Immobilization of Cytochrome P450 BM3 from
Bacillus megaterium on magnetic nanoparticles to
develop an effective biocatalyst for hydroxylation
reactions
Thèse
Atieh Bahrami
Doctorat en génie chimique Philosophie Doctor (Ph.D.)
Québec, Canada
Immobilization of Cytochrome P450 BM3 from
Bacillus megaterium on magnetic nanoparticles to
develop an effective biocatalyst for hydroxylation
reactions
Thèse
Atieh Bahrami
Sous la direction de :
Maria Iliuta, directrice de recherche
Alain Garnier, codirecteur de recherche
Faïçal Larachi, codirecteur de recherche
iii
Résumé
Les catalyseurs chimiques sont utilisés dans différents procédés de synthèse. Cependant, la pollution qu'ils causent sur l'environnement n’est pas prise en considération. Les procédés de synthèse chimique nécessitent généralement un grand volume de solvants organiques, produisant d’énormes quantités de déchets chimiques, souvent toxiques et non dégradables. Le remplacement des catalyseurs chimiques par des biocatalyseurs (enzymes) pourrait donc bénéficier de leur nature écologique et de leur grande sélectivité envers les produits désirés. Néanmoins, la faible activité et stabilité des enzymes ainsi que leurs coûts élevés sont des obstacles majeurs au développement des systèmes enzymatiques. Par conséquent, des études axées sur le développement de systèmes biocatalytiques plus actifs, stables et rentables, pouvant ouvrir les portes vers un environnement plus vert, sont très souhaitables.
Parmi les enzymes qui catalysent des réactions d’importance dans de nombreux procédés de synthèse, le cytochrome P450 BM3 issu de Bacillus megaterium fait l'objet de cette thèse. L'enzyme est capable d’hydroxyler les liaisons C–H des acides gras (C12-C22) à température ambiante et pH physiologique. Pour cette réaction, BM3 n'a
besoin que d’oxygène et de deux électrons habituellement obtenus de son cofacteur naturel, le NADPH. Cependant, pour engager cette enzyme dans les réactions d'hydroxylation, quelques obstacles importants doivent être surmontés : (i) le cofacteur coûteux (NADPH), devrait être remplacé par une source d'électrons moins chère ou régénérée, (ii) la stabilité enzymatique devrait être améliorée et (iii) l'enzyme devrait être facilement récupérable du milieu de réaction pour être réutilisée.
Dans ce contexte, cette étude propose pour la première fois l'immobilisation d'un BM3 sur des nanoparticules magnétiques (NMP) d’oxyde de fer. Ce système enzymatique bénéficie (i) de la préférence de l'enzyme pour les cofacteurs NADH et BNAH (moins chers que le NADPH), (ii) de la réutilisation facile du biocatalyseur et (iii) d’une stabilité significative de l’enzyme lors du stockage. Les NMP synthétisées ont été fonctionnalisées pour permettre l’immobilisation de l'enzyme par adsorption ou liaison covalente. Par conséquent, les BM3-NMP adsorbées / réticulées ou liées de façon
iv
covalente ont été obtenues en immobilisant P450 BM3 (R966D / W1046S) sur Ni2+-PMIDA-NMP ou sur des NMP activés par glutaraldéhyde, respectivement. L'activité de l’enzyme immobilisée a été comparée avec celle de l’enzyme libre dans la réaction d'hydroxylation du 10-pNCA comme substrat modèle. L'acide myristique a également été utilisé comme substrat modèle pour confirmer la capacité d’hydroxylation sélective de l’enzyme sur les atomes de carbone ω-1, -2 ou -3.
Pour les mêmes conditions opératoires, le BM3 adsorbé / réticulé a montré plus de 85% de l'activité de l’enzyme libre, alors que pour les BM3-NMP liées de manière covalente cela représente 60%. La séparation facile des NMP du milieu réactionnel à l’aide d’un aimant a permis de réutiliser le système enzymatique cinq fois consécutives. Après 5 cycles de réaction, l'enzyme réticulée a conservé 100% de son activité initiale. Compte tenu que le recyclage de l’enzyme libre n’est pas faisable, ce résultat est d’une importance considérable dans les applications pratiques. De plus, la stabilité de l’enzyme pendant un mois de stockage à 4 ºC a été évaluée pour chaque système de BM3. Les résultats ont montré que l’enzyme libre n’était plus active après seulement une semaine de stockage dans ces conditions. L'enzyme réticulée n'a montré qu'une activité relative de 41% après un mois de stockage, mais pour le BM3 fixée de façon covalente, la valeur correspondante a été de 80%.
La cinétique de l'hydroxylation du 10-pNCA en présence de l’enzyme libre ou immobilisée a été également étudiée. Sur la base des données expérimentales, un modèle de Hill (coefficient de Hill égal à 2) a été obtenu pour l'enzyme libre. Il a été démontré que les mêmes paramètres cinétiques sont capables de prédire le comportement du système BM3-adsorbé et BM3-réticulé dans la réaction d’hydroxylation, étant donné sa similarité avec celui de l’enzyme libre.
En conclusion, les résultats de cette thèse ont montré qu'un système enzymatique actif, stable et rentable peut être obtenu en immobilisant le BM3 sur des NMP fonctionnalisées. Il bénéficie autant des avantages de l'enzyme que du support. Ainsi, l'immobilisation sur des NMP d’une enzyme spécialement conçue pour remplacer le couteux NADPH par des cofacteurs moins chers mais efficaces (NADH et BNAH)
v
offre en même temps une amélioration significative de sa stabilité et facilite son recyclage.
vi
Abstract
Chemical catalysts are used in different synthetic processes from lab to industrial scales. High reaction yields usually achieved by this type of processes favor their application in many industries without considering the pollution they cause to the environment. Chemical synthesis processes usually require a high volume of organic solvents and produce tons of chemical wastes which are often toxic and not degradable. Replacing conventional catalysts by biocatalysts (enzymes) can benefit from their environmentally friendly nature and high selectivity toward the desired products. Although the advantages of biocatalysts over chemical catalysts have been proven, the application of enzymes in an industrial level is still not considerable. The enzyme low activity, stability, and high cost are the main concerns in developing large-scale enzymatic systems. Therefore, in the context of a greener environment, studies focusing on the development of more active, stable, and cost-effective enzymatic systems are in great demand.
Among several enzymes that can catalyze essential synthesis reactions, cytochrome P450 BM3 from Bacillus megaterium is the subject of this thesis. This enzyme hydroxylates the saturated and unsaturated C–H bonds of medium to long chain fatty acids at room temperature and physiological pH. For this reaction, BM3 only needs molecular oxygen and two electrons usually obtained from its natural cofactor, NADPH. However, to engage this enzyme in hydroxylation reactions, some important obstacles should be overcome: (i) the costly cofactor (NADPH) should be replaced by a cheaper source of electrons or regenerated, (ii) the enzyme stability should be improved, and (iii) the enzyme should be easily recovered from the reaction medium to be reused.
In this context, this study proposes for the first time the immobilization of an optimized BM3 mutant on functionalized iron oxide magnetic nanoparticles (MNPs). This enzymatic system benefits from (i) the enzyme preference towards cofactors like the reasonably priced NADH and the very cheap BNAH, (ii) facile recovery and reuse of the biocatalyst (enzyme-MNPs), and (iii) the enzyme significant storage stability.
vii
MNPs have been synthesized and surface functionalized to attach the enzyme via two different methods, adsorption and covalent binding. Moreover, glutaraldehyde was used to treat the adsorbed enzyme molecules on MNPs (crosslinking-adsorption). Therefore, adsorbed, crosslinked-adsorbed, or covalently bound BM3-MNPs were obtained by immobilizing P450 BM3 on synthesized Ni2+-functionalized MNPs or glutaraldehyde pre-activated MNPs, respectively.
The immobilized enzyme activity was compared to its free counterpart in hydroxylation reaction of 10-pNCA (10-(4-Nitrophenoxy) decanoic acid) as a substrate model. Myristic acid was also used as a substrate model to confirm the enzyme selective hydroxylation at ω-1, -2, or -3 carbon positions. The effect of cofactor (NADH and its analogue, BNAH) on the enzyme activity was also investigated. The adsorbed/crosslinked-adsorbed BM3 showed more than 85% of the free enzyme activity while the covalently bound BM3-MNPs presented 60% of the free enzyme activity under the same reaction conditions. An important feature of BM3-MNPs system is the possibility of recycling the biocatalyst. Facile separation of the magnetic nanoparticles from the reaction medium by applying a magnet provided the opportunity of reusing the enzymatic system for five times. After 5 cycles of reaction, the crosslinked-adsorbed enzyme retained 100% of its initial activity. Although the covalently bound enzyme showed, only half of the crosslinked-adsorbed enzyme activity, its storage stability was more significant. Taking into account that the enzyme reuse is an essential concern in many large-scale applications and the free BM3 cannot be recovered and reused, this result is noteworthy. Storage stability tests revealed that the free enzyme became inactive after one-week while the crosslinked-adsorbed enzyme and the covalently attached BM3 on MNPs showed 41% and 80% relative activity after one month, respectively.
Finally, the steady-state kinetics of 10-pNCA hydroxylation by free and immobilized BM3 was investigated. Based on the experimental data, a non-Michaelis-Menten, Hill model (Hill coefficient of 2) was obtained for the free enzyme which could also predict the adsorbed and crosslinked-adsorbed BM3-MNPs system performance. This sigmoidal behavior was found to be independent of enzyme concentration and type of
viii
cofactor. However, since the enzyme activity was only 60% of the free enzyme for covalently bound BM3, further studies are necessary for a better understanding of this system.
In summary, the results of this thesis show that an active, stable, and cost-effective BM3-MNPs system can be obtained by immobilizing an engineered BM3 on functionalized MNPs. Such systems benefit from the advantages of both enzyme and support. An engineered enzyme can fulfill the desired targets including the replacement of costly NADPH by less-expensive, yet effective cofactors namely NADH and BNAH. Furthermore, immobilization of this enzyme on MNPs improves its stability and facilitates the recycling process.
ix
Table of Contents
Résumé ... iii
Abstract ... vi
Table of Contents ... ix
Index of Tables ... xii
Index of Figures ... xiii
Abbreviations ... xviii
Acknowledgement ... xxi
Preface ... xxii
1. Introduction ... 1
1.1. Background ... 1
1.2. Cytochrome P450 BM3 from Bacillus megaterium ... 3
1.2.1. Monooxygenase domain ... 4
1.2.2. Reductase domain ... 14
1.3. Catalytic mechanism of P450 BM3 ... 17
1.4. P450 BM3 and the source of electrons in the reaction ... 20
1.4.1. Cofactor regeneration ... 22
1.4.2. Biomimetic nicotinamide cofactors ... 27
1.4.3. Chemical reducing agents ... 28
1.4.4. Peroxides ... 29
1.4.5. Light induced systems ... 30
1.4.6. Electrodes ... 31
1.5. Hydroxylation by P450 BM3 using different electron sources ... 33
1.6. Kinetics of reactions hydroxylated by P450 BM3 ... 35
1.7. Enzyme Immobilization ... 39
1.7.1. Methods of enzyme immobilization ... 40
1.7.2. Supports for enzyme immobilization ... 45
1.8. Immobilized P450 BM3 systems ... 49
1.9. Conclusion ... 53
1.10. Objective of the work ... 54
2. Chapter 1: Methodology ... 57
2.1. Bare magnetic nanoparticles synthesis ... 57
2.1.1. Ni2+-PMIDA-MNPs preparation ... 58
x
2.1.3. MNPs characterization ... 60
2.2. P450 BM3 immobilization on functionalized MNPs ... 62
2.3. Enzyme quantification assay ... 63
2.3.1. Free enzyme ... 64
2.3.2. Immobilized enzyme ... 64
2.4. Substrate-cofactor-enzyme reaction system ... 65
2.4.1. Myristic acid-cofactor reaction assay ... 65
2.4.2. 10-pNCA-cofactor reaction assay ... 67
2.5. Enzyme activity assay ... 69
2.5.1. Cofactor consumption ... 69
2.5.2. p-Nitrophenolate production ... 72
2.5.3. Omega-hydroxylated myristic acids production ... 74
2.6. Kinetics of P450 BM3-hydroxylated reaction ... 75
2.6.1. Free enzyme ... 75
2.6.2. Immobilized enzyme ... 77
2.7. Immobilized enzyme recycling process ... 77
2.8. Enzyme storage stability ... 78
3. Chapter 2: Non-covalent immobilization of optimized bacterial cytochrome P450 BM3 on functionalized magnetic nanoparticles ... 81
Résumé ... 81
Abstract ... 82
3.1. Introduction ... 83
3.2. Experimental ... 86
3.2.1. Chemicals ... 86
3.2.2. Magnetic nanoparticles synthesis ... 87
3.2.3. P450 BM3 immobilization ... 88
3.2.4. Immobilized BM3 reuse ... 90
3.2.5. Storage stability of BM3 ... 90
3.3. Results and discussion ... 91
3.3.1. Ni2+-PMIDA-MNPs characterization ... 91
3.3.2. BM3 immobilization on Ni2+-PMIDA-MNPs ... 95
3.3.3. Free and immobilized BM3 catalytic activity ... 96
3.3.4. Immobilized BM3 reuse ... 100
3.3.5. Storage stability of free and immobilized BM3 ... 102
xi
4. Chapter 3: Covalent immobilization of cytochrome P450 BM3 (R966D/W1046S) on
glutaraldehyde activated SPIONs ... 107
Résumé ... 107
Abstract ... 108
4.1. Introduction ... 109
4.2. Experimental ... 112
4.2.1. Enzyme and chemicals ... 112
4.2.2. SPIONs synthesis ... 112
4.2.3. SPIONs modification and characterization ... 112
4.2.4. Enzyme immobilization ... 113
4.2.5. Enzymatic reaction ... 114
4.2.6. BM3-SPIONs recycling process ... 117
4.2.7. P450 BM3 storage stability ... 117
4.3. Results and discussion ... 117
4.3.1. SPIONs characterization ... 117
4.3.2. BM3 immobilization on the SPIONs ... 122
4.3.3. Free and immobilized P450 BM3 activity ... 122
4.3.4. BM3-SPIONs recycling process ... 126
4.3.5. P450 BM3 storage stability ... 128
4.4. Conclusion ... 129
5. Chapter 4: Kinetics of enzymatic hydroxylation in the presence of free and immobilized NADH-dependent cytochrome P450 BM3 from Bacillus megaterium 132 Résumé ... 132
Abstract ... 133
5.1. Introduction ... 134
5.2. Experimental ... 136
5.2.1. Enzyme and chemicals ... 136
5.2.2. 10-pNCA hydroxylation kinetics ... 137
5.2.3. Enzyme activity ... 139
5.3. Results and discussion ... 140
5.3.1. Enzymatic hydroxylation kinetics ... 140
5.4. Conclusion ... 153
6. General Conclusions and Suggestions for Future Work ... 156
References ... 161
xii
Index of Tables
Table 1.1. P450 crystal structure bond distances at the heme (Lewis et al. 2005). ... 7
Table 1.2. Turnover rates of P450 BM3 using various electron sources/cofactor regeneration system. ... 34
Table 1.3. Kinetic parameters for hydroxylation of different substrates by P450 BM3. In all examples, the enzyme consumes NADPH as source of electrons. ... 37
Table 1.4. Immobilization methods used for P450 BM3. ... 52
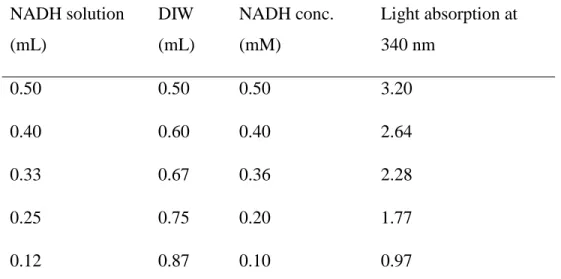
Table 2.1. NADH samples for the related standard curve...70
Table 2.2. BNAH samples for the standard curve. ... 71
Table 2.3. p-Nitrophenolate samples preparation for the standard curve. ... 73
Table 3.1. Free and immobilized enzyme yield; reaction performed in 0.1 M Tris buffer, pH 8.2, with 10-pNCA (350 μM) as the substrate and NADH/BNAH (1 mM) as a cofactor. ... 102
Table 4.1. Production yield (ratio of nanomoles of product to nanomoles of enzyme) by free and immobilized BM3 (reaction system: 350 μM 10-pNCA and 1 mM cofactor in Tris buffer 0.1 M pH 8.2). ... 127
Table 5.1. Kinetic parameters for 10-pNCA hydroxylation reaction by free BM3. . 146
Table A.1. List of amino acids, their full names and abbreviation (Abr). ... 173
xiii
Index of Figures
Figure 1.1. M151odular arrangement of P450 BM3 domains (Shehzad 2013). ... 2 Figure 1.2. Schematic model of enzyme-substrate complex in P450 BM3 active site; B is the binding site for terminal methyl group of the substrate, B' is the biding site for the polar group of the substrate and A indicates the hydroxylation site (Miura and Fulco 1975). ... 5 Figure 1.3. Schematic representation of relative positions of heme and key amino acid residues in the active site of palmitoleate-bound P450 BM3 (Noble et al. 1999). ... 6 Figure 1.4. Key amino acids in the X-ray atomic structure of palmitoleate-bound P450 BM3 (Ost et al. 2000). ... 6 Figure 1.5. A) Crystal structure of P450 BM3 monooxygenase from Bacillus
megaterium (Urlacher 2012) and B) Heme structure of cytochrome P450 (Lewis et al.
2005). ... 7 Figure 1.6. Model for substrate interaction in P450 BM3; (A) Electrostatic forces orientate the carboxylate group of the substrate towards Arg47 and by further hydrogen bonding of the Tyr51 and the ‒COOH group, the binding is stabilized, (B) the carbon chain is drawn to the active site channel and the amino acids inside the channel provide favorable hydrophobic interactions, and (C) final catalytically competent orientation is achieved, and the heme is ready for reduction and initiating the catalytic cycle (Ost et al. 2000). ... 9 Figure 1.7. Schematic representation of Thr268 hydrogen bonding network with the water molecule which can serve as proton donor to the ferrous-dioxygen complex. This hydrogen bonding network is shown in the heme domain of P450 BM3 with arachidonic acid (C18) (Truan and Peterson 1998). ... 12
Figure 1.8. Crystal structure of the substrate free and palmitoleate bound wild-type P450 BM3. By binding the susbtrate Ala264 and iron bound water are moved, then Thr268 and Ala264 provide a hydrogen binding pocket for the displaced water (Clark et al. 2006). ... 13 Figure 1.9. Electron transport chain in monooxygenation reaction by P450 (King et al. 1988). ... 14 Figure 1.10. (A) Ribbon diagram of reductase domain: FMN domain in blue, the hinge in red, the FAD and NADPH binding domains in green. They are also shown as ball and sticks: FMN (light blue), FAD (yellow), NADP+ (orange) (Wang et al. 1997) and (B) FMN domain motion in flavocytochrome P450 BM3 (Munro et al. 2002). ... 15 Figure 1.11. Schematic hypothetical interaction of P450 BM3 domains for electron transfer (working as dimers) (Kitazume et al. 2007). ... 17 Figure 1.12. Catalytic cycle of cytochrome P450 (Cook et al. 2016). ... 19 Figure 1.13. Structure of natural nicotinamide cofactors (NADPH and NADH) in reduced form (left) and oxidized form (right) (Paul and Hollmann 2016). ... 21
xiv
Figure 1.14. The electrochemical regeneration of NADH in the presence of a mediator and LiDH. The electrode reduces the mediator which can reduce the enzyme. Then, the reduced enzyme gives electron to NAD+ and produce NADH. The cofactor will be consumed in the reaction with LDH and the cycle continues (Fry et al. 1994). ... 24 Figure 1.15. Reaction scheme for P450 BM3-catalyzed oxygenation linked by a two-step cofactor regeneration using soluble transhydrogenase (STH) and GLD (Mouri et al. 2009). ... 25 Figure 1.16. Different immobilization patterns for enzymes and cofactor on MNPs. (a) separated immobilization of enzymes and cofactor, (b) co-immobilization of enzymes on the support and separated immobilization of the cofactor, and (c) co-immobilization of both enzymes and the cofactor (Zheng et al. 2011). ... 26 Figure 1.17. A) Structure of biomimetics of nicotinamide cofactors and B) 1a: BNAH, and 1b: para-methoxy-BNAH (Ryan et al. 2008). ... 28 Figure 1.18. X-ray crystal structure analysis of rhuthenium-FeΙΙΙP450 heme region.
Examination of different mutations of P450 BM3 by light induced systems acting on lauric acid indicates that less distance between the Ru(ΙΙ) photosensitizer and the heme results in higher total turnover number (Ener et al. 2010). ... 31 Figure 1.19. Schematic representation of cytochrome P450 on an electrode surface (Bistolas et al. 2005). ... 32 Figure 1.20. Schematic analysis of P450 BM3 electron transfer pathways (Mutants: F87Y: deactivated monooxygenase, W1046A: deactivated FAD domain, G570D and A264H: The enzyme does not bind FMN) (Kitazume et al. 2007). ... 35 Figure 1.21. Categories of enzyme immobilization: A) adsorption, B) covalent bonding, C) entrapment, and D) crosslinking (Sirisha et al. 2016). ... 42 Figure 1.22. Crystal structure of (a) hematite and (b) magnetite (Teja and Koh 2009). ... 47 Figure 2.1. The scheme of the 250 mL three-neck flask used to synthesize the magnetic nanoparticles. ... 58 Figure 2.2. a) PMIDA structure (Demin et al. 2018) and b) schematic of MNPs surface functionalized by PMIDA and Ni2+ (Sahu et al. 2011). ... 59 Figure 2.3. The scheme of a glutaraldehyde activated iron oxide particle... 60 Figure 2.4. The scheme of the enzyme immobilization process: 1) 40 mg of the MNPs is added to the enzyme solution (100 or 200 nM enzyme in 0.1 M PBS pH 7.4); 2) the enzyme and the MNPs were incubated for 4 hours at 4 ºC; 3) after 4 hours, the enzyme is immobilized on the particles; 4) the enzyme-MNPs are separated from the buffer solution by applying a magnet; the supernatant is analyzed to calculate the amount of enzyme immobilized on the MNPs. ... 63 Figure 2.5. Reaction scheme of p-nitrophenoxycarboxylate (p-NCA) conversion by P450 BM3 F87A mutant (Schwaneberg et al. 1999). By receiving help from oxygen and the cofactor, the enzyme hydroxylates the substrate that leads to an instable hemiacetal which dissociates into ω-oxycarboxylic acid and a yellow chromophore
p-xv
nitrophenolate. The formation of this latter can be directly monitored in a
spectrophotometer at wavelength of 435 nm. ... 67
Figure 2.6. NADH standard curve………..……….66
Figure 2.7. BNAH standard curve. ... 72
Figure 2.8. p-Nitrophenolate standard curve. ... 73
Figure 2.9. A) Free enzyme-NADH-10-pNCA reaction system and B) immobilized enzyme-NADH-10-pNCA reaction system (MNPs are separated by putting a magnet under the Erlenmeyer). Yellow color of the reaction medium is the result of p-nitrophenolate formation. ... 74
Figure 2.10. The reaction medium of myristic acid-NADH: A) the enzyme-MNPs in the reaction medium and B) the enzyme-MNPs are separated using a magnet. Samples were taken from the supernatant to analyze the reaction medium. ... 75
Figure 2.11. 10-pNCA-NADH reaction system: the Erlenmeyer on the left contained 1 nmol of enzyme while the Erlenmeyer on the right side has 2 nmol of enzyme. More product was formed in the reaction conducted with a higher amount of enzyme according to the color of the reaction medium (the Erlenmeyer on the right). ... 76
Figure 2.12. 10-pNCA-NADH reaction system containing 50, 100, 150, 200, 250 μM substrate (left to right); as the initial substrate concentration increases in the samples, the intensity of the yellow color of the reaction enhances. ... 76
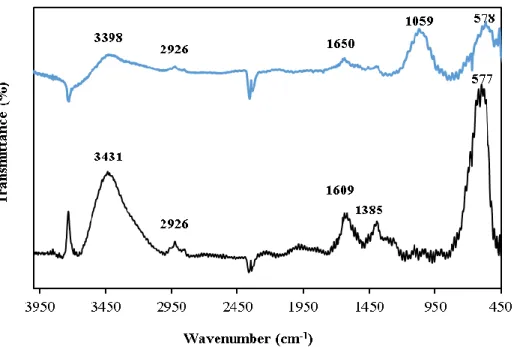
Figure 3.1. Transmission FTIR spectra of functionalized MNPs before (black) and (blue) after enzyme immobilization. ... 92
Figure 3.2. Weight loss analysis from TG curves of Ni2+-PMIDA-Fe3O4. ... 93
Figure 3.3. X-ray diffraction (XRD) patterns for bare Fe3O4 nanoparticles (red) and functionalized Fe3O4 nanoparticles (black). ... 94
Figure 3.4. Magnetization curves of (a) bare and (b) functionalized MNPs. ... 94
Figure 3.5. Dynamic light scattering (DLS) result for size distribution of functionalized MNPs. ... 95
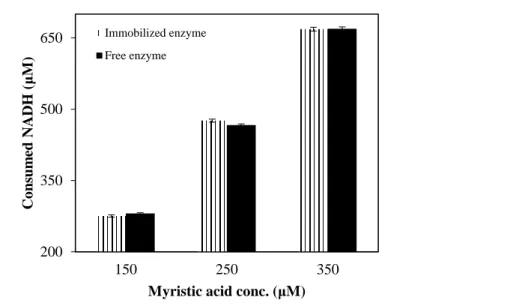
Figure 3.6. NADH consumption by free and immobilized enzyme versus initial myristic acid concentration (reaction performed in 0.1 M PBS, pH 7.4). ... 97
Figure 3.7. BNAH consumption by free and immobilized enzyme versus initial myristic acid concentration (reaction performed in 0.1 M Tris buffer, pH 8.2). ... 97
Figure 3.8. Chromatographic analysis attesting to the formation of ω-1, ω-2 and ω-3 products (reaction catalyzed by immobilized P450 BM3 hydroxylating myristic acid using NADH). ... 98
Figure 3.9. Cofactor consumption by free and immobilized enzyme (substrate concentrations: 150, 250, and 350 μM, using 1 mM cofactor in 0.1 M Tris buffer, pH 8.2). ... 99
Figure 3.10. p-Nitrophenolate production by free and immobilized enzyme (substrate concentrations: 150, 250, and 350 μM, using 1 mM cofactor in 0.1 M Tris buffer, pH 8.2). ... 100
xvi
Figure 3.11. Effect of glutaraldehyde concentration on enzyme activity (350 μM
10-pNCA and 1 mM cofactor in 0.1 M Tris buffer, pH 8.2). ... 100
Figure 3.12. Enzyme residual activity versus number of cycles for (□) adsorbed enzyme consuming NADH and (◊) adsorbed enzyme oxidizing BNAH (350 μM 10-pNCA in 0.1 M Tris buffer, pH 8.2). ... 101 Figure 3.13. Relative activity of immobilized enzyme towards NADH versus storage time (days) for (□) crosslinked-adsorbed enzyme and (■) adsorbed enzyme (350 μM 10-pNCA and 1 mM NADH in Tris buffer 0.1 M pH 8.2). ... 103 Figure 4.1. Reaction scheme of the conversion of 10-pNCA by the P450 BM3 double mutant in the presence of cofactor (NADH) and molecular oxygen; the products of this reaction are ω-oxydecanocarboxylic acid and yellow p-nitrophenolate. The formation of the latter was directly monitored in a spectrophotometer at wavelength of 435 nm to determine the enzyme activity. ... 116 Figure 4.2. BM3-SPIONs: suspended in buffer solution (a); separated by a magnet after overnight reaction with (b) myristic acid and (c) 10-pNCA (the yellow color attests the formation of p-nitrophenolate). ... 116 Figure 4.3. SPIONs activation for P450 BM3 immobilization. By adding AEAPTMS to the bare iron oxide particles suspension, covalent bonds are formed between its alkoxy groups and the hydroxyl groups on the particles surface. In the next step, ‒NH2
group of the coated particles bind to one of the aldehydes in glutaraldehyde while leaving the other one available for the enzyme. ... 118 Figure 4.4. FT-IR spectra of functionalized SPIONs before (blue) and after (black) enzyme immobilization. ... 119 Figure 4.5. TG curves of synthesized glutaraldehyde activated SPIONs. ... 120 Figure 4.6. X-ray diffraction patterns of (blue) bare and (black) glutaraldehyde activated SPIONs. ... 121 Figure 4.7. Magnetization curves of (blue) magnetite and (black) glutaraldehyde activated SPIONs. ... 121 Figure 4.8. Size distribution of the glutaraldehyde activated SPIONs. ... 122 Figure 4.9. Cofactor consumption by free and immobilized enzyme obtained from Set #2 of the experiments. ... 124 Figure 4.10. p-Nitrophenolate production by free and immobilized enzyme obtained from Set #2 of the experiments. ... 124 Figure 4.11. NADH consumption versus initial myristic acid concentration by free and immobilized enzyme obtained from Set #2 of the experiments. ... 125 Figure 4.12. Chromatographic analysis attesting ω-(1-3) products formation. Reaction catalyzed by BM3-SPIONs using NADH as cofactor; ω-3, -2, and -1, hydroxylated myristic acids were detected at 48, 49, and 51 minutes, respectively. ... 126 Figure 4.13. BM3-SPIONs relative activity towards 10-pNCA versus reaction cycles using the cofactor (■) NADH and (□) BNAH. ... 127
xvii
Figure 4.14. Relative activity of BM3-SPIONs towards (■) NADH and (□) 10-pNCA
versus storage time (days). ... 129
Figure 5.1. The scheme of immobilized BM3 on modified MNPs via i) adsorption, ii) crosslinking-adsorption, and iii) covalent bonding. ... 139 Figure 5.2. Reaction scheme of the conversion of 10-pNCA by the immobilized BM3; the enzyme breaks the substrate into ω-oxydecanocarboxylic acid and p-nitrophenolate which changes the color of reaction medium to yellow. ... 140 Figure 5.3. (a) Time dependent p-nitrophenolate concentration profiles at different substrate concentrations: 10-pNCA hydroxylation by 200 nM free BM3; (b) Hill plot of the initial reaction rate. ... 142 Figure 5.4. Hill plot of the free BM3 initial reaction rate: 10-pNCA-NADH reaction system. ... 143 Figure 5.5. Hill plot of the free BM3 initial reaction rate: 10-pNCA-BNAH reaction system. ... 144 Figure 5.6. Hill plot of the free BM3 initial reaction rate: 10-pNCA hydroxylation by 200 nM enzyme consuming NADH or BNAH. ... 145 Figure 5.7. p-Nitrophenolate concentration versus initial substrate concentration: experimental data and the model obtained from the free BM3 reaction: (a) NADH, time = 5 min; (b) BNAH, time = 7.5 minutes. ... 148 Figure 5.8. p-Nitrophenolate concentration versus time: experimental data and the model obtained from the free BM3 hydroxylating 50 μM substrate in the presence of NADH. ... 149 Figure 5.9. The effect of enzyme immobilization on 10-pNCA hydroxylation yield using NADH; enzyme concentration of: (a) 200 nM and (b) 100 nM. ... 151 Figure 5.10. The effect of enzyme immobilization on 10-pNCA hydroxylation yield using BNAH; enzyme concentration of: (a) 200 nM and (b) 100 nM. ... 152
xviii
Abbreviations
10-pNCA 10-(4-Nitrophenoxy) decanoic acid
12-pNCA 12-(4-Nitrophenoxy) dodecanoic acid
AD Adenosine dinucleotide ADH Alcohol dehydrogenase
AEAPTMS N-[3-(trimethoxysilyl) propyl] ethylenediamine
BNAH 1-benzyl-1,4-dihydronicotinamide
CC Carbon cloth
CLEAs Crosslinked enzyme aggregates
CO Carbon monoxide
CO2 Carbon dioxide
DAAO D-amino acid oxidase DIW Deionized water
DLS Dynamic light scattering Em Reduction potential
EPG Edge-plane graphite
EPR Electron paramagnetic resonance
EXAFS Extended X-ray absorption fine structure FAD Flavin adenine dinucleotide
FDH Formate dehydrogenase FMN Flavin mononucleotide
FT-IR Fourier transform infrared spectroscopy GAC Glutaryl acylase
GDH Glucose dehydrogenase GLD Glycerol dehydrogenase GLDH Glutamate dehydrogenase
xix
GPDH Glyceraldehyde-3-phosphate dehydrogenase H2O2 Hydrogen peroxide
Kcat Turnover rate constant
KM Michaelis-Menten constant
LDH Lactate dehydrogenase LiDH Lipoamide dehydrogenase MDH Malate dehydrogenase MOFs Metal organic frameworks MNPs Magnetic nanoparticles MV Methyl viologen dichloride Na2S2O4 Sodium dithionite
NAA N-terminal amino acids
NADH Nicotinamide adenine dinucleotide
NADPH Nicotinamide adenine dinucleotide phosphate NMR Nuclear magnetic resonance
O2 Oxygen
P450 BM3 Cytochrome P450 monooxygenase from Bacillus
megaterium
P450cam Cytochrome P450 monooxygenase from Pseudomonas putida
PG Pyrolytic graphite pI Isoelectric point
ROS Reactive oxygen species
rR Resonance Raman
SDS Sodium dodecyl sulfate STH Soluble transhydrogenase TTN Total turnover number
xx
xxi
Acknowledgement
I would like to express my special appreciation to Professor Maria Iliuta. This work would have never been accomplished without her support. During the past five years, she has guided me through different steps of this research and I gratefully acknowledge all her contributions of time, ideas, comments, suggestions, and funding that made my PhD possible.
I would like also to thank my co-supervisors, Professor Alain Garnier and Professor Faïçal Larachi. Their advices and comments to improve this study and the related articles have been invaluable.
I am so grateful to Thierry Vincent, PhD student in charge of enzyme mutation and production. I very much appreciate his enthusiasm and willingness to mentor and help me until my last days working in the laboratory. Dr Ion Iliuta was kind enough for his significant effort and guidance in enzyme kinetic studies for which I would like to express my sincere gratitude.
I appreciate collaboration of Professor Bruno Gaillet in sharing his lab facilities (Université Laval), Roxane Bernier for GC-MS analysis (Oleotek), and Professor John Boukouvalas (Université Laval) for synthesis of the substrate model, 10-pNCA, for this research. The exceptional academic opportunities provided by the chemical engineering department of Université Laval and financial support by the Natural Sciences and Engineering Research Council of Canada (NSERC) are gratefully acknowledged.
Above all, my humble appreciation and endless gratitude go to my parents, Maryam and Mohammadreza, and my siblings, Arefeh and Rahim for their love, motivational talks, and support through my life which was worth more than I can express on paper. My very special thanks go to my beloved fiancé, Pier-Luc. The greatest things happened to me within the last four years would have never been possible if he had not been there for me.
xxii
Preface
This dissertation contains 6 sections.
Introduction presents the enzyme, cytochrome P450 BM3 from Bacillus
megaterium, its structure, catalytic cycle, and different enzymatic reactions catalyzed
by this enzyme including the studies investigating the kinetic models. A review is presented on this enzymatic system, the importance of enzyme immbilization and the enzyme immobilization methods. Since magnetic nanoparticles have been chosen for enzyme immobilization in this work, they are also introduced. Finally, the immobilized P450 BM3 systems available in the open literature are reviewed. The ‘Introduction’ ends with conclusions and the objectives of this work. Chapter 1 explains all the methods and techniques used for synthesis, surface-functionalization, and characterization of magnetic nanoparticles, enzyme quantification, and activity assays used for free and immobilized enzyme, and enzyme recycling and storage stability tests. Chapters 2-4 present the main part of this thesis (results and discussions), corresponding to the papers mentioned below. Finally, the general conclusions and suggestions for future work are given.
This thesis was prepared based on the following published and submitted papers in/to scientific journals:
1- Bahrami, A., Vincent, T., Garnier, A., Larachi, F., Boukouvalas, J., and Iliuta, M.
C., Noncovalent immobilization of optimized bacterial cytochrome P450 BM3 on functionalized magnetic nanoparticles. Industrial Engineering & Chemistry Research (2017), 56, 10981-10989 (Chapter 2).
2- Bahrami, A., Garnier, A., Larachi, F., and Iliuta, M. C., Covalent immobilization
of cytochrome P450 BM3 (R966D/W1046S) on glutaraldehyde activated SPIONs. Canadian Journal of Chemical Engineering (2018), DOI:10.1002/cjce.23208 (Chapter 3).
3- Bahrami, A., Iliuta, I., Garnier, A., Larachi, F., Vincent, T., and Iliuta, M. C.,
xxiii
NADH-dependent cytochrome P450 BM3 from Bacillus megaterium, (submitted) (Chapter 4).
The author has main contribution in all stages of the work presented in papers 1 to 3, including planning and performing experiments, as well as writing the papers by considering the supervisor’s comments. Enzyme kinetic modelling performed in article 3 (Chapter 4) was done by Dr Ion Iliuta.
The results of the present thesis were also presented in the following academic national conferences:
1- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., Non-covalent immobilization
of a cofactor-dependent enzyme on iron oxide magnetic nanoparticles for organic synthesis reactions. 65th Canadian Chemical Engineering Conference (CSChE),
Calgary, October 4-7, 2015 (oral presentation).
2- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., Evaluation of immobilized
cytochrome P450 BM3 from Bacillus megaterium for hydroxylation of 10-pNCA. 66th Canadian Chemical Engineering Conference (CSChE), Quebec, October
16-19, 2016 (oral presentation).
3- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., Synthesis and characterization
of surface functionalized Fe3O4 and its application for immobilization of
cytochrome P450 BM3 from Bacillus megaterium. 66th Canadian Chemical Engineering Conference (CSChE), Quebec, October 16-19, 2016 (poster).
The results of the present thesis have also been presented in annual meetings of Centre en Chimie Verte et Catalyse (CCVC):
1- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., Cofactor-dependent enzyme
immobilization for organic synthesis reaction using cofactor regeneration system. 6th annual conference CCVC, Montreal, May 2014 (poster).
2- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., Non-covalent
immobilization of a cofactor-dependent enzyme on iron oxide magnetic nanoparticles. 7th annual conference CCVC, Quebec, May 2015 (poster).
xxiv
3- Bahrami, A., Garnier, A., Larachi, F., Iliuta, M. C., A study of the efficiency of
immobilized cytochrome P450 BM3 from Bacillus megaterium in hydroxylation of 10-pNCA. 8th annual conference CCVC, Montreal, June 2016 (poster). It should be noted that within the framework of a collaborative NSERC research project, the research group of Professor Alain Garnier, who worked on the enzyme mutation and production, provided the enzymes for this study. The enzyme production and mutation are therefore not the subject of this thesis.
1
1. Introduction
1.1. Background
Regio- and stereoselective hydroxylation of C‒H bonds is a very difficult transformation in chemistry. However, these kinds of reactions are deftly catalyzed by a variety of metalloenzymes including many members of a superfamily of enzymes named cytochrome P450s. The defining reaction of these enzymes is the reductive activation of molecular oxygen (O2) through which the enzyme inserts one atom of O2
into an activated or non-activated aliphatic or aromatic R‒H bonds (R: C, N, and S) while reducing the other atom to a water molecule. P450s can decrease the activation energy of such reactions to 38-71 kJ·mol-1 which is 418-460 kJ·mol-1 for an uncatalyzed reaction. The exceptional feature of this reaction is that the electrons required for reducing the molecular oxygen are provided from reduced pyridine nucleotides such as nicotinamide adenine dinucleotide phosphate (NADPH) (Lewis et al. 2001, Maurer et al. 2005, Ortiz de Montellano 2010, Shuler and Kargi 2002). The overall reaction equation is shown as follows (King et al. 1988):
RH + O2 + H++ NAD(P)H → ROH + NAD(P)+ + H2O (1.1) RH stands for a substrate (e.g. fatty acid molecule) and ROH is the mono-oxygenated product of the reaction.
Cytochrome P450 enzymes exist in all type of organisms, bacteria, fungi, plants, insects, and mammals. However, mammalian, fungal, and plant P450s are membrane-bound proteins with restricted applications in industrial bio-transformations. Degradation of products, toxicity of the products for the cells, and complicated recovery of products from the fermentation medium are the main disadvantages of such systems. According to these drawbacks, employing non-membrane associated microbial P450s in enzymatic processes can be beneficial. Particularly appropriate for enzymatic processes are bacterial P450s such as P450 BM3 from Bacillus megaterium. This soluble, self-sufficient enzyme has its reductase domain naturally fused to the
2
monooxygenase domain (Heme) (Figure 1.1) through a polypeptide linker. This arrangement in the enzyme structure resulted in the highest turnover frequency (more than 1000 min-1 in hydroxylation of archidonate) between members of P450 family (Ferrero et al. 2008, Maurer et al. 2005, Nazor et al. 2008, Shehzad 2013, Tsotsou et al. 2002).
Figure 1.1. Modular arrangement of P450 BM3 domains (Shehzad 2013).
Based on these remarkable properties, since its discovery, BM3 has been an ideal model for investigation on P450s behavior from different aspects. The wild-type enzyme was mutated to act on unnatural substrates/cofactors and improve its co-solvents tolerance. Hydroxylation activity of BM3 (wild-type or mutated) was evaluated in an enzymatic reaction with a cofactor regeneration system, or where the cofactor was replaced by an electrode or a reducing agent. However, reducing the process cost and improve the enzyme stability by immobilizing the enzyme was only the subject of a few reports.
The present thesis focuses on the immobilization of an optimized BM3 (which consumes less expensive cofactors than costly NADPH) on functionalized magnetic nanoparticles, with the aim of improving the enzyme stability, and facilitating the enzyme recovery and reuse. The literature review will therefore be limited to reaction systems involving cytochrome P450 BM3 from Bacillus megaterium.
First, the information about the enzyme, P450 BM3, its catalytic reaction cycle, structure, and active site is provided. Second, the cofactor regeneration and alternative electron supplies used in hydroxylation reactions by BM3 are reviewed. In addition, kinetic studies of the hydroxylation reactions by BM3 and related kinetic parameters are provided. Finally, the necessity of enzyme immobilization, the common methods used to immobilize enzymes, the advantages of using magnetic nanoparticles (MNPs)
3
as appropriate support for enzyme immobilization, and examples of immobilized BM3 systems (BM3 immobilization via different methods and on various supports) are discussed.
1.2. Cytochrome P450 BM3 from Bacillus megaterium
Cytochrome P450 BM3 from Bacillus megaterium (CYP102A1) was discovered in 70s by Miura and Fulco (1975). It was identified as a soluble medium- to long-chain fatty acid hydroxylase that required only oxygen and cofactor (NADPH) to function. In its name, P stands for pigment and 450 reflects the P450s special property: carbon monoxide (CO) bubbling through a preparation of a P450 results in a spectrum with maximum absorbance at 450 nm. As BM3 was the third isolated enzyme from Bacillus
megaterium, its name was designated P450 BM3 (Kim and Oh 2013, van Bogaert et al.
2011, Whitehouse et al. 2012). The physiological substrate of BM3 is unknown; however, Wolf and coworkers have speculated that this enzyme is responsible to protect Bacillus megaterium from toxic fatty acids of the environment (Kitazume et al. 2007).
This enzyme can catalyze hydroxylation and/or epoxidation of a broad range of substrates including alkanes, alkenes, alcohols, fatty acids, amides, polyaromatic hydrocarbons, and heterocycles. The natural substrates for P450 BM3 are the long chain fatty acids (C12 to C22). They can be hydroxylated at the subterminal ω-1, ω-2,
and ω-3 positions, partially with high enantioselectivity. The artificial mutants of P450 BM3 can also hydroxylate non-natural substrates like short chain fatty acids (C8 to C10),
indole, polycyclic aromatic hydrocarbons, alkanes, and styrenes (Ferrero et al. 2008, Maurer et al. 2003).
The soluble 119 kDa cytoplasmic protein, P450 BM3 contains both monooxygenase (55 kDa) and reductase domain (65 kDa) on a single polypeptide chain (naturally fused). Monooxygenase contains the substrate and oxygen binding sites of the enzyme while reductase domain serves as an electron carrier and shuttles electrons from the cofactor to the enzyme active site. This structure led to the turnover frequency
4
of 17000 min-1 hydroxylating arachidonate which is the highest catalytic activity reported for a P450 monooxygenase to date. BM3 is also more thermostable and easier to handle compared to other P450s (Munro et al. 2002, Roh et al. 2012, Tsotsou et al. 2002, Whitehouse et al. 2012). The structure and function of monooxygenase and reductase domains are explained in §1.2.1 and §1.2.2, respectively.
1.2.1. Monooxygenase domain
1.2.1.1. P450 BM3 active site
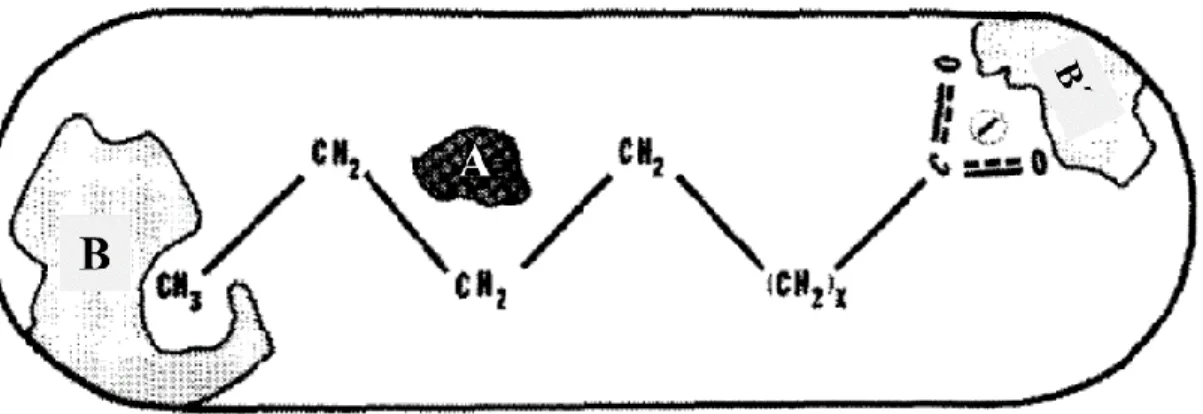
Omega hydroxylation of fatty acids by P450 BM3 results in isomeric mixtures of the ω-1, ω-2, and ω-3 derivatives. The distribution of these three isomers depends on chain length and type of the substrate, however, generally the ω-2 position is favored. Hydroxylation of terminal methyl (ω) group of these substrates has never been reported and it did not appear to be significant hydroxylation of methylene carbons beyond the ω-3 position. Considering these information on hydroxylation activity of BM3, first hypothesis on the enzyme active site structure was made by Miura and Fulco (Miura and Fulco 1975). They hypothesized that the enzyme should have a polar group binding site (B' in Figure 1.2) for effective attachment of substrate via its carboxyl (‒COOH) group. As the terminal methyl group of the alkyl chain is never hydroxylated, there should be also a methyl-group binding site (B in Figure 1.2) to which the methyl group is tightly bound. Dependence of enzyme hydroxylation activity on substrate chain length should be related to these two sites. For example, pentadecanoic acid has the most favorable chain length for the enzyme active site (C‒H bond: 1.09 Å). Fatty acids longer than 15 carbons would require additional folding to fit in this region whereas shorter substrates need additional extension. Distribution of omega isomers can be ascribed to slight positional shifts of the last three methylene groups of the substrate which require the methylene carbon (ω) to be free to rotate.
Later, this primary model and the role of key amino acids in enzyme activity were explained in more details by other reports. It was found that P450 BM3 has a long, hydrophobic, large non-aromatic, funnel shape substrate access channel (Figure 1.3). It is approximately 20 Å long and 10 Å in diameter extending from the surface of the
5
enzyme molecule down to the heme. The access channel would reshape in response to substrate binding, producing a closed conformation and consequently enhancing the protein-substrate interactions (Oliver et al. 1997, Whitehouse et al. 2012).
Figure 1.2. Schematic model of enzyme-substrate complex in P450 BM3 active
site; B is the binding site for the terminal methyl group of the substrate, B' is the biding site for the polar group of the substrate and A indicates the
hydroxylation site (Miura and Fulco 1975).
Position of heme and important amino acid residues in the substrate access channel (active site) of P450 BM3 are shown in Figure 1.4. The structure and function of heme and amino acid residues involved in the substrate association and regioselective oxidation are explained as follows.
6
Figure 1.3. Schematic representation of relative positions of heme and key
amino acid residues in the active site of palmitoleate-bound P450 BM3 (Noble et al. 1999).
Figure 1.4. Key amino acids in the X-ray atomic structure of
palmitoleate-bound P450 BM3 (Ost et al. 2000).
• Heme
P450s require cofactors, such as flavins, metal-ions, and hemes for their catalytic activity. Sometimes cofactors are tightly or covalently bound to the enzyme (like heme in P450s (Figure 1.5, A)) and form part of the enzyme structure. The heme cofactor and the surrounding residues are deeply buried inside the P450s. In all members of this
7
enzyme family, a strictly conserved cysteine is found in the active site that acts as the fifth ligand of the heme-iron center which is responsible for activating the metal complex (Torres Pazmiño et al. 2010). The heme moiety environment in P450 and the pyrrole ring nomenclature are illustrated in Figure 1.5, B. Bond distances between key atoms and the heme iron are reported in Table 1.1 (Lewis et al. 2005).
A B
Figure 1.5. A) Crystal structure of P450 BM3 monooxygenase from Bacillus
megaterium (Urlacher 2012) and B) Heme structure of cytochrome P450
(Lewis et al. 2005).
Table 1.1. P450 crystal structure bond distances at the heme
(Lewis et al. 2005).
Type Bond Distance
(Å) Comments
Iron to cysteinate Fe-S 2.15-2.52 Ranges for several structures
Iron to bond water Fe…..OH2 2.09-2.35 Ranges for several structures
Iron to porphyrin Fe-N 1.95-2.05 Ranges for several structures
Iron to oxygen Fe-O 1.81-1.67 Values for dioxygen and oxene,
8
Fatty acids initially bind relatively far from the heme with a distance of ⁓ 7.5 Å between the terminal methyl group and the heme iron. Conformational changes occur when the heme is reduced. It drives the fatty acid to the position of 6 Å nearer to the heme, proper position for hydroxylation at the ω-1, ω-2, and ω-3 positions (Oliver et al. 1997, Ost et al. 2000, Munro et al. 2002). Key amino acid residues involved in substrate association and oxidation and enzyme active site conformational changes are introduced as follows. Table A.1 provides the list of amino acids, their full names and abbreviations.
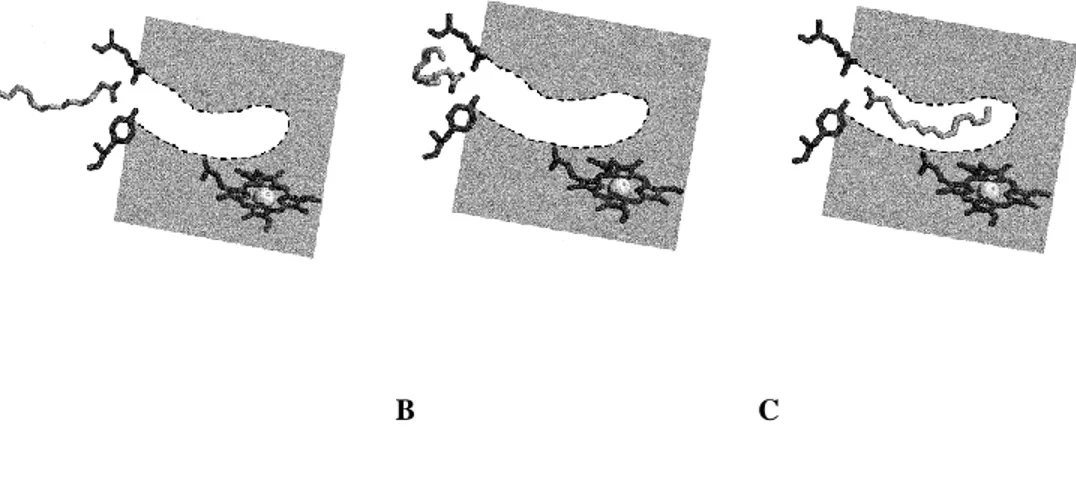
• Arginine47 and Tyrosine51
Two hydrophilic polar residues close to the mouth of the substrate access channel are arginine (Arg) at position 47 and tyrosine at position 51. Arg47 lies in the enzyme surface and Tyr51 tether the carboxylate group of fatty acid substrate many ÅngstrÖms
from the heme (Figure 1.6). Together, they control the admission of substrate, water molecule and co-solvents, therefore controlling substrate specificity. P450 BM3 is the only CYP102A subfamily member with a polar residue at position 51 and the only one with a positively charged residue at position 47. Tyr51 and perhaps Arg47 can form hydrogen bonding pairs with the substrate carboxylate. It was observed that the Arg47 substrate interactions may be flexible transient and perhaps not very critical for substrate binding (Li and Poulos 1997, Noble et al. 1999, Shehzad 2013, Whitehouse et al. 2012).
9
A B C
Figure 1.6. Model for substrate interaction in P450 BM3. (A) Electrostatic
forces orientate the carboxylate group of the substrate towards Arg47 and by further hydrogen bonding of the Tyr51 and the ‒COOH group, the binding is stabilized, (B) the carbon chain is drawn to the active site channel and the amino acids inside the channel provide favorable hydrophobic interactions, and (C) final catalytically competent orientation is achieved, and the heme is
ready for reduction and initiating the catalytic cycle (Ost et al. 2000).
Removing these amino acids, arginine47 and tyrosine51 from the P450 BM3 active site disfavored binding of all fatty acids and resulted in 4.3 and 37.1-fold decrease in catalytic efficiency (kcat/KM) for laurate and archidonate, respectively. This reduction
in enzyme activity indicates the important role of the arginine and tyrosine in the initial recognition of substrate. Fatty acid chain length selectivity depends on these two amino acids. As can be seen in Figure 1.6, substrate cannot enter directly into the long hydrophobic active site channel until carboxylate group of fatty acid is attracted to the guanidinium group of Arg47 at the mouth of active site by electrostatic forces. Afterwards, formation of hydrogen bond to the hydroxyl group of Tyr51 occurs. Once the hydrogen bond to this residue formed, there is a strong driving force for the movement of the long hydrophobic tail of the fatty acid from the polar solvent phase into the substrate binding channel (Ost et al. 2000). It was reported that substitution of arginine at position 47 by phenylalanine increases hydrophobicity at the entrance of the binding pocket and further enables the diffusion of shorter chain substrate like 8-, 10-, and 12-(4-nitrophenoxy)dodecanoic acid (pNCA) into the active site (Urlacher et al. 2004).
10
• Phenylalanine87
Phenylalanine (Phe) at position 87 is located directly over the heme. It extends from the lumen of the substrate access channel to the heme iron. It is known to undergo a dramatic movement on substrate binding and responsible for control of the regio-specificity of substrate oxidation. This amino acid blocks the substrate from approaching too close to the heme. In fact, it rotates from a position nearly perpendicular to the heme in the substrate-free structure to a more parallel position in the substrate complex which effectively blocks the ω-terminal methyl group from approaching the heme (by forming a small hydrophobic pocket within the active site). Therefore, the terminal methyl group of the fatty acid is stuck in a hydrophobic patch. This can explain why wild-type P450 BM3 never hydroxylates the ω-terminal methyl group of fatty acids (Brühlmann et al. 2014, Li and Poulos 1997, Munro et al. 2002, Noble et al. 1999, Whitehouse et al. 2012). Phe87 is one of the most studied active-site residues. It was mutated to amino acids with small nonpolar side chains such as glycine (Gly), alanine (Ala), leucine (Leu), isoleucine (Ile), and valine (Val) and the amino acid residues with only two uncharged polar amino acids like serine (Ser) and tyrosine. Catalytic property of such mutants of BM3 depends on the substrate and the amino acid that replaced phenylalanine. Changes in active-site volume can restrict or improve the access to the reactive oxygen species at the heme center (Vottero et al. 2011). The bulky phenylalanine can restrict the access of organic co-solvents to the heme and increase the enzyme organic solvent resistance. For example, the amino acids residues like alanine (less bulky than Phe) at this position shows lower organic solvent resistance (Wong et al. 2004). Furthermore, when alanine is found at position 87, the fatty acid adopts another conformation in the binding pocket than in the wild type enzyme. The terminal methyl group is brought closer to the heme and subsequently, the fatty acid is hydroxylated at the ω-position. In addition, when phenylalanine was substituted by valine (F87V), the enzyme mutant preferentially hydroxylates the substrate at the ω-3 position. This might be due to an enlargement of the binding pocket caused by the amino acid replacement close to the heme. Therefore, the substrate moves deeper into the binding pocket, leaving the ω-3 position closer to the heme where the hydroxylation takes place (Lentz et al. 2001). In another report, tyrosine at this position
11
restricted the heme accessibility of substrates (fatty acids) and subsequently resulted in full uncoupling (Uncoupling will be explained in §1.3). However, the same mutant was active towards alkoxyresorufines and testosterone (Vottero et al. 2011).
• Threonine268
Threonine (Thr) residue at position 268, available in the distal Ι helix, has a key role in oxygen binding and activation (Urlacher et al. 2004). It was proposed that, Thr stabilizes the oxygen-bound intermediate by a hydrogen bond to the iron-bound oxygen. Therefore, the loss of hydroxyl group reduces the stability of oxygen-bound intermediate, thus releasing superoxides and decreasing the catalytic activity. Furthermore, it involves in proton delivery, substrate recognition, electron transfer, prevent solvent water molecules to access the active site, and the positioning of heme-ligand water (Clark et al. 2006, Truan and Peterson 1998). This amino acid residue might prevent the opening of a proton channel to the substrate binding pocket which is located around the heme iron, therefore, it blocks the protons from water to attack the iron-dioxygen complex. If protons attack the iron-oxygen intermediates, hydrogen peroxide (H2O2) will be generated and uncoupling of NADPH oxidation from the
hydroxylation of the organic substrate occurs. For instance, substitution of this amino acid residue by alanine in P450cam (at position 252 for this enzyme) resulted in
uncoupling of NADH and formation of H2O2. The same mutation in P450 BM3 led to
a decrease in fatty acid hydroxylation and an increase in H2O2 formation when lauric
acid was used as a substrate (Truan and Peterson 1998, Yeom et al. 1995). A hypothesis made by Ishimura and coworkers suggested that the oxygen in Thr creates a hydrogen bond network with a water molecule which could serve as the proton donor to the ferrous-dioxygen complex (Figure 1.7). As can be seen, the position of arachidonic acid (AA) close to the heme iron prevents access of the water molecule to the proximal oxygen. However, if Thr is replaced by Ala, more space will be available for AA resulting in moving the ω-end of the substrate upward and preventing the hydrogen bonding network. In this situation, any proton that find access to the oxygen can lead to H2O2 formation (Truan and Peterson 1998).
12
Figure 1.7. Schematic representation of Thr268 hydrogen bonding network
with the water molecule which can serve as proton donor to the ferrous-dioxygen complex. This hydrogen bonding network is shown in the heme domain of P450 BM3 with arachidonic acid (C20) (Truan and Peterson 1998).
Mutation of Thr268 to an asparagine retained the hydrogen binding capability of the enzyme, however, removed the proton donating possibilities, thus resulted in a highly uncoupled enzyme, a decrease in first electron transfer rate, and a destabilized oxy-ferrous species (Clark et al. 2006).
• Other residues
In the access channel, the amino acid residue, leucine (Leu) at position 437 prevent rotation of the side-chain to the substrate-bound position when there is no substrate in the channel and therefore, acts as a safety catch for Phe87 (Whitehouse et al. 2012). Phenylalanine at position 42 forms a hydrophobic lid over the active site. Once the substrate has entered and bound, it caps the access channel (Munro et al. 2002, Noble et al. 1999, Whitehouse et al. 2012). It was reported that replacing this amino acid by alanine, decreased the first electron transfer from flavin mononucleotide (FMN) to heme by 90% and thereby decreasing the catalytic efficiency with lauric acid 15 times. Other residues worthy of note are alanine (Ala) at position 264 and Phenylalanine at
13
position 393. Ala264 is responsible to draw away the water ligand from the axial binding site when the substrate binds to the heme iron. In collaboration with Thr268, Ala264 provides a hydrogen binding pocket for the water molecule which was displaced by substrate binding (Figure 1.8). The water molecule is in equilibrium between the two positions explained in Figure 1.8 and replacement of Thr268 by an alanine would weaken the hydrogen bonding interaction and direct the equilibrium in favor of water bound to the iron (Clark et al. 2006). Phe393 control the rate of heme reduction to optimize the oxygen binding rate (Whitehouse et al. 2012). It establishes the equilibrium between the reduction rate of the heme and the rate of the ferrous-heme binding and subsequently reduce molecular oxygen (Urlacher and Schmid 2004). Substitution of Phe393 with a histidine resulted in a positive shift in the reduction potential (Em) which consequently increased the rate of first electron transfer and the
stability of oxy-ferrous complex (Clark et al. 2006).
Figure 1.8. Crystal structure of the substrate free and palmitoleate bound
wild-type P450 BM3. By binding the susbtrate Ala264 and iron bound water are moved, then Thr268 and Ala264 provide a hydrogen binding pocket for
the displaced water (Clark et al. 2006).
The amino acids Phe87, Ala82, Val78, and Arg47 have been the subject of different studies aiming to facilitate substrate diversification. Phenylalanine, alanine, and valine are internal residues while arginine interacts with the carboxylate group of the fatty acid at the protein surface. Mutations of Phe87 affect the regioselectivity of fatty acid oxidation by moving the hydroxylation position of lauric acid from ꞷ (1-3) to ꞷ-5 in F87A/S/G mutants (Butler et al. 2013).
14 1.2.2. Reductase domain
Although P450s contain substrate and oxygen binding sites, they are not capable of accepting electrons directly from NADPH since the heme is deeply buried within the enzyme active site. Therefore, they need an electron transport chain to shuttle the electrons from the cofactor, NADPH, to the heme. P450 BM3 from Bacillus
megaterium is unique as it is catalytically self-sufficient owing to two distinct domains,
monooxygenase and reductase domains fused on the same polypeptide chain. Reductase domain consists of two flavin groups, FMN and flavin adenine dinucleotide (FAD) which transfer the electrons from the cofactor to the heme by going through oxidized and reduced states (Cook et al. 2016, King et al. 1988, Truan and Peterson 1998, Whitehouse et al. 2012). Electron transport chain in monooxygenation reaction by P450 is schematically shown in Figure 1.9.
Figure 1.9. Electron transport chain in monooxygenation reaction by P450
(King et al. 1988).
A backbone structure of reductase domain composed of four structural domains as shown in Figure 1.10 (A). From N to C terminus, they are the FMN-binding domain, the hinge, the FAD-domain, and the cofactor binding domain. The reductase domain is in shape of an oval with approximate depth, width, and height of 50, 70, and 60 Å, respectively. The cofactor lies in the middle of this bowl-shape domain. The two flavin groups and the heme are in equimolar ratio (1:1:1). As it can be seen in Figure 1.10 A, the two flavin groups do not overlay each other and communicate through the hinge. It was proposed that the electrons are transferred (one at a time) from the oxidized
15
NADPH to FAD and then FAD is brought to FMN by the connecting domain (the hinge) (Figure 1.10 B). After FMN received the electron (closed conformation: two flavin domains juxtaposed for inter-flavin (FAD-FMN) electron transfer), it moves ~10Å (open conformation) to interact and reduce the heme center which is required to activate the molecular oxygen (Munro et al. 2002, Peterson and Boddupalli 1992, Wang et al. 1997, Whitehouse et al. 2012). For wild type BM3, the electron transfer rate from FMN to heme was reported 111 s-1 (Neeli et al. 2005).
A B
Figure 1.10. (A) Ribbon diagram of reductase domain: FMN domain in blue,
the hinge in red, the FAD and NADPH binding domains in green. They are also shown as ball and sticks: FMN (light blue), FAD (yellow), NADP+ (orange) (Wang et al. 1997) and (B) FMN domain motion in flavocytochrome
P450 BM3 (Munro et al. 2002).
The reductase domain has many possible oxidation-reduction states and it is difficult to evaluate which predominates during the reaction cycle. However, there is evidence to indicate that the reductase cycles primarily between FAD∙/FMNH2, FAD∙/FMNH
forms in reducing ferric P450, and probably between FADH∙/FMNH2 and
FADH∙/FMNH∙ in reducing the Fe2+‒O2 (Figure 1.12) (Guengerich 2010) as it will be
discussed in §1.3.
Size exclusion chromatography has shown that P450 BM3 is oligomeric in comparison to standard proteins with an estimated molecular weight of 360 kDa. Based on this analysis, it is presumable that the protein is essentially globular and if it is
16
considered ellipsoidal, the apparent molecular size will be larger (Kitazume et al. 2007).
P450 BM3 exists in oligomer forms (under some conditions): monomers, dimers, trimers, and higher order oligomers. Kitazume and coworkers (Kitazume et al. 2007) showed that BM3 was only active as dimeric species. Therefore, electron transfer between the FAD domain of one monomer to the FMN domain of the other subunit is obligatory in hydroxylation reaction by the enzyme (Figure 1.11). Since the linker between FAD and FMN is not long enough for electron transfer, then FAD domain of one molecule of BM3 might be required to accept the electrons from the other monomer. This can explain why BM3 as monomer lacks activity. Based on shared amino acids, monooxygenase and reductase domain of P450 BM3 segregate into two groups. There is a significant gap in the sequence of the fusions in the region joining the FMN and the hinge region of the reductase. FMN is beyond 20 Å which required the proposed rotation and translation. Furthermore, the docked form of the flavodoxin domain to heme domain in the X-ray crystal structure of this bidomain complex is like the right-hand portion of Figure 1.11 and is only compatible with rotation and translation of the flavodoxin-like domain. To deliver the second electron from NADPH, FMN domain should return to the FAD binding domain (Kitazume et al. 2007).
17
Figure 1.11. Schematic hypothetical interaction of P450 BM3 domains for
electron transfer (working as dimers) (Kitazume et al. 2007).
1.3. Catalytic mechanism of P450 BM3
All cytochrome P450s originate from one single ancestor CYP51. Therefore, they all share a common protein fold, contain the same heme prosthetic group (Fe-protoporphyrin IX) attached to cysteine as the fifth ligand, and they all use a similar catalytic cycle to activate O2 or H2O2 (Mak and Denisov 2018). The overall catalytic
cycle of P450 BM3 that is generally accepted is shown in Figure 1.12. In the resting state, the ferric iron atom (in the heme center) is six coordinates: four positions occupied by nitrogen atoms, one with cysteine and the sixth by water molecule. In the first step, the substrate is bound near (but not to) the iron atom of the heme and it results in loss of axial water ligand. Substrate binding to the heme shifts the heme iron spin from low spin (S=1/2) to high spin (S=5/2) and induces a positive shift of 130-140 mV in heme iron reduction potential (more oxidizing state) (for example: +138 mV when archidonate binds to the BM3 active site). The increase in redox potential allows efficient electron transfer from redox partners and ensures the rapid electron transfer to the heme iron which occurs only when the substrate is trapped in the active site (Urlacher 2012). It should be noted that binding the substrate and displacing the water
18
molecule result in a shift of Soret absorption maximum from ⁓418 nm to ⁓392 nm as well (Butler et al. 2013). The high spin iron II with four unpaired electrons provides a spin favored interaction with ground state O2. The second step is transferring one
electron from the flavoprotein NADPH-P450 reductase to the substrate-bound P450. Then, O2 binds to the ferrous enzyme (third step). The product of one electron reduction
of the oxy P450 complex is a nucleophilic ferric peroxo intermediate (Fe–O–O-). It is
a short-lived intermediate to which a proton can be added and produce the ferric hydroperoxo intermediate (Fe–O–O–H). Additionally, Fe–O–O- complex can accept
the second electron to produce a ferric peroxy anion which is unstable and can be decomposed to generate superoxide anion (O2·¯) or if protonated, H2O2. The rate of
first electron transfer in BM3 is significantly faster than other bacterial P450s, in range of 130-220 s-1 with various substrates. In step 4, the second electron enters the system.
Usually, this step is the rate-limiting step of the catalytic cycle. After this step, all the intermediates are unstable. The complex formed in step 4 must be protonated for function (step 5). This complex can be decomposed at this point and generate as well, H2O2. In step 6, the heterolytic scission of the O‒O bond occurs and generates a formal
(FeO)3+. By releasing the product (ROH) in step 9, the reaction cycle is completed. After formation and releasing the product, the enzyme arrives at its resting state (ferric state) with a water molecule as the sixth ligand of the heme (Cysteine is the fifth ligand) (Clark et al. 2006, Guengerich 2010, Lewis et al. 2005, Mak and Denisov 2018, Ortiz de Montellano 2010, Torres Pazmiño et al. 2010, Whitehouse et al. 2012).
19
Figure 1.12. Catalytic cycle of cytochrome P450 (Cook et al. 2016).
After O2 binds to the heme iron, cessation of catalysis can occur in three ways: (1)
auto-oxidation shunt pathway by which the anion superoxide radical is formed after degradation of the peroxo-ferrous intermediate, (2) peroxidase shunt pathway which leads to protonation of the hydroxyferric intermediate and releasing H2O2 and
dissociation of the catalytic cycle, and (3) oxidase shunt pathway: water molecule is released and the enzyme returns to resting state at the beginning of the cycle (Cook et al. 2016).
Spectroscopic methods have been used to study and provide information about each step of the P450s catalytic cycle. For example, cryoreduction technique and nanodisc technology have been applied to study the ferrous and ferric states of cytochromes