (p=0.0004). Genetic testing of this marker may identify patients who might benefit from increased irinotecan dosing. In combined cohorts, a two-marker ABCC5 rs3749438 and rs10937158 haplotype (T–C) predicted a lower risk of severe diarrhea (odds ratio (OR) of 0.43; p=0.001). The co-occurrence of ABCG1 rs225440T and ABCC5 rs2292997A predicted an increased risk of severe neutropenia (OR=5.93; p=0.0002), which was further improved when incorporating the well-known risk marker UGT1A1*28 rs8175347 (OR=7.68; p<0.0001). In contrast, carriers of one protective marker (UGT1 rs11563250G) but none of these risk alleles experienced significantly less severe neutropenia (8.2% vs. 34.0%; p<0.0001). This combination of predictive genetic markers could lead to better risk assessment and may thus enhance personalized treatment.
Table of Contents
Résumé ... iii
Abstract ... v
Table of Contents ... vii
List of Tables ... ix
List of Figures ... xi
List of Abbreviations ... xiii
Acknowledgements ... xvii
Foreword ... xix
Chapter I Introduction ... 1
1. Colorectal cancer... 1
1.1 Disease Prevalence and Incidence ... 1
1.2 An overview of treatment options ... 2
2. Irinotecan, an antineoplastic agent used in first-line treatment of mCRC . 4
2.1 Mechanism of Action ... 62.2 Pharmacokinetic pathways of irinotecan: metabolism and transport ... 7
2.3 Tissular & Cellular transport of irinotecan and its metabolites ... 12
2.4 Pharmacodynamic pathways of irinotecan ... 19
2.5 Toxicities induced by irinotecan ... 22
3. Pharmacogenomics (PGx) ... 25
3.1 Established pharmacogenomics tests : a few examples ... 26
3.1.1 Warfarin ... 26
3.1.2 Clopidogrel ... 28
3.2 Irinotecan Pharmacogenomics ... 29
3.2.1 Molecular and clinical impact of UGT1A1*28 ... 29
3.2.2 Other UGT1A variants ... 31
3.2.3 UGT1A1*28 genotyping test ... 33
4. Hypothesis & Objectives ... 39
4.1 Identify UGT1 markers to better predict risk of severe toxicity ... 39
4.2 Identify novel markers to help predict toxicity induced by irinotecan in drug transport pathways ... 40
5. Methodologies & Approaches ... 41
5.1 Study cohorts: ... 41
5.1.1 Discovery cohort ... 41
5.1.2 Validation cohort ... 42
5.2.1 Severe neutropenia ... 43
5.2.2 Severe diarrhea ... 43
5.2.3 Bilirubin levels ... 44
5.3 Selection of candidate genes and single nucleotide polymorphisms (SNPs) .. 45
5.4 DNA Sample preparation and Genotyping ... 46
5.5 Statistical analysis ... 46
5.5.1 Hardy-Weinberg equilibrium ... 46
5.5.2 Genetic Associations ... 47
5.6 Functional studies and in-silico analyses ... 48
5.6.1 3’ Rapid Amplification of cDNA Ends (RACE) ... 48
5.6.2 Resequencing of UGT1 locus ... 49
5.6.3 In-silico predictive bioinformatics analysis ... 49
Chapter II ... 51
Identification of a novel genetic marker in UGT1A locus with better tolerance
against severe neutropenia in metastatic colorectal cancer patients ... 51
Chapter III ... 81
Identification of ABCC5 and ABCG1 polymorphisms that predict
irinotecan-induced severe toxicity in metastatic colorectal cancer patients ... 81
Discussion ... 125
Conclusion ... 131
List of Tables
Table 1. Common treatment strategies used in the management of colorectal cancer. ... 2 Table 2. Irinotecan-based regimens for treatment of metastatic colorectal cancer (mCRC) 5 Table 3. Dose-limiting toxicities experienced by irinotecan-receiving patients ... 6 Table 4. UGT1A polymorphisms found to influence SN-38 glucuronidation rates in vitro .. 11 Table 5. Selected drug transporters and their substrates (non-exhaustive) ... 12 Table 6. Major transporter gene polymorphisms and clinical outcomes studied previously
in irinotecan-treated cancer patients ... 16 Table 7. Pharmacodynamic genes associated with SN-38 pharmacokinetics, toxicity and
response in irinotecan-receiving colorectal cancer patients. ... 21 Table 8. Dosing recommendations according to current CPIC guidelines based on
VKORC1 and CYP2C9 genotype ... 27 Table 9. UGT1A gene polymorphisms and associations with clinical outcomes studied
previously in irinotecan-treated cancer patients ... 32 Table 10. Demographic and Clinical characteristics studied in Discovery and Validation
cohorts ... 42 Table 11. NCI-CTACE v3.0 criteria for severe neutropenia and diarrhea toxicities ... 44 Table 12. Genes involved in the metabolism and transport of SN-38 studied in the
discovery cohort ... 45 Table 13. In silico bioinformatic databases and tools used to evaluate the potential
functionality of novel markers ... 49
List of Figures
Figure 1. Global cancer incidence, death and prevalence rates according to cancer types
from 184 countries. ... 1
Figure 2. Metabolism, biotransformation and inactivation reactions of CPT-11 ... 7
Figure 3. UGT1A enzymes involved in the metabolism of CPT-11 and its metabolites ... 10
Figure 4. Commonly known SNPs in the UGT1 locus ... 11
Figure 5. Schematic representation of ABC and SLCO transporter members involved in the transport of irinotecan and its metabolites from hepatocytes to the intestine. ... 13
Figure 6. Distribution of Irinotecan and its metabolites excreted in the bile and urine ... 18
Figure 7. SN-38 mechanism of action on Top1-DNA complex and activation of pharmacodynamic pathways ... 19
Figure 8. Timeline of Pharmacogenomics over the last decade. ... 25
Figure 9. Schematic representation of UGT1A1*28 on UGT1 locus and its effect on SN-38 glucuronidation ... 30
List of Abbreviations
3’UTR 3’Untranslated region 5-FU 5-FluorouracilABC ATP-binding cassette transporters ADR Adverse Drug Reaction
ANC Absolute Neutrophil Count
APC 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxycamptothecin AUC Area under the curve
CAP Capecitabine
CDC45L Gene encoding for CDC45-related protein (cell-division cycle 45) CES Carboxylesterase I or II
CEU Caucasian Europeans from Utah
ChIP-seq Chromatin Immunoprecipitation Sequencing
CPIC Clinical Pharmacogenomics Implementation Consortium CPT Camptothecin
CYP Cytochrome P450 enzyme DLT Dose-limiting toxicity
DPWG Dutch Pharmacogenomics Working Group ECOG Eastern Cooperative Oncology Group
EGAPP US Evaluation of Genomic Applications in Practice and Prevention EGFR Epidermal Growth Factor Receptor
ENCODE Encyclopedia of DNA Elements
ERCC1 Excision repair cross-complementation group I protein FDA United States Food and Drug Administration
G-CSF Granulocyte-Colony Stimulating Factor GI Gastrointestinal
GSTP1 Gene encoding for Glutathione-S-transferase P enzyme GWAS Genome-Wide Association Study
htSNP Haplotype-Tagging SNP HWE Hardy-Weinberg Equilibrium INDEL Insertion/Deletion
IRI Irinotecan
IV Intravenous
KRAS GTPase KRas protein LD Linkage Disequilibrium
LV Leucovorin
M4 Metabolite of irinotecan MAF Minor Allele Frequency mCRC metastatic Colorectal Cancer MMF Mycophenolate Mofetil MPA Mycophenolic Acid
MPAG Mycophenolic Acid Glucuronide
MS Mass Spectrometry
MTD Maximum Tolerated Dose
NCBI National Center for Biotechnology Information NCCN National Comprehensive Cancer Network
NCI-CTCAE National Cancer Institute Common Toxicity Criteria for Adverse Events NGS Next Generation Sequencing
OR Odds Ratio OS Overall Survival
OX Oxaliplatin
PARP1 Poly(ADP-ribose) polymerase enzyme PCR Polymerase Chain Reaction
PD Pharmacodynamic
PFS Progression-free Survival PGx Pharmacogenomics
PK Pharmacokinetics
RACE 3’ Rapid Amplification of cDNA Ends SLCO Solute-carrier organic anion transporter SNP Single Nucleotide Polymorphism SSB/DSB Single/Double-Strand Breaks
TDP1 Tyrosyl-DNA phosphodiesterase I enzyme TOP1 Topoisomerase I
UGT UDP-glucuronosyltransferase ULN Upper Limit Of Normal Range
VEGF-A Vascular Endothelial Growth Factor A VKORC1 Vitamin K Epoxide Reductase Complex C1 XRCC1 X-ray repair complementation group I protein
May all sentient beings have happiness and its causes, May all sentient beings be free of suffering and its causes, May all sentient beings never be separated from bliss without suffering, May all sentient beings be in equanimity, free of bias, attachment and anger. - The Four Immeasurables To all sentient beings, may they be free of diseases and sufferings
Acknowledgements
First, I would like to express my utmost gratitude and appreciation to my doctoral advisor, Dr. Chantal Guillemette, for giving me the opportunity to be mentored and trained in your laboratory. Despite a rocky start, I grew to understand and appreciate your motivation to expect the best from your students. I have learned excellent organization skills, the rigors of scientific investigations, developed critical thinking as well as how to function as a team player. My time under your mentorship has allowed me to evolve not only personally but also enhance my communication skills and work ethics professionally. Your ability to be firm and yet patient during moments of self-doubt and poor self-confidence has enabled me to persevere and complete my studies. This dissertation would not have been completed without your constant encouragement and critical feedback. My goal was to work and learn from the best in the field of Pharmacogenomics and I will cherish every little bit of my time here. I strive to use my training in this field into my future endeavors and make you proud!
Next, I thank all the members of the jury committee for spending time to review this dissertation. I am honored to be given the opportunity to present my work to you for your comments.
Naturally, every successful laboratory is made up of a team of cohesive members. I would like to thank especially Isabelle Laverdiere, an excellent fellow PhD student who helped make my transition into the lab a smooth one. Thank you for spending your precious time showing me the ropes, providing constructive comments and giving me pep talks. In part, this PhD would not have been successfully completed without your support. Another person who deserves many thanks is Lyne, who has been a delight to work with. Thank you for guiding me with my experiments, questioning my rationales and most importantly helping me with my Français! Merci beaucoup! Also, a special mention should be given to Anais, whose arrival brought much joy and vivacity into the lab. Thank you for your candor and positive spirit, you often made my frown into a smile! Thank you Michele for entertaining my questions, they helped me to put things in perspective, especially when writing this thesis; I thoroughly enjoyed our scientific conversations! To other members, former and present, thank you for your presence and friendship, and also tolerating my broken French! You all helped me to assimilate into Quebec fast and furious, without any official French courses!
My cross-border move from Boston would not have been smooth-sailing if it were not for my group of dedicated friends who never failed to encourage and support me mentally and physically during times of need. They convinced me that I would overcome all challenges in front of me and never doubted that I would succeed. To Kelly, Rachel, Minu (and family), Casey (and family), Ieva, Joey (and Rory) and Myra, I am truly blessed to have you all in my life. Thank you for being there for me always.
To all the wonderful people I met here in Quebec who became my close friends, namely, Joannie, Adriana and Ricardo, thank you for making my life here not all about work. I am fortunate to explore some of Quebec’s beauty and culture in spite of my hectic student life with you. I express my sincere gratitude towards new friends, Aimee, Ewan, Fiona and Tessa, who took me into their family, cared and nurtured me during these past months of difficulties. I am eternally grateful for your love, generosity, kindness, support, keeping my spirits up, dispensing advice and helping me regain confidence. Thank you seems insufficient in expressing how much I am indebted to your family, but I hope to repay you some day.
Last but not least, I want to thank my family for their endless love and support, albeit from afar in Singapore. I was first inspired to study Pharmacogenomics as a result of my father’s condition and his medication, warfarin. I am proud to declare that I have succeeded in this goal and I plan to apply this knowledge acquired to benefit more people in the future. To my mother, who showered me with love and concern, always making sure that I focused and never strayed from my goal. I did it! Also, thank you to my sisters, Joy and Charlene, for holding the fort down at home and keeping me sane. I hope you both would achieve similar success in your careers.
Thank you to all the wonderful people around me, I am excited to explore the next stage in my life and cannot wait for you to be my witnesses!
Foreword
This thesis, entitled, “Novel pharmacogenomic markers of irinotecan-induced severe toxicity in metastatic colorectal cancer patients,” embodies the work achieved during my doctoral training under Dr. Chantal Guillemette’s mentorship in the Pharmacogenomics Laboratory at CHU de Quebec Research Center. The purpose of this thesis is for submission to Université Laval for the degree of Philosophiae doctor, encompassing two original articles, “A novel UGT1 marker associated with better tolerance against irinotecan-induced severe neutropenia in metastatic colorectal cancer patients” and “ABCC5 and ABCG1 polymorphisms predict irinotecan-induced severe toxicity in metastatic colorectal cancer patients”
The first article, “A novel UGT1 marker associated with better tolerance against irinotecan-induced severe neutropenia in metastatic colorectal cancer patients“ was completed in collaboration with multicenters in Canada and Italy, comprising of 167 and 250 Caucasian metastatic colorectal cancer patients. This article was recently published in the Pharmacogenomics Journal (Impact factor: 5.513) in March 2015. My role has been to participate in the selection of candidate genes and SNPs, together with another Ph.D. student Isabelle Laverdière. I also participated in sample preparation, statistical analysis, interpretation of results and further in-silico analyses while Isabelle Laverdière and Lyne Villeneuve performed and validated all statistical analyses, with support by Alan Tourancheau for bioinformatic scripts. Mario Harvey participated in the initial data analysis. I participated in the draft of the manuscript while Dr. Chantal Guillemette wrote the complete version of the paper with the help of Dr. Lévesque. All authors performed a critical review of the manuscript. All tables and figures presented in the article were ensembled by Isabelle and myself. Dr. Derek Jonker, Dr. Félix Couture, Dr. Èric Lévesque and their team recruited Canadian patients and provided clinical information while Dr. Erika Cecchin and Dr. Giuseppe Toffoli coordinated the Italian cohort. Dr. Michael H. Court provided the human liver tissue samples. The conception, design and supervision of the study was by Dr. Chantal Guillemette.
The second article, “ABCC5 and ABCG1 polymorphisms predict irinotecan-induced severe toxicity in metastatic colorectal cancer patients” has been accepted in the journal Pharmacogenetics and Genomics (Impact factor: 3.481) in July 2015. It was also performed with our Italian collaborators and their roles were the same as in the first study.
My role was essentially the same as in the first study with regards to SNPs selection, data analysis and drafting parts of the manuscript. Statistical analyses and their validation were performed by Lyne Villeneuve and Isabelle Laverdière. Dr. Chantal Guillemette wrote the complete version of the paper. All authors performed a critical review of the manuscript. The conception, design and supervision of the study was by Dr. Chantal Guillemette.
Chapter I Introduction
1. Colorectal cancer
1.1 Disease Prevalence and Incidence
Colorectal cancer (CRC) is currently the third most common cancer in the United States and Canada and the third leading cause of death in men and women. As of 2014, the incidence rates projected amongst men and women in North America are approximately 51 per 100,000 in men and 40 per 100,000 in women. These North American trends are in line with the global cancer statistics whereby CRC was the third most diagnosed type of cancers (10%) in 2012, after breast and lung cancers while the mortality rate (9%) caused by CRC deaths was ranked fourth. The 5-year global prevalence rate of CRC remains at 11%, a drastic improvement from the mid-20th century (1930-1950s) as a result of improved treatment options and early detection. In particular, a 3% decrease in incidence rates per year during the period of 2001 and 2010 has been observed.1-3 (Figure 1)
Figure 1. Global cancer incidence, death and prevalence rates according to cancer types from 184 countries. A) Overall cancer incidences ranked by cancer types in 2012. B) Mortality rates segregated by cancer types in 2012. C) 5-year prevalence rates of cancer types. (Adapted from GLOBOCAN project 2012, http://globocan.iarc.fr accessed Oct 15, 2014)
These trends correlate with better early detection and stage diagnosis of CRC, improving the 5-year relative survival rate for patients with all tumor stages from 65% to 90% for those with localized tumors.1-3
1.2 An overview of treatment options
The improvement of 5-year relative CRC survival rate is in part due to a greater adoption of colorectal screening during annual medical examinations, advances in surgical and radiation therapy and the use of efficacious chemotherapeutic agents. Treatment strategies of diagnosed CRC are largely dependent on type of tumor, location, disease stages, the extent of tumor progression, curative intention, genetic mutations and tolerance to toxicities. Table 1 lists the common clinical treatment options used in the management of CRC.
Table 1. Common treatment strategies used in the management of colorectal cancer. Treatment Option Clinical Goal/Intention Disease Stage
Surgery
- Open
- Laparoscopy
Resect metastases and localized tumor, only option of long-term survival
I-III, neoadjuvant therapy candidates
Radiation therapy
- short course - long course
Shrink tumor size and prevent recurrence, relieve cancer symptoms (ex. pain)
I-III, candidates who reject surgery
Chemotherapy Prolong survival and prevent tumor
progression
IV, liver/lung metastases, candidates for resection - monotherapy 5-fluorouracil/leucovorin (5-FU/LV)
irinotecan oxaliplatin capecitabine metastatic CRC patients - combination therapy (regimens)
FOLFIRI (5-FU/LV, irinotecan) FOLFOX (5-FU/LV, oxaliplatin) CAPOX (capecitabine, oxaliplatin) CAPIRI (capecitabine, irinotecan)
FOLFOXIRI (5-FU/LV, irinotecan, oxaliplatin)
metastatic CRC patients - targeted therapy (administered with another regimen) bevaxizumab cetuximab1 panitumumab1 aflibercept sorafenib2 regorafenib metastatic CRC patients with drug resistance to regimens
Abbreviations: IRI = irinotecan, 5-FU, 5-fluorouracil, LV, leucovorin, CAP, capecitabine, OX, oxaliplatin, CRC, colorectal cancer
2 Currently in clinical trials for treatment of mCRC
Unfortunately for the majority of patients who are presented with metastases at time of diagnosis (almost 40%-50%)9,10, the main intention is to prolong survival and prevent further tumor progression. Hence, intensive treatment with chemotherapeutic agents becomes the best logical option. Patients who were not previously considered for resection may also benefit from neoadjuvant chemotherapy to become candidates for surgical removal options. Furthermore, the risk of local recurrence in Stage III patients is 15-50%, hence adjuvant chemotherapy is also employed in an effort to prevent regrowth of resected tumors.2
Current combination therapies have been favored over monotherapy strategies as a result of improved response rates, overall survival (OS) and progression-free survival (PFS) rates. The two most common combination regimens used in medical centers in North America and Europe are FOLFIRI (irinotecan-based) and FOLFOX (oxaliplatin-based). The preference to use one regimen over the other often depends on clinicians’ decisions, type of medical center, availability of chemotherapeutic agents, toxicity occurrence, tumor resistance and patients’ physical condition and economic circumstances.11-14 In fact, one major advantage irinotecan-containing regimens have over oxaliplatin-based ones is the absence of neurotoxicity after several months of therapy. In lieu of a dose-reduction, patients may choose to switch to an irinotecan-based treatment in order not to compromise therapeutic objectives. Irinotecan has also been observed to incur a 1.2-1.8 fold increase in PFS and OS when administered concurrently with 5-FU/leucovorin compared with non-irinotecan containing treatments.15-17 The most frequently used irinotecan-containing treatments are available in Table 2.
In recent years, novel biologics have been developed to improve cytotoxic targeting of tumors and are undergoing clinical trial testing in conjunction with existing regimens. Despite early promising results, the clinical implementation of targeted therapy is still nascent and a majority of medical centers continue to use either irinotecan-based or oxaliplatin-based regimens.12,18,19
Several trials have also assessed the risk-benefit characteristics of FOLFOXIRI (irinotecan+oxaliplatin+5-fluorouracil+leucovorin) but have yet to demonstrate a superior efficacy and decreased toxicities over existing FOLFIRI treatment. Until further evaluative
studies on the added benefits of oxaliplatin or novel biologics, FOLFIRI is still one of the best regimens for the treatment of metastatic colorectal cancer (mCRC).
2. Irinotecan, an antineoplastic agent used in
first-line treatment of mCRC
The introduction of irinotecan for use as an anticancer drug has tremendously improved the current state of treatment options in metastatic colorectal cancer patients. The efficacy of irinotecan in the treatment of mCRC is far superior over 5-FU/LV treatment. Irinotecan-based regimens indicate a two-fold improvement in overall survival, progression-free survival and response rates, with less adverse toxic events. In fact, a 50% reduction in chemotherapy-induced gastrointestinal toxicity was observed with irinotecan monotherapy compared to 5-FU/leucovorin.16
Irinotecan (CPT-11) is a water-soluble camptothecin analog synthesized by Yakult Honsha Co Ltd (Tokyo, Japan), displaying antitumor activity in mouse and human carcinoma cell lines.20-24 The anticancer drug class, camptothecins are plant alkaloids first isolated from the bark of the Chinese tree Camptotheca acuminata and were found to have antileukemic properties in mice cell lines.25,26 Characterization studies later confirmed this cytotoxic effect was a result of specific inhibition of DNA topoisomerase I (Top1) enzyme activity, by induction of single-strand DNA breaks and premature termination of replication.27,28
Unlike traditional camptothecins, irinotecan was found to exhibit favorable physical properties, such as solubility, stability and reduced side-effects.29,30 Furthermore, as a prodrug, CPT-11 can be safely administered intravenously without drug delivery problems. Therefore, irinotecan has emerged as the best camptothecin-based drug currently used for treatment of colorectal and lung cancers.
Table 2. Irinotecan-based regimens for treatment of metastatic colorectal cancer (mCRC)
Regimen Irinotecan Leucovorin
5-Fluorouracil Oxaliplatin Frequency Monotherapy 350mg/m2 IV (90 min) Once every 3 weeks 125mg/m2 IV (90 min) Once per week FOLFIRI 180mg/m2 IV 400mg/m2 IV 400mg/m2 IV Bolus + 2400mg/m2 IV (46hrs) Once every 2 weeks FOLFOXIRI1 165mg/m2 IV 200mg/m2 IV 3200mg/m2 IV (48hrs) 85mg/m2 IV Once every 2 weeks 1 Recently completed in clinical trials, actual use in practice unknown
Abbreviation: IV, intravenous
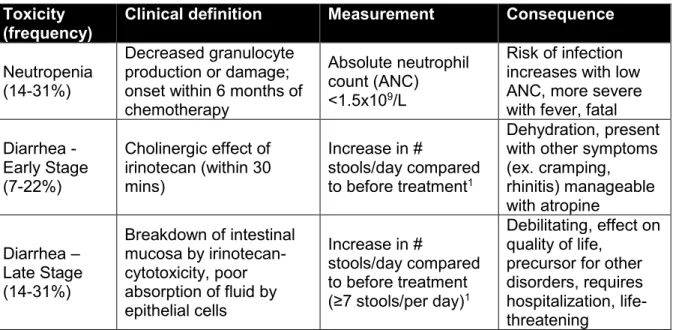
A major limitation to any irinotecan-based treatment is the occurrence of two dose-limiting toxicities (DLT) - diarrhea and severe myelosuppression (ex. neutropenia). Numerous studies have reported that neutropenia and late-stage diarrhea were the most common forms of toxicities experienced by patients receiving irinotecan. Approximately 10-54% patients were found with severe Grade 3-4 neutropenia while 9-30% had severe Grade 3-4 diarrhea.15-17,31,32 (Table 3)
The clinical implication for severe neutropenia is the inability to fight infections as a result of decrease neutrophil production and a compromised immune system. Similarly, gastrointestinal toxicities are a common adverse effect of chemotherapy. In particular, the most severe type of chemotherapy-induced diarrhea is late-onset at 24 hours-3 days after treatment with irinotecan. This event is often debilitating, life-threatening as well as socially-challenging to manage. Patients may not accurately report their bowel patterns, as a result of embarrassment, leading to ineffective prophylactic use. Overall, both toxicities could result in hospitalization and delay in chemotherapy treatment. Loperamide, an anti-diarrheal drug may be given to ameliorate late-onset diarrhea episodes. Almost half of the patients undergoing irinotecan-based regimens will experience a DLT. Even though a dose reduction can likely reduce these events, it is performed at the expense of chemotherapeutic tumor response. Therefore, the ability to predict patients who would be at risk for severe neutropenia and diarrhea would greatly benefit from dose-management decisions in mCRC treatment, without compromising therapeutic response.
Table 3. Dose-limiting toxicities experienced by irinotecan-receiving patients Toxicity
(frequency)
Clinical definition Measurement Consequence Neutropenia
(14-31%)
Decreased granulocyte production or damage; onset within 6 months of chemotherapy
Absolute neutrophil count (ANC)
<1.5x109/L
Risk of infection increases with low ANC, more severe with fever, fatal Diarrhea - Early Stage (7-22%) Cholinergic effect of irinotecan (within 30 mins) Increase in # stools/day compared to before treatment1 Dehydration, present with other symptoms (ex. cramping, rhinitis) manageable with atropine Diarrhea – Late Stage (14-31%) Breakdown of intestinal mucosa by irinotecan-cytotoxicity, poor absorption of fluid by epithelial cells Increase in # stools/day compared to before treatment (≥7 stools/per day)1 Debilitating, effect on quality of life,
precursor for other disorders, requires hospitalization, life-threatening
1 It is necessary to determine the baseline levels of bowel patterns before evaluation
2.1 Mechanism of Action
As a synthetic analog of camptothecin, irinotecan exerts antitumor activity by targeting Top1 to disrupt DNA replication. The prodrug (CPT-11) is converted into its active metabolite (SN-38) and other minor metabolites (M4, APC, NPC) by carboxylesterase (CES) and CYP3A4/5 enzymes respectively. (Figure 2) The active metabolite SN-38 is 100-1000x more potent than CPT-11 in part due to its specificity as an intercalating molecule between Top1-DNA single-strand interactions.33-36 Like other camptothecins, SN-38 binds to Top1-DNA cleavage complexes (Top1-cc) formed momentarily during DNA unwinding and prevents re-ligation of the template strand.37-39 This binding is reversible upon changes in physiological pH and absence of active compound. Hence, the effect on tumor death and drug response is strongly dependent on the bioavailability of this active metabolite.40,41
Figure 2. Metabolism, biotransformation and inactivation reactions of CPT-11 (Adapted from Mathijssen RH et al, 200142)
In contrast, the prolonged exposure of toxic SN-38 in the systemic circulation and intestinal cells can result in onset of myelosuppression and gastrointestinal toxicities.43,44 These adverse events are likely to occur more frequently due to inadequate biotransformation (CES) and inactivation (UGT1A) caused by altered protein function.
2.2 Pharmacokinetic pathways of irinotecan: metabolism and
transport
As an antineoplastic agent, the metabolism of CPT-11 is indeed crucial in delivering therapeutic concentrations of SN-38 to tumor cells. The efficacy of CPT-11 on tumor response and induced toxicities relies on the efficiency of various pharmacokinetic pathways that transform, inactivate and metabolize CPT-11. The biotransformation of CPT-11 is performed by carboxylesterases, enzymes that hydrolyze the piperidino side chain to form active SN-38.45 (Figure 2) Human CES2 (hCES2) was discovered to have a 12.5-fold higher specificity for CPT-11 and is 26-fold more active than hCES1.46,47 hCES1 and hCES2 are commonly expressed in the liver, kidney and gut, amongst other tissues, where their biotransformation efficiencies are dependent on the tissue type. hCES2 is highly expressed and performs 99% of CPT-11 hydrolysis in the small intestine while hCES1 is present in the liver and is responsible for hydrolyzing at least 50% of CPT-11 levels.48,49
Many studies have also observed an interindividual variability of hCES2 expression in liver, colon and plasma specimens, indicating underlying phenotypic differences in CPT-11 conversion.49-52 A moderate significant correlation between CES2 mRNA expression and irinotecan-induced toxicities (severe neutropenia and diarrhea) was also confirmed, with 35% of CRC patients expressing high mRNA levels, who had experienced severe grade 3-4 toxicities.53 Hence, it is clear that interindividual differences, most likely due to genetic variations, can account for the observed variability in tumor response and toxicities.
Other CPT-11-derived metabolites such as APC, NPC and M4 are derived from CYP3A-mediated metabolism. CYP3A4 and CYP3A5 catalyze the Phase I reactions of CPT-11 to form APC by hydrolyzing the terminal piperidine ring whereas the N-dealkylation of the distal piperidine ring forms NPC.54-56 These CYP450-converted metabolites do not display strong antitumor effect, unlike SN-38, and are negligible in predicting irinotecan efficacy or toxicity.29 In addition to irinotecan, the CYP3A family is responsible for the oxidative conversion of numerous drugs. Studies involving drug-drug interactions between irinotecan and CYP3A4 inhibitors have clarified that there was no change in the pharmacokinetic measures and induced-toxicities of irinotecan-receiving patients, in the presence of omeprazole.57 Conversely, ketoconazole was suggested to increase SN-38 exposure but failed to identify a significant effect on irinotecan clearance and SN-38 glucuronides.58 A haplotype-based CYP3A4 investigation also observed the absence of positive associations between CYP3A4 polymorphisms, clearance and irinotecan-induced toxicities.59 The contribution of concomitant medications towards these toxicities60 may indeed be important when considering irinotecan treatment efficacy. In fact, some have demonstrated a significant association between the administration of other drugs with irinotecan-related toxicities while others have clarified patients’ comorbidities, although indirectly indicate the use of multiple medications, do not affect toxicities.61,62 The mechanistic explanations of drug-drug interactions influencing irinotecan efficacy remain confounding. Differences in cohort size, methodological approaches and cancer types make it challenging to determine the impact of drug substrates of CYP3A4. For now, clinicians prefer to avoid co-administering irinotecan and CYP3A4 inhibitors.63
Interestingly, even though NPC was found to be 100x less potent than SN-38, it can be readily converted into active SN-38 by hCES2. The catalytic efficiency of hCES2 is more
pronounced for NPC than APC.64 Thus, it is no surprise that the majority of existing irinotecan-related pharmacokinetic and pharmacogenomic studies have investigated the effect of carboxylesterase-mediated formation of SN-38.
High circulating levels of toxic SN-38 often result in gradual degradation of epithelial cells in the intestine and liver. Histological and pharmacokinetic findings have correlated the increased exposure of SN-38 and CPT-11 to decreased absolute neutrophil counts (ANC)43,65,66, severe damage of intestinal mucosa 67,68 and higher biliary indexes.69,70
The inactivation of SN-38 is mediated by a Phase II reaction catalyzed by UDP-glucuronosyltransferase enzymes (UGTs) whereby the transfer of a glucuronic acid (G) moiety from uridine diphosphoglucuronic acid (UDPGA) forms a more polar SN-38 glucuronide (SN-38G). UGTs are widely expressed in drug-metabolizing tissues, glucuronidating numerous endogenous compounds (ex. steroid hormones) and xenobiotics (ex. drugs). The formation of these glucuronides enables compounds to be readily eliminated in the bile or urine.71
Human UGT enzymes are classified into four families, UGT1, UGT2, UGT3 and UGT8, with only UGT1 and UGT2 being involved in drug metabolism.72,73 Both UGT1 and UGT2 families possess distinct substrate specificity and account for the majority of glucuronidation activities. The UGT1A protein family consists of 13 members, which are derived from alternatively spliced first exons and 4 common exons (exons 2-5) on the gene UGT1 locus.74 In particular, in vitro studies have confirmed UGT1A proteins, namely UGT1A1, UGT1A6, UGT1A7 and UGT1A9 to be responsible for the inactivation of SN-38.75-78 (Figure 3)
Figure 3. UGT1A enzymes involved in the metabolism of CPT-11 and its metabolites (from Guillemette, C et al, 201474)
Furthermore, strong interindividual variability of UGT1A1 mRNA expression detected in mice correlated with irinotecan-induced toxicities.79 As the only detoxification enzyme of SN-38, functional UGT1As are necessary to alleviate toxic effects in the liver, intestine and circulation. The UGT1A family is composed of transmembrane-bound proteins that are responsible for the detoxification of numerous drug substrates and exhibit distinct substrate specificity according to the type of drug metabolizing tissue.74 The tissue distribution of UGT1A enzymes emphasizes their important glucuronidating function and their ability to impact the clinical consequences of drug metabolism. The substrate specificity of multiple UGT1A enzymes in different tissues may infer a compensatory mechanism present in the event of a malfunction protein and/or suggest a cooperative effort in drug detoxification. Indeed, the co-occurrence of functional UGT1A variants was found to potentiate irinotecan-induced toxicity80 and indicate the need to systematically investigate these UGT1A enzymes together. The primary cellular location of UGT1A enzymes is in the endoplasmic reticulum where they are docked within the membrane with the substrate-binding domain exposed on the lumen.81 UGTs have also been detected in other subcellular compartments such as mitochondria, nucleus but their functions in these organelles have yet to be determined.82,83
Genetic variations in the UGT1 locus have been associated with changes in glucuronidation rates of SN-38 and inferred reduction in UGT1A function. These in vitro studies summarized in Table 4 demonstrate the impact of polymorphic changes on
UGT1A glucuronidating ability of SN-38. Figure 4 depicts the location of these commonly known UGT1 SNPs, of which several have been studied in relation to SN-38 metabolism.
Table 4. UGT1A polymorphisms found to influence SN-38 glucuronidation rates in vitro UGT1
Gene
Variant rs # Effect on SN-38
glucuronidation
Ref UGT1A1 promoter TA repeat 7/7
(*28) c.-3156G>A (*93) c.-3279T>G (*60) G71R (*6) Y486D (*7) P229Q (*27) L233R (*35) rs8175347 rs10929302 rs4124874 rs4148323 rs34993780 rs35350960 rs72551344 SN-38G formation SN-38G formation SN-38G formation SN-38G formation SN-38G formation SN-38G formation SN-38G formation 84-86 84 84,78 78,87 78,87 78,87 78 UGT1A7 N129K, R131K (*2) N129K,R131K,W208R (*3) W208R (*4) G115S (*5) E139D (*6) rs17868323 rs17863778 rs11692021 rs61261057 rs114052958 - SN-38G formation SN-38G formation SN-38G formation SN-38G formation SN-38G formation 78,88 88 88 88 88 UGT1A9 M33T (*3) intronic C399T -118dT[9/10] (*22) rs72551330 rs2741049 rs3832043 SN-38G formation SN-38G levels SN-38G levels 88 84 84
Figure 4. Commonly known SNPs in the UGT1 locus
(*) Represents variants from Table 4 that have been previously identified to regulate SN-38 glucuronidation levels. (Modified from Guillemette, C et al 201474)
2.3 Tissular & Cellular transport of irinotecan and its
metabolites
The ATP-binding cassette (ABC) and solute-carrier (SLC) families of membrane transporter proteins are responsible for the uptake and efflux of CPT-11, SN-38 and its glucuronide, SN-38G. Together, they play a critical role in modulating the irinotecan pharmacokinetics and the bioavailability of the drug.89-95 The entire family of ABC transporter is divided into seven subfamilies of proteins, A-G. Each subfamily consists of transporters that have unique substrate specificity and structure. Currently, certain gene members of each subfamily, namely, ABCB1, ABCC1, ABCC2 and ABCG2 have been linked to drug transport and resistance to irinotecan. (Table 5) Similarly, the SLC superfamily of membrane-bound transporters facilitates the transport of a diverse group of molecules, including drug substrates. In particular, SLCO1B1 from solute-carrier organic anion transporter (SLCO or OATP) family is well characterized by its cellular disposition of drugs in the liver.96
Table 5. Selected drug transporters and their substrates (non-exhaustive) Gene Drug substrate
ABCB1
colchicine, doxorubicin, VP16, adriamycin, vinblastine, digoxin, saquinivir, paclitaxel, doxorubicin, daunorubicin, vincristine, indinavir, topotecan, CPT-11
ABCC1 VP16, colchicines, etoposide, rhodamine, adefovir, indinavir, CPT-11
ABCC2 vinblastine, cisplatin, sulfinpyrazone, SN-38, active
metabolite of CPT-11
ABCC3 methotrexate, etoposide, tenoposide ABCC4 nucleoside monophosphates
ABCC5 nucleoside monophosphates, mitoxantrone, topotecan, doxorubicin
ABCG1 mitoxantrone, doxorubicin
ABCG2 daunorubicin, doxorubicin, rosuvastatin, sulfasalazine, CPT-11, rhodamine
SLCO1B1
atrasentan, atorvastatin, bosentan, ezetimibe, fluvastatin, glyburide, rosuvastatin, simvastatin acid, pitavastatin, pravastatin, repaglinide, rifampin, valsartan, olmesartan SN-38, active metabolite of CPT-11
Modified from 97 and U.S. Food and Drug Administration (FDA) website (accessed November 10, 2014)
In addition, each transporter member from the ABC and SLCO family is tissue-specific and tends to be localized on either the apical or basolateral side of the cellular membrane. As
assumption is that clearance of drug metabolites is a result of a combined, cooperative effort of transporters present in a cellular location. Indeed, pharmacokinetic evidence depicting the role of various transporters in irinotecan disposition confirms that each transporter type has different substrate affinities.98 Figure 5 illustrates a schematic representation of drug transporters involved in active efflux and movement of irinotecan and its metabolites. As these transporters make up the entire cellular transport system, they are also speculated to work in sync with drug metabolizing enzymes to mediate overall drug transport.99-102
Figure 5. Schematic representation of ABC and SLCO transporter members involved in the transport of irinotecan and its metabolites from hepatocytes to the intestine. (Modified from Pancyzk, M. 2014103)
The adequate expression and function of these membrane-bound proteins is necessary for the movement of drugs across tissue compartments.97,104,105 It is unsurprising that these proteins are associated with drug resistance, often a result of impaired or enhanced transporter function, which can invariably change metabolite concentrations and impact tolerance levels. The interindividual variability of drug resistance in cancer treatments has
been suggested to be the result of variable expression of transporter proteins and Phase I, Phase II enzymes.106-108
The majority of this variability exists as polymorphisms in transporter genes that alter expression and function. The impact of these genetic variants on drug disposition has been widely evaluated in numerous in-vivo and in-vitro models, while associations to clinical outcomes in different diseases have been made in clinical studies.106,109,110 As a prototypic drug for mCRC treatment, the efficiency of irinotecan transport to its target tissue has largely been the focus of most investigations: outlining evidence of transporter dysfunction, tendency to acquire drug resistance and develop related adverse events. (Table 6)
The ABC transporter ABCB1, also known as multidrug resistance protein 1 (MDR1/p-glycoprotein) is located on the biliary canalicular side in the liver and the apical surface of gastrointestinal tract epithelial cells.111 Iyer et al. have found that ABCB1 has a stronger affinity for irinotecan, compared to its active metabolite, 38 or its glucuronide, SN-38G.92 Numerous findings regarding polymorphisms in ABCB1 that influence irinotecan efflux and toxicities so far have been mixed and therefore necessitate further validations. The most frequently studied polymorphisms are three variants (1236>T, 2677G>A/T, 3435C>T) that are in strong linkage disequilibrium with one another.112 These variants have been linked to an increase in active metabolite concentration113, increase/decrease in tumor response114,115 and inconsistent associations with toxicity outcomes115-118 while others have yet to be verified in other patient cohorts. The potential mechanistic impact of these polymorphisms has also yet to be clarified. The variant, ABCB1 3435T correlated with decreased gene expression in normal human duodenum samples119 whereas another study found this variant associated with elevated ABCB1 mRNA levels in SN-38-resistant human colon tumor colonies.120
Compared to ABCB1, ABCC1, the multidrug resistance-related protein 1 (MRP1) transporter is less efficient in the secretory efflux of irinotecan and SN-38. Despite being located in the basolateral side of liver cell membrane, the ABCC1 transporter protein is more efficient in transporting SN-38 in an apical-to-basolateral transport manner than vice-versa.121 Since ABCC1 has also been detected in CPT-11-resistant human epidermoid carcinoma cells and SN-38-resistant HeLa cells,94,122 its function in irinotecan transport has
also been supported by two genetic studies linking SN-38-related pharmacokinetic and toxicity outcomes.114,123 More recently, an in vitro study confirmed that ABCC1 variant 2012G>T had no functional effect on overall ABCC1 expression in embryonic kidney cells, contradictory to clinical observations known.124
The ABCC2 protein, a canalicular multispecific organic anion transporter (cMOAT) also located in the hepatic biliary canicular membrane, transports SN-38 and its glucuronide SN-38G more efficiently than its prodrug, irinotecan.90,93 Lower metabolite uptake and clearance levels measured in ABCC2-deficient rats indicate the importance of this transporter in irinotecan metabolism. Several genetic reports have investigated at least two variants (-24C>T, -3972T>C) in multiple independent patient populations receiving irinotecan-based treatments without similar findings.116,123,125,126 Other ABCC2 variants evaluated against toxicities and response have not yet been validated elsewhere.
Amongst all the types of transporters responsible for irinotecan efflux, the breast cancer resistance protein (BCRP) or ABCG2 has been most studied. ABCG2 is located on the canalicular side and apical membrane of enterocytes, where it transports SN-38 and its glucuronide across the biliary membrane.127,128 Many have shown the ability of ABCG2 to induce irinotecan resistance in overexpressed cellular models89,129-132 or have increased activity in SN-38-resistant cells.122 Pharmacogenetic studies of mCRC and lung cancer patients have since identified five polymorphisms that are associated with irinotecan pharmacokinetics, toxicities and tumor response.114,126,133,134 In fact, the variant 421C>A was found to reduce protein expression and decrease irinotecan resistance in in vitro models135, indicating that ABCG2 is critical in the transport of irinotecan. This observation was confounding in Asian subjects who were heterozygous for this variant whereby an increase in ABCG2 activity was implied with greater SN-38G clearance levels.
The human solute carrier organic anion family member 1B1 (SLCO1B1) gene encodes for the protein SLCO1B1 that is predominately expressed on the basolateral membrane of hepatic cells and exclusively absorbs 38 from blood, instead of irinotecan and SN-38G.95 Two most studied variants, 521T>C and 388A>G have been respectively linked to increases in severe neutropenia and gastrointestinal toxicity.126,136,137 These associations with clinical toxicities also appear to correlate with other pharmacokinetic studies measuring irinotecan, SN-38 concentrations and clearance.123,136,138
Even though ABCG1 and ABCC5 are not known to be involved in irinotecan transport, two variants, ABCC5 (3’UTR 1243T>C) and ABCG1 (c.974-898C>G) were associated with severe gastrointestinal toxicities in a small mCRC patient cohort.137 An increase in ABCC5 mRNA expression has also been observed in SN-38-resistant HeLa cells122, possibly implicating the role of ABCC5 in SN-38 efflux or clearance.
Table 6. Major transporter gene polymorphisms and clinical outcomes studied previously in irinotecan-treated cancer patients
Gene Variant Main findings Ref
ABCB1 1236 C>T (G412G) CPT-11 AUC SN-38 AUC SN-38 clearance 113 Asthenia 117
2677G>A/T (A893T/S) PFS, OS 115
OS 114
Grade 4 neutropenia in T or A
carriers 116
3435C>T (I1145I) Early toxicities 115 CPT-11 plasma concentration
in T carriers 134
Diarrhea 117
Grade 3 diarrhea in TT carriers 116 Diarrhea, Combined
neutropenia + diarrhea in TT
carriers 118
IVS9 -44A>G SN-38 AUC 123
IVS4-25G>T OS in T carriers 114
IVS13+24T>C OS in T carriers 114
IVS14+38G>A OS in T carriers 114
ABCC1
IVS11 -48C>T ANC nadir levels 123
1684T>C (L562L) SN-38, SN-38G/SN-38 123 Nonhematological toxicity in T carriers Hematological toxicity in TT carriers 114 2012G>T (G671V) Response rate in T carriers 114 IVS18-30C>G Hematological toxicity in CC carriers 114
ABCC2 -24C>T CPT-11 for T carriers 123 Response rate PFS CC carriers 125 Response rate, PFS in TT carriers 116 Grade 3-4 neutropenia in CT carriers 139 -3972T>C SN-38G
APC for T carriers 123
Grade 3 diarrhea in CC carriers 126 1249G>A (V417I) Response rate in A carriers 114 IVS23+56T>C Response rate in CC carriers 114 -1023G>A Nonhematological toxicity in A carriers 114 -1774delG SN-38 AUC/dose Neutropenia (*1A/*1A) 138 ABCC5 3’UTR 1243T>C GI toxicity in CC carriers SN-38G in C carriers 137 139 ABCG1 c.974-898C>G GI toxicity in GG carriers 137
ABCG2
34G>A (V12M) CPT-11 plasma concentrations Grade 3 diarrhea in A carriers 134 126
421C>A SN-38G in CA carriers 133
-1203_-1200
delCTCA SN-38 AUC REC 134
-15622C>T (Novel) Response rate in T carriers 114 -15994G>A Response rate in A carriers Non-hematological toxicity(first
cycle) 114 SLCO1B1 521T>C (V174A) SN-38 AUC/dose 138 Toxicity 140 CPT-11 AUC 123 SN-38 AUC, SN-38 clearance Grade4 neutropenia 136 Grade 4 neutropenia in C carriers 126 SN-38 AUC in C carriers 139 -11187G>A SN-38 AUC, SN-38 clearance in AA carriers Grade4 neutropenia 136 Response rate in A carriers
PFS 114 388A>G (N130D) ANC nadir 123 RRR, PFS, OS, TTF in A carriers 141 Grade3 diarrhea 136
GI toxicity in GA carriers 137 Oral mucositis in GA carriers 139
AUC, area under curve; ANC, Absolute Neutrophil Count; PFS, progression-free survival; REC, relative extent of conversion; OS, overall survival; GI, gastrointestinal; RRR, relative response rate; TTF, time to treatment failure.
Irinotecan, like many other xenobiotics, is excreted via the biliary and renal systems. Early studies conducted in rodents detected its active compound, SN-38 in bile, urine and feces while 33% and 58% of irinotecan was found in bile and urine, respectively. A majority of the dose excreted in the bile was detected in the form of inactivated SN-38.142,143 Studies in human patients revealed that over 90% of detected compounds in the bile and urine were composed of CPT-11 and its metabolites, SN-38, SN-38G and APC.42,144 High CPT-11 and SN-38 concentrations in the bile and urine were identified analytically as inactive carboxylate forms.54,145-148 Specifically, CPT-11 accounts for 3-30% and 10-30% of the
dose administered found in the bile and urine respectively. SN-38 and SN-38G amounts excreted through both biliary and urinary systems are low, accounting for 1-3% of the effective dose. On the other hand, SN-38G, despite being most abundant in the blood plasma showed a wide interindividual variability across samples investigated;69,146,149 suggesting differential SN-38 metabolism rates may be implicated. (Figure 6)
Minor metabolites of irinotecan, APC and NPC that do not display any drug activity are also present in bile, urine and feces in trace concentrations.29,146,148 The cumulative low amounts of APC and SN-38G in the urine also confirm that the primary route of irinotecan metabolism is not mediated by the renal system.150,151, therefore reiterating that irinotecan elimination is conducted via biliary and fecal pathways.
Figure 6. Distribution of Irinotecan and its metabolites excreted in the bile and urine
(Adapted from Slatter, JG. 2000144)
More importantly, conjugated SN-38 molecules can be subjected to deconjugation by intestinal beta-glucuronidase activity. These de-conjugated entities, observed as rebound peaks in pharmacokinetic AUC curves 30 min to 1 hour after drug administration have been presumed to undergo enterohepatic recycling.69,152-154 Enterohepatic recycling enables the prolongation of active compounds’ bioavailability as de-conjugated compounds re-enter the systemic circulation and liver from the intestine. Several factors, including genetic variations in transporters have been noted to have an impact on the efficiency of this recycling.155 Particularly, further exposure of cytotoxic SN-38 in the plasma, liver and intestine translates into increased efficacy and higher rates of toxicities such as severe neutropenia and diarrhea.
2.4 Pharmacodynamic pathways of irinotecan
Current research has described the pharmacodynamic action of SN-38 to implicate cellular response from Top1-induced DNA damage. As the sole molecular target of SN-38, the Top1 protein acts in concert with the unwound single-strand DNA to form the protein-DNA complex and momentarily halts DNA replication. SN-38 specifically binds to this complex and disrupts further synthesis processes. This breakage also induces the activation of three distinct pathways - apoptosis, DNA damage repair and cell-cycle arrest. (Figure 7)
Figure 7. SN-38 mechanism of action on Top1-DNA complex and activation of pharmacodynamic pathways (Adapted from Hurley, LH et al, 2002 156 and Pharmagkb.org)
As the primary molecular target of SN-38, the cellular levels, gene expression and catalytic activity of Top1 protein are crucial in determining the efficacy of SN-38-induced cytotoxicity. So far, resistance to SN-38 has been attributed to genetic mutations, reduced quantities and activity of Top1.157-161 Further experiments describe the diminished expression of Top1 in SN-38-resistant cell lines157,158 while others have associated this resistance to copy number variations and point mutations.162-164 Accordingly, certain variants have been proposed to alter the binding affinity of SN-38 at the catalytic site of Top1, with clear evidence of their impact on the formation of Top1-cleavage complex and double-strand breakage.165
Even though these specific point mutations were not detected in CRC patients, intra- and inter-variation in Top1 activity and protein levels have been observed in different untreated tumor tissues.166-168 Therefore, other genetic polymorphisms may be involved in modulating Top1 expression. More importantly, colon tumor tissues were found to have a higher Top1 heterogeneous activity than other tissues evaluated (64-fold variation compared to 16-fold in cervix tumors).167 Together, these observations show that inter-individual differences in Top1 can affect levels of unbound cytotoxic SN-38 but also indirectly influence SN-38-induced tumor response and toxicities.
The formation of Top1-cc-induced DNA single-strand breaks (SSB) triggers the response of DNA repair pathways, where proteins are recruited during the repair process. The presence of Top1-cc-induced SSB determines the state of DNA repair and its subsequent sensitivity to the Top1 inhibitor. For example, poly(ADP-ribose) polymerase (PARP1) is stimulated by DNA breakage events and activates its catalytic function by acting as an intermediary to destabilizes Top1-cc. At the same time, it works with tyrosyl-DNA phosphodiesterase I (Tdp1) to remove Top1 from the DNA strand. This allows base-excision repair proteins, including X-ray repair complementation group 1 (XRCC1) to access the region of damage.169-171
Genetic deficiencies of both PARP1 and TDP1 debilitate the DNA repair system and result in accumulation of unrepaired DNA breaks.172,173 PARP1 and excision repair cross-complementation group 1 (ERCC1) have also been implicated in alternative repair pathways of Top1-cc with endonucleases.174,175 Presence of XRCC1 gene also conferred a higher resistance to campthotecin while at the same time XRCC1 interacts closely with Tdp1 to activate the repair process.176 The breakdown of the DNA repair system will likely lead to unrepaired DNA lesions and eventual cell-cycle arrest and apoptosis. In addition to SSB, double-strand breaks (DSB) converted from single strand lesions occur during collision of Top1-cc and moving replication forks. DSB-mediated repairs incorporate two distinct processes – homologous recombination and non-homologous end joining. Variants in genes (ex. checkpoint proteins and tumor suppressor) from these signaling pathways can render the dysfunction of their supposed function and prevent tumor cell death or halt DNA synthesis. The overall impact of these downstream genes can affect intrinsic SN-38-induced cytotoxicity and thereby influence tumor response and accumulated toxicities.
Interestingly, further investigations with clinical samples have associated higher Top1 protein levels with longer OS and PFS in mCRC patients177 while TDP1 and ERCC1, amongst other genes, were upregulated in CRC samples.178 ERCC1 expression was also higher in responders of CPT-11 treatment but was less than that of the reference gene.179 Thus, the regulatory contributions of these genes in irinotecan-induced toxicities should be considered as well. The importance of these signaling pathways is obvious by differential gene expression detected in colorectal cancer samples. Transcriptional RNA/DNA profiling expression methods have identified the expression of certain genes with functions in DNA repair, cell cycle, apoptosis, to be higher in tumoral or SN-38-treated tissues.178,180 Other reports using proteomic methods also revealed levels of such proteins to vary in SN-38-resistant cell-lines.181,182
To date, few cohort studies have collectively studied the biological relevance of all genes in relation to CPT-11-mediated tumor response and toxicities. The majority of them have focused on few genes or only known polymorphisms. Table 7 outlines the genes and/or specific variants related to SN-38 pharmacodynamic pathways previously investigated in mCRC patients receiving irinotecan-based regimens. Unfortunately, these studies failed to clearly define the precise association of pharmacodynamics genes with predicting clinical outcomes. Moreover, the majority of these reports only studied specific putative causal variants from certain genes, not considering other variants and potentially functional SNPs present in the gene locus. Hence, in order to clarify their roles, it is necessary to perform a comprehensive study incorporating all known polymorphisms from both pharmacokinetic and pharmacodynamic genes.
Table 7. Pharmacodynamic genes associated with SN-38 pharmacokinetics, toxicity and response in irinotecan-receiving colorectal cancer patients.
Genes studied Type of chemotherapy Cohort Size Major findings Ref MLH1/MSH2, p53, TOP1, ERCC1, MGMT, COX2 irinotecan-containing regimens 1313 mCRC pts
High Top1 expression OS, PFS
(from immunohistochemistry) 177 TOP1, ERCC1, GSTP1 irinotecan-containing regimens 56 mCRC pts
Higher ERCC1 expression in non-responders vs non-responders, no
association with toxicities 179 Specific Variants
studied Type of chemotherapy Cohort Size Major findings Ref MLH1 -93G>A XRCC1 R339Q GSTP1 I105V irinotecan-containing regimens 982-1002
ERCC2 K751Q MLH1 -93G>A XRCC1 R339Q GSTP1 I105V ERCC2 K751Q irinotecan-containing regimens 633-1136 mCRC pts XRCC1 Toxicity-induced dose delay/reduction ERCC2 , GSTP1 irinotecan-related Grade ≥3 toxicity
183
HNF1A 79A>C Irinotecan 85 pts CPT-11 AUC levels 123
ERCC2 -1989A>G, D711D, K751Q GSTP1 A114V, I105V XRCC1 R399Q irinotecan-containing regimens 520
mCRC pts None significant with survival outcomes or toxicities 184 14 SNPs* from CDC45L, NFKB1, PARP1, TDP1, TOP1, XRCC1 irinotecan-containing regimens 89-107 mCRC pts
TOP1 and PARP1 diplotype neutropenia risk, TDP1 and XRCC1
diplotype objective response 185 6 htSNPs from TOP1,
PARP1, TDP1,
XRCC1 irinotecan 85 pts
None associated with ANC or
neutropenia risk 186 XRCC1 R399Q ERCC2 K751Q irinotecan-containing regimens 43 mCRC
pts XRCC1 and ERCC2 OS, 187
10 SNPs from TOP1, TDP1, PARP1, XRCC1, CDC45L irinotecan-containing regimens 61 mCRC
pts CDC45L IVS-24C>TPFS, Other variants not significant 125 ERCC1 N118N, XRCC1 R399Q ERCC2 N312N, K751Q, XRCC3 T214M irinotecan-containing regimens 146 mCRC pts XRCC3 PFS 188
GSTP1 I105V irinotecan-containing regimens
267
mCRC pts Not associated with toxicities, PFS 189 ERCC2 K751Q,
GSTP1 1105V
irinotecan-containing
regimens 48 mCRC
ERCC2, GSTP1 not associated with
response, OS, PFS 190
*includes 10 haplotype-tagged SNPs (htSNPs) and 4 putative causative SNPs
AUC, Area Under the Curve; ANC, Absolute Neutrophil Count; PFS, progression-free survival; OS, Overall Survival
2.5 Toxicities induced by irinotecan
As alluded earlier, prolonged exposure to both CPT-11 and SN-38 is significantly associated with reduced absolute neutrophil counts (severe neutropenia) in a sigmoidal fashion.43,65,66 Early Phase I trials have identified a strong correlation between absolute neutrophil count, CPT-11 levels, clearance rates and SN-38 plasma concentrations, while similar observations were also reported for SN-38G associations with diarrhea events.69,152,191-193 Indeed, higher concentrations of circulating SN-38 may on one hand improve efficacy against rapidly dividing tumor cells but at the same time cause dose-limiting toxic events and prove to be fatal.