HAL Id: dumas-01400733
https://dumas.ccsd.cnrs.fr/dumas-01400733
Submitted on 22 Nov 2016HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de
Sensibilité au Cetuximab en première ligne de
chimiothérapie palliative dans les carcinomes
épidermoïdes des VADS
Vianney Bastit
To cite this version:
Vianney Bastit. Effet de la mutation KRAS LCS6 variant sur la Sensibilité au Cetuximab en première ligne de chimiothérapie palliative dans les carcinomes épidermoïdes des VADS. Médecine humaine et pathologie. 2016. �dumas-01400733�
&'
( ) *) + ,
*
-
" .
'
'
/( )
(0 & ,
1 !
/2 (3(
. .4
& ) *) +
5!
43 -.
'3
!
$" /0 6 2'
1
5 ' 1'3
*
-. 57
'/8)
3(
! " #
!" # $ $ % & $ ' !'# ( )* &+,"'% - ) ) . /$ ! " # $ % $ * ! " # $ & $ / $ " ' # $ $ " " " # " %( " # ' % ) $ " # " * %+ $ $) . % ,),), ,), , )- . " / & $ 0 % " % * $ * % %+ %( " " " ! ) !! % %" % 0 " 1 2 % %" % " " % " % %" % ) $ )% ) 0 1 ! 3 % %" ' % "
% 2 ' % ' 5 ! " # % %" % " " $ % $ * $ " ! ) !! ! " " %" %6 ( " ! # 5 " # % " " ! # " " " # $ " % ) % " " % " % 4% ) 5 & $ / % " # ) % " ' % %+ % " %" " * % ) ) ! & " $ ! , / ) ) & ) !! % # ! * )) ) & " " . ( $ " 7 ( )* % 8 % ) ( )* " 2 % %% ) . % " " &" ! " # 2 % %" ) $ % " " % " % ' 9 ) )) %( %" * ) $ $ % % " " # 5 & ) . $ " ) $ " " % %% ) ) & * ) . $ % %" % %4 ) 0 %( %" ) * ) 0) # $ " % " 3 & 3 &" % " #
$ % %" % ' % ) " % &% 7 : /&% " " # " ! % ) " 2 % # ) ! ) ) ! " " %" ' % %+ $ )% ! " ) !! " 2 % " ! )) % " % ' % " ! ) $ " 2 ) * / 9" % % % $ % % " $ * !! %" " ! " % ' % " ! % " % % 8 % 2 % %" ) /&% " " # ' % " ! $ % " ) " / * ! " " % * - )) ) ) " " * %% % " % 7 $ )) )% " % )) 0 $ ) * ))* $ " " " % ( % * ; ' % ' # " * ' % ) ) " % " # $ % <" $ % " % " % ) $ $ ! & # " " " %" 8
' # 1 $ ) & - ) 9 $ $ $ $ " = 2 ' & $ )) ) $ ) ) $ 2 $ 2 *$ " * / $ " ) " " ) & ! ) * $ " : " " % " ! % * % %( " " " ! ( . % ' 9 ) ! 3 % %" ' % $ !" " ) $ 5 0 $ " . )) $ " " ) )% ) $ / % " # %% $ ( % " % # ) 0 % 5 & 1 $) % " %
% 0) ) 1 $ ! & # ' % 0) ( " 1 ) $ $ 7 : 0) ) 1 ) " ' % ) )) 5 ; $ 5 ; ) 2 )) ) & " ) 5 ; " % ) 0) ) 1 " ) % 0) ) 1 ) % # ' % ) ) !! % &" # $ $ / % " % % # $ ) $ ) / % # ) & " % $ 7 " % ! " # " % %" > " $ " % # $ $ $ ! & # " ) $ )
7 ) $ " % # * $ ! & # " " " # 7 " " ) ) " % % ) $ % % # / $ ) % # 6 / $ ) % # " % " ! " # 0 ) $ . ) (( % ' % %+ ) (( % " % ) ' % 3 & 0 / $ ) " %+ )) / % # % ) ) % # & % $ ) 0 0 $ :"
$ $ / % " % 5 & $ " ! " # % $ ! & # ' % " " # " % $ 7 " % " % %" " $ " % # ' % ' # $ ) &% $ $ 7 : ) " $ 5 ; %+ % # ) 2 )) ) & " % ) " ) % / $ ) % # ) !! % &" #
4 4 4 4 ) ' % 7 ! ) % % % ) $ % % ) !! * % % ) $ ) % % " % % 7 )) % % * % % % 5 4 2 % %
0 1 ) & 0 1 ) ) 0! 1 % %" 0 1 / % %( " # 0 1 $ " 0 % " % @AB1 / 9 $ * )) 0 1 $ " 0 CAD@5 % ) 0! 1 % 0 * 1 ) % ) 0! 1 / % " # % 0 CAD@1 0! 1 ;! %" %" % 0 1 ) & 0 % " % CADE1 . % % 3 " ! % 0! 1 $ 0 CAD@1 2 % # ) !"# $%"&&'%()
opinions émises dans les dissertations qui lui seront présentées, doivent
être considérées comme propres à leurs auteurs et qu’elle n’entend leur
donner aucune approbation ni improbation.
A Monsieur le Professeur Jean-Paul Marie : vous me faîtes l’honneur de présider ce Jury ; merci pour votre enseignement et votre bienveillance tout au long de mon internat, pendant lequel vous m’avez appris les secrets et la richesse de notre belle spécialité. Je vous remercie également pour votre confiance réitérée à plus d’une occasion ; j’espère en être digne. Recevez le témoignage de mon admiration et de ma gratitude.
A Monsieur le Docteur Florian Clatot : tu as accepté de diriger les tâtonnements scientifiques d’un « simple chirurgien » ; merci pour ta confiance, ta rigueur scientifique et tes nombreux conseils. Ce fut un plaisir de faire avec toi ce travail qui n’est j’espère qu’un commencement.
A Monsieur le Professeur Dominique Chevalier : vous me faîtes l’honneur de juger ce travail ; merci pour votre accueil et votre gentillesse lors de mon inter-CHU avec vous ; j’ai pu y apprécier, outre vos qualités humaines, votre rigueur clinique et chirurgicale (et les aphorismes de la chirurgie parotidienne). Merci également pour votre sens de l’humour dont j’espère ne pas avoir trop abusé, et vos nombreux conseils professionnels. Recevez le témoignage de mon admiration et de ma gratitude.
A Monsieur le Professeur Frédéric Di Fiore : merci pour l’honneur que vous me faîtes de juger cette thèse et pour votre accueil chaleureux lors de mon passage en oncologie médicale. Merci également de votre confiance et de votre soutien au sein l’IRON pour la réalisation de ce travail.
A Monsieur le Docteur Nicolas Bon-Mardion : bien plus que pour l’honneur que tu me fais en acceptant de juger ce travail, je te remercie pour ta présence, ta gentillesse et ton soutien tout au long de mon internat. Chacun de mes projets, même les plus farfelus, ont trouvé en toi une oreille attentive et des conseils avisés. Merci aussi pour ton aide et ta disponibilité dans les moments difficiles. C’est une grande joie pour moi de pouvoir travailler avec toi.
A Monsieur le Docteur Jean-Michel Picquenot : je vous remercie pour l’honneur que vous me faîtes de juger cette thèse ainsi que pour votre aide et votre accueil au sein du service d’anatomopathologie qui ont permis la réalisation de ce travail.
Le Dr Arnaud François, les docteurs Ducastelle, Tourré, de Kergat, de Mauroy, Boivin, Ziade, Vaquer, Landréat, et le personnel de la tumorothèque du CHU de Rouen, qui m’ont aidé et donné accès au prélèvement anatomopathologique à la base de cette étude ;
L’unité de recherche U918 du Pr Jardin et l’ensemble du service d’anatomopathologie du centre Henri Becquerel qui m’ont accueilli et aidé pour la partie technique de ce travail ; je tiens tout particulièrement à remercier Vinciane Marchand, Phillipe Ruminy, Marie Cornic, Marine Carbourg et la plus grande collection nationale de pyroséquenceurs pour leur aide précieuse et leur patience ;
Le service d’oncologie médicale du Centre Henri Becquerel qui a financé cette recherche.
Le service d’ORL et Chirugie Cervico-faciale du centre Henri Becquerel, qui m’accordé le temps et la liberté nécessaires à la réalisation de ce travail de recherche et accepté mes évasions régulières au laboratoire.
Cette thèse étant l’aboutissement de longues années d’étude, comment ne pas adresser ces remerciements à ceux qui, tout au long de mon internat, m’ont prodigué leurs enseignements et partagé leur savoir et leurs conseils, et plus particulièrement :
Au Dr Olivier Choussy : tu as confirmé en moi le goût de la chirurgie cervicale et carcinologique ; merci pour ton enseignement et ton soutien dans l’apprentissage de l’oncologie, ainsi que ton aide et ton appui lors des démarches en ce sens (Stage hors filière, Inter-CHU, DESC d’oncologie etc…).
Au Dr Angélique de Barros : merci de ta gentillesse, de ta disponibilité et de ton soutien infaillible (et les garden-party).
Au Dr Yanick Lerosey : je te remercie de ton enseignement et de ta confiance, notamment au bloc opératoire et pour avoir « décomplexifié » la chirurgie otologique.
Au Dr Isabelle Amstutz-Montadert : merci pour ces six mois d’audiophonologie, ton partage du savoir, ton appui et pour nos longues discussions.
Aux Dr Pierre Bouchetemble et Aurore Marcola : merci pour votre accompagnement en otologie et en pédiatrie.
Au Dr Philippe Lelion, qui a à la fois simplifié et compliqué ma vision du vertige ; merci de ton enthousiasme lors de mon mémoire.
Aux Dr Philippe Brami et Adrien Marronnier, (je n’aurais malheureusement jamais participé à la chorale…) et tous les médecins attachés qui m’ont ouvert les portes de leurs consultations spécialisées et fait découvrir des pans insoupçonnés de l’ORL.
Aux ORL du CHU de Lille et particulièrement les docteurs F. Mouawad, X. Pasquesoone et G. Mortuaire, un grand merci pour tout ce que vous m’avez appris pendant cet interCHU.
Aux docteurs C. Coudray et P.Y. Lienhardt qui ont guidé mes premiers pas en chirurgie ORL. Je garderai un souvenir ému de mon passage dans votre service.
Aux docteurs Dorel Manu, Faissal El Ouakif, Frédéric Decourselle, et Elena Ilies, merci pour ce dernier semestre, je suis ravi de rejoindre votre équipe.
A tous les médecins des services d’orthopédie et de chirurgie maxillo-faciale dans lesquels j’ai fait mes premières armes et plus particulièrement le Pr. O. Trost et le Dr B. Guichard.
A l’ensemble des médecins anesthésistes, radiologues, radiothérapeutes et anatomopathologistes du CHU de Rouen et du Centre Henri Becquerel.
A mes chefs de clinique vénérés de Rouen et d’ailleurs : Nathalie (toujours sur son 31), Elise (on va se marreeeer ! (et pour un diner des patrons extraordinaire !)), Quentin (on notera que le fumé n’est qu’une expression de la tourbe), Bénédicte (Mother of the dragons), Aurore (l’anatomiste), Laurent (à toute vitesse !) Camille (et la soupe de la mère Pétrau).
A toutes les équipes médicales et paramédicales, des blocs opératoires, consultations et différents services qui m’ont supporté, moi et mon humour simplet, tout au long de ces années. Merci de votre aide et de votre accompagnement.
A tous mes co-internes et amis, merci pour tous ces bons moments !! :
Les Rouennais ORL et CMF : Anaïs (enceinte jusqu’aux yeux), Anne (et son cachalot), Emilie (qui m’a ouvert la voie), Ivan (et le journal l’Equipe), Majeed (very good very good), Nadia (hâte d’être co-chef ça va être trop bien !), José (kung-fu panda), Mathilde (Globe-trotter), Marie (la Parisienne), Chacha (envoie nous des photos de New-York), Fred (et la bataille rat-trach), Pierre (et les pédiatres en détresse), Raphou, Clarinette et François et tous les internes de CMF, Michaël (le recruteur de Minor), Fanny (party organizer ; désolé pour les jeux de mots pourris, un jour j’arrêterai…. en fait non...), Paupau, Saïd, Delphine (et son parachute), Philippine, Kevin et Anne (on viendra vous voir !!!), Charles (et son insatiable curiosité), Mathieu (un jour on arrivera à trouver une date ou tu ne seras pas de garde !), Hélène, Mohammad (et 9gag) et tous les nouveaux et futurs internes à qui j’espère savoir transmettre le goût et la passion de l’ORL que mes aînés ont su m’inspirer.
Les oncologues : Marie, Marine (merci pour ton aide), Michaël (l’homme à tout faire, n’oublie pas ta perceuse pour réparer le Clinac), Anna (la musicienne), Imène, Lucie, Romain (vive les mariés), Simon (l’ORL refoulé), Emilie, Laureline, Carole, Edgard (je finirais Dark Soul !), Cyril (attention aux cyberattaques, les virus ça enrhume !), Mathilde, Gwen (sous amphet’), Anne (la rebelle).
Les Lillois : Hélène (Truc) un binôme hors-pair !! merci pour ces 6 mois où on s’est franchement bien amusé (je crois qu’il est de coutume de mentionner ton caractère… c’est fait), Mika et ces soirées qui ont du sens !! (même si on peut y perdre quelques neurones), Isabelle (et nos longues conversations), Tac (ou Monoï au choix ; un burritos ?), Anne-Sophie et Ichizan (le chef), Céline, Juma, Elodie, les membres de « The otologists » (on va conquir le monde !), tous les internes de Salengro, de Jeanne de Flandres et d’ailleurs.
Comment ne pas mentionner tous ceux qui ont été présents depuis le début et tout particulièrement : Mes grands-parents et parents : vous m’avez soutenu tout au long de ma vie et il n’est pas possible d’exprimer toute la gratitude que je vous porte. Merci de m’avoir accompagné, de m’avoir donné l’amour de la musique et du chant, de m’avoir donné le goût du travail bien fait, de m’avoir appris à avoir de grandes aspirations et de grands idéaux et se donner les moyens pour y parvenir, et pour votre exemple.
A mes oncles, tantes, cousins et cousines que je ne vois malheureusement pas assez et qui m’ont donné l’exemple d’une famille unie dans la diversité.
A ma belle-famille : les voyages dans le sud-ouest et en Espagne, les fêtes de Bayonne, les corridas, les bons moments passés ensemble pendant toutes ces vacances.
A Jean-François et Catherine, qui ont recueilli un exilé pendant 6 mois.
Et tous mes amis
Louis-Marie et Claire : merci pour cette grande amitié indéfectible !! (on arrivera à partir au ski un jour ! (avec une valise pleine de jeux évidemment))
Ocelot et Mapie : merci pour votre présence à nos côtés depuis tant d’années, malgré la distance vous êtes toujours avec nous !
Monsieur Coquin et Mariem : merci pour tous ces bons moments (et aussi les haricots surgelés, les inondations…), les jeux de société, les balades en forêts, les cours de géopolitique…
Adrien et Florence (bienvenue chez les ch’tis) ; PF et Théthé (le chocolat festif, les diners, les révisions) ; Bertrand (la moto, les hurons, le Bowmore et tellement plus encore) et Philippine que je ne connais pas encore ; Maxime et sa franchise à tout épreuve (ou est-ce une absence de filtre ?) et Clémence ; Gerardo mon marchand de sommeil attitré ; Joseph qui l’a été pendant longtemps (et pour nos grandes discussions philosophiques que j’ai toujours aimées (surtout quand on est pas d’accord ! Moi ?! un esprit de contradiction ?????)) ; Brigitte et Pierre le sudiste (et le coussin-chien par qui tout a commencé) merci pour votre exemple ; Father Donal, le père Brétéché, le père Jozan, le père Laurent ; et tous ceux avec qui on a passé tant de bons moments : Sophie, Jean, Jean-Eudes (Ooooh Danieeeella), Alexis, Marie, Constance, les rennais, la 1ère Orvault-Sautron et la 13ème Nantes, Minnie-Mouse, MV,
m&m’s et le pélé CDF ; mes co-externes de choc : Marie-Céline et Simon, Blandine, Maëlle, Simon et tous les autres
Et les Rouennais : Tiphaine et Hervé (c’est nous qu’on l’a la maison maintenant !!) ; Claire et Ludovic (une consultation pour un spectacle), Mathilde (j’espère que tu auras autant besoin de mes services que moi des tiens) et Ludovic, Loïc et Claire, Clémence et Antoine, Marcos et Pilar (merci pour votre confiance), Yves, Jean, Pierre, Théophile et Mathilde, Baudoin, Jean-Do, Fred, Yann et Denis, François-Xavier, Paul et Gwen, Maëlla et Olivier, Clémence et Nicolas, Marie Sophie et Edouard, Marine et Antoine, Laurence et Thibaut, Jérôme,
A tous les autres et ceux que j’aurais pu oublier, merci pour votre amitié.
Et surtout, surtout … à ma chère et tendre Aude, comment te remercier pour ton amour qui m’abreuve au quotidien. Tout cela n’aurait pu avoir lieu sans ton soutien sans faille, ta présence à mes côtés, ta compréhension… Je t’offre de tout cœur ce travail qui est bien plus le reflet de ta tendresse que de mes efforts. Et à nos crapautins Sibylle, Mayeul et Azilis…
Mais ce tain sous l’une et l’autre Peut-il être toléré ? Cher lecteur déjà tu juges Là de nos difficultés… »
Francis Ponge
Fable, Le parti pris des choses
« Que dites-vous ?... C’est inutile ?... Je le sais ! Mais on ne se bat pas dans l’espoir du succès ! Non ! non ! c’est bien plus beau lorsque c’est inutile ! »
Edmond Rostand
! " # $ % & '()* & '()* ) +,-*. '()*. )/,-&0*11+1&)-*. ! " # 2 3 4 2 3 4 5 ! $ %& ' ( ) *&'+ , -% $ $ .. / . & ' ( 0 67-*..)(/ 2 $ " 0(7)* 8* 9:/*.3 +,&,)(/ 8+ -;0*7,*+-+,&,)(/ +,&,)(/ ! 4 1,;-&,)(/ 8* 1& '()* " 4 &-)&,)(/ 2-. 43 4 PATIENTS ET METHODES
!< !< = = $ 5 = > 1& ! . 2 / ! " RESULTATS $ 4 # $ % & DISCUSSION ' " ( ) " > 3 . 2 4 # 3 . 4 5 3 . . & ' (4 * 5 ! 5 ! $ ! CONCLUSION BIBLIOGRAPHIE
1 EPIDEMIOLOGIE
1.1 Incidence et mortalité
Selon les données de l’INCa, les cancers des voies aéro-digestives supérieures (VADS) représentaient 14638 cas pour 4098 décès en 2012. Bien qu’ils touchent de manière prépondérante les hommes (74%), leur incidence augmente chez la femme (+1% par an depuis 2005), tandis qu’elle diminue chez l’homme (-5% par an depuis 2005)1. Concernant la mortalité, cette évolution inverse ne se retrouve pas, avec une diminution nette chez les hommes (-6.5% par an) et plus faible chez les femmes (-2.2% par an). Les modélisations réalisées à partir des recueils de données du réseau FRANCIM de 1990 à 2005 montraient déjà cette tendance qui se poursuit encore actuellement (Figure 1). Le pic de fréquence se situe entre 50 et 65 ans. Les carcinomes épidermoïdes (CE) représentent 90% de ces cancers2.
1.2 Facteurs de risque
Les principaux facteurs de risque des cancers des VADS sont le tabagisme, avec une relation dose-effet, le risque étant majeur à partir de 20 PA (RR 10 - 12) et la consommation éthylique, elle aussi avec une relation dose-effet (RR 2 à 4)4. Les deux ont un effet synergique
avec un risque majeur chez les grands fumeurs et grands buveurs (RR = 15)5.
L’évolution de la prévalence est principalement liée aux modifications de comportements vis-à-vis de ces deux facteurs. On note en effet, en France, une diminution de la consommation d’alcool constante au cours des ans (diminution de 50% depuis 1960)6. La consommation tabagique quotidienne reste quant à elle stable chez les hommes, mais en augmentation significative chez les femmes et plus particulièrement entre 45 et 64 ans, âge où l’incidence des cancers des VADS est la plus forte7. Cette recrudescence peut expliquer les changements épidémiologiques précédemment cités.
D’autres facteurs sont parfois évoqués : carences vitaminiques, noix de Bétel, immunosuppression, exposition au nickel ou à l’amiante, mais l’évolution majeure au niveau mondial reste l’émergence des cancers oropharyngés induits par le Human Papilloma Virus (HPV)8. Les sérotypes 16 et, dans une moindre mesure, 18, sont les principaux impliqués. Dans certains pays comme les Etats-Unis, la prévalence de carcinomes oropharyngés viro-induits serait proche de 80%8. En France, seule une étude multicentrique a été réalisée, retrouvant la
présence d’HPV dans 46,5% des cancers oropharyngés et 10% des carcinomes épidermoïdes buccaux, avec une prédominance chez la femme9. Cette prévalence varie entre régions et est
plus faible dans les zones à forte intoxication alcoolo-tabagique telles que la Normandie9.
Il semble que ces cancers induits par HPV soient une entité à part entière : profil clinique différent (patients plus jeunes, non alcoolo-tabagiques, meilleure survie globale et sans récidive, meilleure radio et chimio sensibilité), profil génétique différent (moins de mutations au sein des cellules tumorales, inhibition de p53 et de pRb par les protéines virales E6, E7), histoire clinique différente avec une atteinte ganglionnaire plus fréquente mais également un meilleur pronostic global10. Néanmoins, il n’existe pas de protocole de prise en charge particulière de ces
1.3 Evolution et pronostic
Les carcinomes épidermoïdes ORL sont des cancers de mauvais pronostic. Le taux de survie globale, tous stades confondus, est de l’ordre de 50% à 5 ans11. Cette survie est évidement
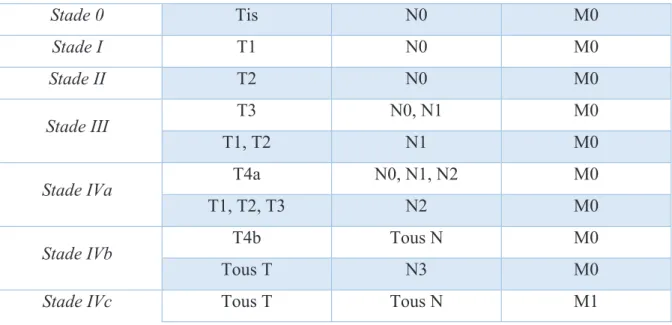
liée à l’extension de la maladie au moment de sa prise en charge initiale. Celle-ci est évaluée grâce au stade TNM et AJCC défini en fonction de l’évolution locale et de l’extension ganglionnaire et métastatique (Tableau 1). Ainsi, d’une survie de 90% à 5 ans pour des stades précoces (Stade I et II)12, celle-ci chute pour les stades avancés non métastatiques (Stade III à IVb) avec une survie à 5 ans variant de 20%12 à 40%13. Le pronostic global est d’autant plus sombre que nombre de patients se présentent d’emblée avec des tumeurs de stade avancé13.
TNM Stade 0 Tis N0 M0 Stade I T1 N0 M0 Stade II T2 N0 M0 Stade III T3 N0, N1 M0 T1, T2 N1 M0
Stade IVa T4a N0, N1, N2 M0
T1, T2, T3 N2 M0
Stade IVb T4b Tous N M0
Tous T N3 M0
Stade IVc Tous T Tous N M1
Tableau 1 : Stade AJCC d'après l'UICC 7ème édition, 2009
Malgré l’importance du stade initial, de récentes études montrent, pour les carcinomes oropharyngés, l’importance du statut HPV. Il serait pour cette localisation le facteur pronostic principal avant même le stade initial14. Les autres facteurs pronostics de haut risque de récidive ou de métastase à distance sont post-opératoires : marges envahies, plus de 2 ganglions métastatiques, rupture capsulaire d’une adénopathie envahie, emboles endo-lymphatiques ou vasculaires, engrainements péri-nerveux15.
Après traitement d’une tumeur localisée, les récidives locorégionales sont fréquentes (30 à 40% des cas) et leur traitement difficile en raison des séquelles des traitements initiaux12.
Le risque de métastase à distance est également proche de 30%, toutes localisations confondues, au cours de la prise en charge12, et de 10% lors du diagnostic initial16. Les sites
métastatiques préférentiels sont pulmonaires, osseux, et plus rarement hépatiques. Le pronostic de ces patients est sombre avec une survie nulle à 5 ans17.
2 NOTIONS DE CARCINOGENESE
L’évolution des connaissances sur la biologie et la génétique des cellules tumorales permet aujourd’hui de comprendre la genèse des cancers comme un ensemble de mutations, parfois sur terrain prédisposé, qui permet à la cellule d’acquérir des caractéristiques physiologiques propres. Hanahan, dans ces célèbres « hallmarks of cancer »18, en recense dix chacune pouvant être à la base de thérapeutiques spécifiques (Figure 3).
Ces caractéristiques sont :
Indépendance vis-à-vis des facteurs de croissance ; Echappement aux signaux inhibiteurs de croissance ; Résistance à la mort cellulaire programmée ;
Réplication illimitée avec perte de la senescence réplicative et immortalisation ; Promotion de la néo-angiogenèse ;
Perte de l’inhibition de contact et capacité à envahir les tissus adjacents et à distance (métastase) ;
Reprogrammation du métabolisme énergétique en vue de promouvoir la croissance et la division cellulaire ;
Echappement aux réponses immunitaires anti-tumorales ; Instabilité génomique ;
Sécrétion par la tumeur elle-même de facteurs péri-tumoraux inflammatoires créant un microenvironnement favorable à la progression locale et à distance.
L’ensemble de ces caractéristiques est issu de processus complexes qui varient selon le type de tumeur et le microenvironnement qui l’entoure.
2.1 Notion de clonalité
Les premiers travaux considérant le tissu tumoral comme provenant d’une anomalie cellulaire débutent avec l’avènement de l’anatomopathologie et notamment les travaux de Müller, puis Vorchow en 1858. Harrison en 1889, puis Fibiger dans ses travaux de 1913 qui lui ont valu le prix Nobel de Médecine en 1926 concluent qu’un facteur déterminé appliqué à un tissu cellulaire peut provoquer la différenciation de celui-ci en tumeur19.
C’est Slaughter qui finalement introduit la notion de « cancérisation en champ » dans une étude de 1953 dans laquelle il montre l’existence d’anomalies histologiques dans 100% des cas à distance de la lésion initiale et dans 10% la présence d’une seconde lésion indépendante de la première20. C’est le principe de la « muqueuse condamnée » où les mêmes contraintes,
comme l’exposition aux carcinogènes, appliquées à un même tissu, vont provoquer une maladie globale de l’ensemble de la muqueuse, ici des VADS. Les lésions précancéreuses comme les leucoplasies en sont le signe visible.
Une meilleure compréhension des anomalies génétiques liées au processus de carcinogénèse a permis que de nombreux travaux s’intéressent à l’application, sur le plan génomique, du principe de cancérisation en champ et à la présence d’anomalies génétiques à distance de la tumeur initiale. Ainsi Voravud et al.21 montraient la présence de polysomie au
niveau des chromosomes 7 et 17 dans le tissu péri-tumoral, anomalie retrouvée au sein de la tumeur, tandis que ces altérations disparaissaient à distance de la lésion. Ces anomalies peuvent se retrouver à plus de 5mm de la tumeur, marges normalement considérées comme satisfaisantes en cas d’exérèse22.
Plusieurs études sont actuellement en cours afin de mieux déterminer l’extension microscopique après exérèse des lésions et les marges de résection nécessaires. Trois voies de recherche sont principalement suivies : l’étude des anomalies moléculaires en immunohistochimie23, l’étude des anomalies de l’expression de certains gènes et l’étude de méthylation de gênes cibles24. Il n’existe actuellement pas d’adaptation thérapeutique ou de surveillance basée sur ces observations.
Comment alors expliquer la présence, au sein de ces « champs de cancérisation », de zones présentant des carcinomes invasifs et d’autres zones présentant de simples altérations génétiques ou épigénétiques n’aboutissant pas à une lésion cancéreuse ? De plus, en cas de seconde localisation, le profil génétique peut parfois différer25. Il faut donc considérer
l’apparition de ces lésions comme une cancérogénèse multi-étapes et parallèle.
2.2 Cancérogénèse multi-étapes
Au fil du temps se produit une accumulation des anomalies génétiques moléculaires favorisée par l’exposition aux carcinogènes avec, notamment, l’altération des gènes de régulation cellulaire de manière séquentielle : altération de p53 (rôle dans l’induction de l’apoptose), de p16 (rôle dans le contrôle la division cellulaire), de la cycline D1 (progression dans le cycle). A chaque étape, chacune des cellules peut ou non progresser dans les anomalies qu’elle présente et faire évoluer le tissu auquel elle appartient, de la dysplasie légère jusqu’au carcinome invasif avec la possibilité de régression à chaque étape. Cette évolution parallèle des cellules tumorales explique l’hétérogénéité des lésions au sein des champs de cancérisation. Leemans26 en 2011 a proposé un modèle séquentiel reliant ces différentes altérations à
Figure 3 : Modèle de cancérogénèse multi-étapes, d'après Leemans et al.
3 PRINCIPALES ANOMALIES BIOLOGIQUES DANS LES CE DES VADS
L’apport des nouvelles techniques de séquençage permettant des analyses parallèles à haut débit sur l’ensemble du génome humain, a permis d’identifier les anomalies génétiques principales dans les carcinomes épidermoïdes de VADS. Ainsi Agrawal et al.27 et Stransky et
al.28 ont rapporté, de manière simultanée, les principales altérations retrouvées dans ces cancers
: TP53, RAS, PIK3CA, CDKN2A, PTEN, NOTCH1 et plus rarement IRF6 et TP63. S’y associent également d’autres spécificités telle que l’hyperactivité de la voie PD1/PDL-1 et l’hyper-expression de l’Epidermal Growth Factor Receptor (EGFR). L’importance et la hiérarchisation de chacune de ces anomalies restent encore incertaines ; néanmoins quelques-unes apparaissent comme prépondérantes : p53, p16, EGFR, PD1/PDL-1 et leurs voies respectives.
3.1 Altération de la voie p53
Plus de la moitié des carcinomes épidermoïdes des VADS est associée à des altérations de TP53, gène suppresseur de tumeur présent sur le chromosome 17 et codant la protéine p5327–
détection d’une altération de l’ADN, l’activation de p53 par séparation du complexe MDM2-p53 va permettre de bloquer le cycle cellulaire et d’induire la réparation de l’ADN ; si les dommages sont trop importants et que la réparation n’est pas possible, p53 va induire l’apoptose cellulaire ou la sénéscence31.
Les principales voies déclenchées par p53 sont résumées dans la figure 4, les deux plus importantes étant p21 qui, par l’inhibition des protéines CDK1 et CDK2 (Cyclin-Dependant Kinase 1 et 2), va bloquer le cycle cellulaire, et BAX (Bcl-2- Associated protein X) qui, lorsqu’elle est exprimée en plus grande quantité que Bcl-2, va induire le relargage de cytochrome-c et ainsi l’apotpose32.
Figure 4 : Principales voies activées par p53 après séparation du complexe MDM2 -p53, d'après Hao et al.
On comprend aisément que les altérations de cette voie (hyperexpression de MDM2, altération de p53) jouent un rôle dans l’acquisition de l’immortalisation de la cellule et de la résistance à l’apoptose. Celles-ci ont été retrouvées de manière précoce dans l’apparition de cancers ORL et ce, dès le stade de dysplasie qui présente alors un risque accru de transformation en lésion invasive. Les mutations de TP53 au sein de la tumeur sont également un facteur de mauvais pronostic concernant la survie et la sensibilité aux traitements de radio et chimiothérapie33.
Dans les tumeurs HPV positives, l’altération de la voie de p53 ne passe pas par une mutation de TP53 mais par la synthèse d’une protéine E6 qui va induire une dégradation de p53 et donc une perte de fonction de celle-ci (Figure 5)26. Agrawal et al. notaient d’ailleurs que le
nombre de mutations des oncogènes par tumeur était bien moindre dans les tumeurs HPV+ par rapport aux tumeurs HPV– (4.8 vs 20,6), relation qui se confirmait selon l’exposition au tabac (9.5 vs 21.6)27.
3.2 CDKN2A, p16 et pRB
Tout comme pour TP53, l’implication du gène codant p16, CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A), survient très tôt au cours de l’oncogenèse et ses altérations sont retrouvées dans les champs de cancérisation indemnes de tumeur34.
p16 a un rôle crucial dans le contrôle du cycle cellulaire. Il s’agit d’un inhibiteur des Cycline-Dependant Kinase 4 et 6 (CDK4/CDK6) dont le rôle est de phosphoryler la protéine Ribosomale (pRb), cette phosphorylation provoque la libération de E2F avec lequel pRb réalise un complexe inactif. Une fois libérée, E2F va permettre le passage de la cellule de la phase G1 à la phase S et favoriser la division cellulaire35. La diminution de l’expression de p16 au cours du processus de carcinogénèse va être responsable d’une levée de cette inhibition et favoriser la prolifération cellulaire. Des mutations de CDKN2A ont été retrouvées dans 9 à 18% des tumeurs mais son expression est abaissée la plupart du temps, indépendamment de lésions génétiques27,36.
A l’instar des altérations de la voie p53, il est important de différencier ici les tumeurs HPV négatives et HPV positives pour lesquelles l’infection virale va aboutir à une libération de E2F par un biais différent. En effet, l’intégration du génome viral au sein de la cellule va induire la synthèse d’une protéine E7 qui va interagir avec le complexe pRb-E2F et être responsable de la libération permanente de E2F et donc le passage en phase S (Figure 5).
Par un mécanisme de rétrocontrôle, on observera une surexpression de p16, marqueur utilisé en immunohistochimie courante pour différencier les tumeurs HPV+ (tumeur p16+) et HPV- (tumeur p16-). Ce marqueur, bien que sensible, n’est que partiellement spécifique puisque 15 à 20% des tumeurs p16+ sont en réalité HPV– lors de recherches de génome viral par PCR ou par hybridation in situ37–39. De plus, ces tumeurs sur-exprimant p16 mais non liées
p16+/HPV+14,37. Il est donc nécessaire pour diagnostiquer ces tumeurs HPV + d’associer une
seconde technique à la détection de p16 en immunohistochimie, que ce soit la recherche d’ADN ou d’ARN spécifique à HPV par PCR ou hybridation in situ39.
Figure 5 : Altération de p53 et de pRB par action des protéines E6 et E7 dans les carcinomes épidermoïdes HPV+ ou par altération de génomique dans les carcinomes épidermoïdes HPV-. D’après Leemans et al.
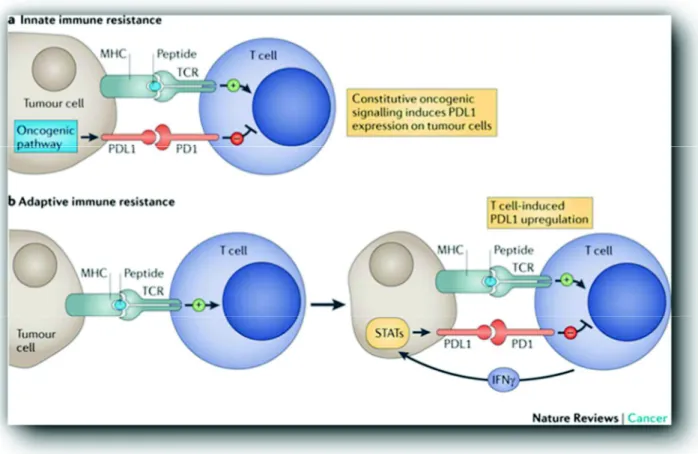
3.3 PD1 / PDL1
L’une des caractéristiques des tissus tumoraux est l’échappement à la réponse immunitaire anti-tumorale18. Il existe de manière physiologique des points de blocage à la
réponse immunitaire médiée par les lymphocytes T et B afin d’éviter les phénomènes d’auto-immunités : les « immune checkpoint ». Certaines cellules tumorales ont la capacité de détourner ces points de contrôle et d’échapper ainsi à la réponse immunitaire cellulaire. L’une de ces voies, actuellement au cœur de recherches dans la thérapeutique de nombreux cancers, est l’interaction PD-1 / PDL-1.
Les lymphocytes T CD8+, CD4+, Natural Killer et B expriment sur leur membrane un récepteur PD1 dont le ligand PDL-1 est, lui, exprimé par la cellule tumorale. L’interaction récepteur-ligand provoque un phénomène de tolérance et bloque l’activation du lymphocyte T
normalement déclenché par l’interaction TCR (T Cell Receptor) - CMH (Complexe Majeur d’Histocompatibilité) (Figure 6)40.
Or PDL-1 est surexprimé dans les carcinomes épidermoïdes dans 45 à 70% des cas41.
Cette surexpression semble notamment liée à l’action d’IFN gamma secrété par les cellules NK ou les lymphocytes CD8+ et le microenvironnement péri-tumoral42 ce qui peut expliquer qu’elle soit plus importante au sein de tumeurs HPV+, l’interféron gamma étant associé à la réponse immunitaire anti-virale43. De plus la production d’IFN gamma par le lymphocyte T lui-même en cas d’activation va induire l’expression membranaire de PDL-1 et créer un échappement immunitaire adaptatif (Figure 6)44.
Figure 6 : Mécanisme d’échappement immunitaire inné et acquis par l’interaction PD1-PDL1. D’après Pardoll
En outre, sur une cohorte de 182 patients présentant un carcinome épidermoïde des VADS, Feldman et al. retrouvaient, en plus de la surexpression PDL-1, une hyperexpression de PD1 sur les membranes des lymphocytes infiltrant la tumeur dans 68% des cas renforçant cet échapement29.
Le développement d’anticorps anti-PD1 et anti PDL-1 vise à supprimer le détournement tumoral de ces « immune checkpoint » en bloquant l’interaction récepteur / ligand afin de restaurer la sensibilité de nombreux cancers aux défenses immunitaires naturelles.
Plusieurs essais cliniques ont été présentés lors du congrès de l’ASCO 2016. Parmi ceux-ci on citera les études CheckMate-141 et KEYNOTE-012 qui ont permis de valider l’intérêt en pratique courante de l’inhibition de la voie PD1-PDL1. Ces inhibiteurs, tel que le Nivolumab ou le Pembrozolumab, permettent un bénéfice en survie globale en 2ème ligne de traitement dans les cancers ORL métastatiques. On peut souligner la présence de longs répondeurs chez les patients qui tirent un bénéfice de ce traitement, dans une population avec une médiane de survie minime avec les chimiothérapies standards de 2ème ligne45,46. Des biomarqueurs de sensibilité au traitement sont actuellement recherchés.
3.4 Epidermal Growth Factor Receptor : EGFR
La voie de l’EGFR est l’une des plus importantes voies impliquées dans la cancérogénèse des carcinomes épidermoïdes des VADS. Son hyperactivation, présente dans plus de 90% des cancers ORL, est le plus souvent liée à la surexpression de l’EGFR sur les membranes cellulaires sans qu’il y ait de mutation retrouvée. Dans leur récente étude, Feldman et al.29 retrouvaient une surexpression du récepteur en immunohistochimie dans 90% des tumeurs testées tandis que seul 1% était muté. La connaissance de cette voie est nécessaire pour appréhender les principes des thérapeutiques actuelles qui visent à la bloquer.
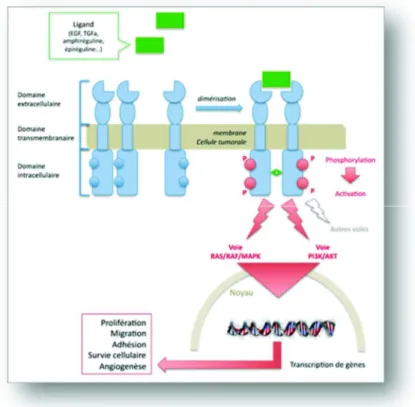
3.4.1 Famille des HER
L’Epidermal Growth Factor Recepteur (EGFR) est un récepteur à tyrosine-kinase appartenant à la famille des récepteurs HER, comprenant 4 récepteurs indépendants : l’EGFR ou HER1, également appelé ErbB 1, HER2 (ErbB2), HER 3, HER 4. Chacun de ces récepteurs est composé d’un domaine extracellulaire présentant le site de réception complémentaire au ligand et un domaine intra-cytoplasmique à activité tyrosine-kinase (Figure 7)47.
Ces récepteurs ErbB sont activés par des facteurs de croissance appartenant à la famille des EGF (Epidermal Growth Factor), qui sont produits, soit par sécrétions autocrines par les
Growth Factor alpha), Epiréguline (EPR), Amphiréguline (AR), Béta-Celluline (BTC), Heparin-Binding EGF-like Growth Factor (HB-EGF)47.
Figure 7: Le récepteur EGFR et son activation, d'après Laurenty et al.#
3.4.2 Activation du récepteur
De manière inactive, l’EGFR forme un monomère. La fixation du ligand va changer la conformation tridimensionnelle de la partie extracellulaire en le faisant passer d’une forme repliée à une forme ouverte. Ce changement de conformation va permettre une dimérisation des récepteurs, soit avec un récepteur EGF activé identique (homo-dimérisation), soit avec un autre récepteur de la famille HER (Hétéro-dimérisation)50. Cette dimérisation met en contact les domaines tyrosines-kinases intra-cytoplasmiques des 2 monomères et déclenche leur phosphorylation de manière réciproque (Figure 7). Des protéines cytoplasmiques, présentant des domaines SH2 (Src Homology domain 2) complémentaires des tyrosines ainsi phosphorylées, peuvent alors s’ancrer sur la partie C-terminale du récepteur. C’est le cas notamment du complexe Grb2/hSos, capable d’activer les protéines RAS et Pi3K et leurs voies respectives51.
3.4.3 Voie de signalisation intracellulaire
Les voies RAS et PI3K, toutes deux intimement connectées52, sont les deux voies
principales activées par le récepteur EGFR. D’autres voies plus accessoires peuvent également être déclenchées tel que JAK/STAT, Phospholipase C (PLC ) ou Src/FAK (Focal adhesion kinase).
3.4.3.1 La voie RAS
La famille RAS, famille de protéines G, inclut 3 protéines isoformes : HRAS, KRAS, NRAS. Ce sont des GTPases dont l’activation par le biais de récepteurs tyrosine-kinases (principalement l’EGFR), va déclencher des signaux de prolifération, de migration et de survie (Figure 8)53.
Figure 8 : La voie de signalisation RAS/MAPK d’après Lièvre et al.
Dans sa forme inactivée Ras est liée à un Guanine Di-Phosphate (GDP). Sa phosphorylation par l’activité tyrosine kinase de l’EGFR permet l’échange de GDP en GTP (Guanine Tri-Phosphate), lui donnant sa forme activée. Les effecteurs de RAS, notamment RAF, interagissent avec sa forme activée et engagent une cascade de phosphorylations intra cytoplasmiques par la voie RAS -> RAF -> MEK1 et 2 -> ERK 1 et 254. La phosphorylation de
c-JUN ou JUN, eux-même responsables de nombreux signaux de croissance (dont l’expression de la cycline D1 et de CDK6), de migration et de division cellulaire55.
Une fois la cascade engagée, une protéine GAP (GTPase Activating Proteins) vient catalyser le GTP en GDP, restaurant RAS dans sa forme inactive56.
Dans les carcinomes épidermoïdes, les mutations de RAS intéresseraient principalement HRAS et représenteraient seulement 5% des patients27–29. Il s’agit d’une grande différence avec les cancers du côlon où les mutations de KRAS (codon 12 et 13) sont présentes dans 30 à 40% des tumeurs57. Elles provoquent l’activation permanente de RAS et donc de sa cascade intracellulaire indépendamment de l’activation des récepteurs tyrosine-kinases. Cette activation permanente est l’une des causes de résistance aux thérapeutiques ciblées contre les récepteurs à tyrosine-kinase, rendant la recherche de la mutation indispensable dans le cancer du côlon avant d’initier un traitement anti-EGFR58. Cette activation va également être responsable d’une suractivation de Pi3K.
3.4.3.2 La voie Pi3K
Pi3K est une kinase qui, tout comme RAS, active une cascade de phosphorylations et d’activations de molécules intracellulaires et notamment la voie Pi3K -> AKT -> mTOR, responsable de signaux de survie cellulaire, d’angiogenèse, de prolifération, de croissance et de migration. Elle est activée par RAS dans une certaine mesure, mais surtout par les récepteurs tyrosine-kinases et une fois encore principalement par l’EGFR. Le principal modulateur de son activité est PTEN, qui exerce sur elle une action inhibitrice31. L’ensemble de cette voie est résumé dans la figure 9.
La voie Pi3K est fréquemment suractivée dans les carcinomes épidermoïdes des VADS et corrélée à un mauvais pronostic59. Les mutations activatrices de PIK3CA, gène qui code Pi3K peuvent être retrouvées dans 10 à 20% des tumeurs ORL29,60, et celles inhibitrices de PTEN dans environ 10%27,29.
Figure 9 : Voie de signalisation PI3K : PI3K est activé soit directement par une récepteur Tyrosine Kinase (RTK) soit par la protéine RAS. PI3K va transformer PIP2, un lipide membranaire, en PIP3 auquel va se lier AKT, une protéine kinase. AKT, après phosphorylation par PDK1, va activer plusieurs facteurs dont mTOR, responsable de signaux de prolifération, de mitose, de survie et d’angiogenèse. PTEN est le principal inhibiteur de PI3K par déphosphorylation de PIP3 en PIP2. d’après Faivre et al61
3.4.3.3 Autres voies intracellulaires
D’autres voies plus accessoires peuvent également être activées par l’EGFR :
Voie JAK/STAT capable d’activer la transcription de gènes impliqués dans la résistance à l’apoptose (Bcl-XL, Mcl1, AKT, survivine), la migration (MMP3, MMP9), la prolifération cellulaire (c-myc, c-fos, cycline D1, cdc25A, télomérase) et l’angiogenèse (VEGF, EPAS1)62 ;
La voie Phospholipase C (PLC ) qui a un rôle important dans la mobilité cellulaire et donc l’invasion tumorale63. Par ailleurs elle peut activer une protéine DAG qui elle-même peut activer RAS et sa voie propre, PKC (Protéine Kinase C), et IP3 (Inositol Triphosphate) aboutissant à des signaux de survie et de prolifération cellulaire64 ;
La voie Src/FAK (Focal adhesion kinase) qui diminue l’adhésion cellulaire et favorise la migration et l’invasion tumorale65.
3.4.4 Inactivation de l’EGFR
La régulation négative des récepteurs EGFR se fait par son internalisation dans des vésicules de Clathrine. Le signal d’endocytose des récepteurs est principalement le fait de l’ubiquitination des récepteurs bien que celle-ci ne soit pas systématique. Cette ubiquitination est réalisée par une E2 Ligase activée par Cbl (Casitas B-lineage Lymphoma) qui peut se fixer soit directement sur les résidus tyrosines du récepteur, soit via le domaine SH3 de la protéine Grb2. Ainsi Grb2 possède à la fois un rôle pro-oncogène, via l’activation de la voie RAS, et suppresseur de tumeur via l’endocytose des récepteurs. Une fois les vésicules de clathrine internalisées, celles-ci sont adressées, soit aux lysosomes où les protéines sont digérées, soit recyclées vers la membrane51.
3.5 Micro-environnement tumoral
L’ensemble de ces anomalies ne doit pas être conçu uniquement à l’échelle cellulaire et il est impératif d’intégrer ce modèle au niveau tissulaire. En effet, le rôle du micro-environnement tumoral apparaît crucial tant dans les phénomènes de cancérogénèse que dans la progression et l’évolution métastatique des cancers et les différents éléments qui composent le tissu péri-tumoral participent tous à ces phénomènes.
Nous avons précédemment vu les interactions avec les cellules immunitaires via l’interaction PD1-PDL1, mais plus encore il semble que les macrophages activés au sein des tumeurs sécrètent des facteurs anti-inflammatoires et pro-oncogènes66. De même, les
fibroblastes infiltrant le tissu tumoral possèdent des caractéristiques propres. Ces FAC (Fibroblastes Associés aux Cancers), qui pourraient se différencier en myofibroblastes et participer ainsi à l’invasion tumorale, sécrètent de nombreux facteurs de croissances et pro-angiogéniques. Ils participent également au modelage de la matrice extracellulaire (MEC)66.
Cette MEC péri tumorale est en perpétuel renouvellement notamment sous l’action de métalloprotéases de la matrice (MMP) sécrétées par les cellules qui s’y trouvent. L’hypoxie, particulièrement importante dans les carcinomes épidermoïdes ORL, est un élément-clé de l’activation des MMPs qui vont eux-mêmes stimuler l’activité de facteurs de croissances, de facteurs pro-angiogéniques et de chémokines66.
Il se crée de cette manière une homéostasie particulière, spécifique au tissu tumoral et indépendant du tissu sain adjacent, où la cellule tumorale dispose d’un milieu favorable à sa croissance et à l’acquisition de potentiel invasif et métastatique.
4 PRINCIPES THERAPEUTHIQUES
Longtemps considérée comme le seul traitement curatif, la chirurgie n’est actuellement que l’une des options thérapeutiques avec la radiothérapie seule ou en association à un traitement systémique. A ce jour, le choix du traitement dépend avant tout de données cliniques : stade initial et localisation anatomique.
4.1 Stades précoces (I et II)
La plupart des études thérapeutiques sont des études de faible niveau de preuve et il est difficile de conclure à la supériorité d’un traitement par rapport à l’autre. Il semble que pour les stades précoces (T1, T2 et N0), la radiothérapie plus ou moins potentialisée par la chimiothérapie ait des résultats similaires à la chirurgie pour certaines localisations (oropharynx, hypopharynx)67,68. Le choix du traitement dépendra donc des séquelles
potentielles des traitements envisagés et du choix du patient.
En cas de chirurgie première, la réalisation d’une radiothérapie post-opératoire sera guidée par la présence de facteurs de gravité constatés sur la pièce opératoire : la taille de la tumeur, les marges de résection atteintes, plus de 2 ganglions envahis, la présence d’emboles endo-lymphatiques ou vasculaires ou d’engaînements périnerveux, adénopathie en rupture capsulaire69,70. L’indication de la potentialisation de la radiothérapie par une chimiothérapie est formelle en cas de marges envahies ou de rupture capsulaire chez les patients de moins de 70 ans. Le standard dans ce cas est l’utilisation de Cisplatine 100mg/m² à J1, J22 et J43 de la radiothérapie.
4.2 Stades avancés (III et IV)
précédemment cités. Néanmoins, les avancés récentes sur la connaissance des tumeurs oropharyngées HPV+ et leurs radio et chimio sensibilités devraient changer prochainement ce paradigme en permettant de proposer une radio-chimiothérapie seule, y compris pour des stades localement avancés71. Actuellement le traitement par radio-chimiothérapie est basé, comme
dans la situation post-opératoire, sur l’association radiothérapie-Cisplatine 100 mg/m² J1-J22-J43. A noter que dans la situation de tumeur non opérée, le Cetuximab constitue une alternative au Cisplatine, en particulier en cas d’insuffisance rénale ou de polyneuropathie72.
On notera également le cas particulier des lésions laryngées ou hypopharyngées classées T3 et pouvant bénéficier au cas par cas d’un protocole de préservation laryngée associant une chimiothérapie d’induction par Taxotere - Cisplatine - 5FU (TPF) permettant de sélectionner les patients bons répondeurs carcinologiques et fonctionnels, suivie, selon la qualité de cette réponse initiale, d’une radio-chimiothérapie ou d’une chirurgie radicale11,73,74.
Malgré ces traitements, le risque de poursuite évolutive ou de métastase à distance reste élevé.
4.3 Patients métastatiques
Malgré l’évolution des thérapeutiques, le pronostic des patients porteurs de métastases reste faible, faisant considérer ces patients comme relevant d’emblée d’un traitement palliatif. Celui-ci associe soins de support (prise en charge antalgique, nutritionnelle, psychologique, sociale etc.) déjà partie intégrante du traitement curatif, et une chimiothérapie à visée palliative ou « retardatrice ». Le standard de première ligne métastatique était, à partir des années 1990, l’association de Sel de platine (Cisplatine ou à défaut Carboplatine si l’état général ou les comorbidités l’indiquent) à du 5’FluoroUracile (5FU) avec une médiane de survie globale d’environ 6 mois.75 Cette association permettait, au prix de plus de complications, un meilleur
contrôle local sans pour autant obtenir un grand bénéfice en termes de survie globale. Les associations Sel de Platine - Taxanes ont également montré leur efficacité76.
Le développement des thérapies ciblées, et notamment du Cetuximab en ORL, a permis d’accroitre cette survie avec un taux de réponse d’environ 30%, et une médiane de survie globale de 10 mois17. L’association Platine - 5FU - Cetuximab est, depuis 2009, devenue le standard de chimiothérapie palliative de première ligne. Les patients présentant des carcinomes épidermoïdes récidivants et ne relevant pas d’un traitement curatif, bénéficient également de ce
protocole. Les principales difficultés résident dans les comorbidités, l’état général et l’âge de ces patients qui rendent souvent difficile la mise en place et le suivi d’un traitement optimal, et grèvent d’eux-mêmes le pronostic. D’autres traitements systémiques peuvent alors être proposés tels que l’association Paclitaxel - Cetuximab, ou encore le Méthotrexate en monothérapie77. Les soins de support seuls demeurent parfois l’unique prise en charge possible ou raisonnable chez des patients altérés.
Les anticorps anti-PD1 et antiPDL1 semblent, comme évoqué précédemment, une voie prometteuse. Elle pourrait devenir prochainement le standard de chimiothérapie de 2ème ligne si les prochaines études en cours confirment son efficacité.
Une attitude plus agressive, visant à éradiquer les foyers tumoraux secondaires, a par ailleurs été décrite chez certains patients oligométastatiques bien sélectionnés et contrôlés au niveau local. Une méta-analyse portant 403 patients de 13 études rétrospectives publiées de 1986 à 2011 montrait ainsi une probabilité de survie à 5 ans de 29,1% après métastasectomie78. Cette étude présente néanmoins de nombreux biais (hétérogénéité des traitements, hétérogénéité des patients, différence entre métastase unique et primitive pulmonaire parfois non évidente, différence de suivi…), et il existe peu de données actuellement sur les techniques de métastasectomie non-chirurgicales telles que la radiothérapie stéréotaxique en ORL. L’étude OMET (GORTEC 2014-04) est actuellement en cours afin d’évaluer la place de la radiothérapie stéréotaxique sur les localisations secondaires, chez les patients oligométastatiques.
L’adjonction des thérapies ciblées aux chimiothérapies cytotoxiques classiques en ORL reste actuellement le principal élément ayant permis d’améliorer le pronostic de ces patients.
5 APPORTS DES THERAPIES CIBLEES
5.1 Généralités
Les thérapies ciblées, à l’inverse des chimiothérapies cytotoxiques classiques, ont pour but d’agir de manière spécifique sur les cellules tumorales en ciblant un marqueur distinctif de la cellule tumorale. La surexpression de l’EGFR dans la plupart des cancers et en particulier dans les cancers du côlon et des VADS en a fait l’une des voies les plus précocement étudiées. Outre les thérapeutiques dirigées vers les protéines situées en aval du récepteur (par exemple l’Evérolimus, une molécule anti-mTor79), deux groupes de molécules ont été développés contre l’EGFR : les inhibiteurs de tyrosine kinase et les anticorps monoclonaux anti-EGFR.
Les inhibiteurs de tyrosine-kinase ne sont pas spécifiques de l’EGFR. L’inhibition réalisée passe par leur action compétitive vis-à-vis de l’ATP. Les deux principaux étudiés en cancérologie ORL sont l’Erlotinib et le Lapatinib qui peinent tous deux à démontrer leur efficacité79–81.
Les anticorps monoclonaux anti-EGFR se fixent sur la partie extracellulaire de l’EGFR et bloquent son activation par le ligand. Le seul anti-EGFR ayant démontré une certaine efficacité dans les carcinomes épidermoïdes des VADS et ayant l’AMM en France reste le Cetuximab.
5.2 Cetuximab
Le Cetuximab est un anticorps monoclonal dirigé vers la partie extracellulaire du récepteur EGFR. Il s’agit d’une immunoglobuline chimérique associant les parties constantes d’une immunoglobuline humaine à des fragments variables murins82. Après fixation de
l’immunoglobuline par son fragment Fab, trois modes d’action entrent en jeu : compétition avec les ligands naturels et blocage du récepteur dans sa forme repliée (inactive) ; augmentation de l’internalisation du récepteur ; promotion de l’ADCC par activation des lymphocytes T CD8 + et Natural Killer grâce à la fixation des cellules immunitaires par leurs récepteurs Fc rIIa ou Fc rIIIa sur les fragments Fc de l’immunoglobuline (Figure 10)82.
Figure 10 : Mode d’action du Cetuximab :
1 - compétition avec les ligands naturel et blocage du récepteur dans sa forme repliée ;
2 - promotion de l’ADCC et de l’immunité cellulaire ;
3 - augmentation de l’internalisation du récepteur.
Si les deux premiers mécanismes aboutissent au blocage de la voie intracellulaire pro-tumorale induite par l’EGFR, la promotion de l’ADCC est probablement quant à elle l’un des facteurs importants expliquant la différence d’efficacité entre les différents anticorps anti-EGFR. Dans une étude in vitro sur cellules tumorales de cancer ORL, Trivedi et al.83 ont
comparé les mécanismes d’action du Panitumumab (un autre anti-EGFR) et du Cetuximab. Ils concluaient que, bien que les deux aient la capacité de bloquer les voies intracellulaires de l’EGFR, le Cetuximab induisait de manière bien plus importante l’immunité cellulaire anti-tumorale. Cela est probablement lié à la composante murine de l’immunoglobuline.
Actuellement le Cetuximab est utilisé en pratique courante dans deux indications : en association avec la radiothérapie à visée curative sur tumeur non-opérée ou en association avec d’autres chimiothérapies à visée palliative.
5.2.1 Association Radiothérapie-Cetuximab (RTCx)
L’étude princeps ayant permis l’obtention de l’AMM en 2006 pour le Cetuximab (Cx) en association à la radiothérapie dans les carcinomes épidermoïdes des VADS localement avancés a été réalisée par Bonner et al.84 Elle comparait l’efficacité de l’association radiothérapie-Cx vs radiothérapie seule retrouvant un bénéfice significatif pour le bras RT-Cx, que ce soit en termes de contrôle locorégional, de survie sans récidive ou de survie globale. Ces bénéfices n’étaient pas retrouvés chez les patients de plus de 65 ans. Les résultats actualisés en 2010 montrent un bénéfice de survie globale à 5 ans passant de 36,4% pour le bras RT seule, à 45,6% pour le bras RT-Cx (HR 0,7 IC95[0,6 – 0,95] ; p=0·02) pour les patients ayant bénéficié de l’association radiothérapie-anticorps monoclonale72.
Malgré ces résultats, il faut noter que cette étude ne comparait pas l’association RT-Cx au gold standard qu’est l’association Radiothérapie - Cisplatine (RT-Cis). Il n’existe pas à ce jour d’étude prospective de niveau de preuve suffisante pour conclure à l’équivalence de ces deux traitements. Bien qu’elle comporte de nombreux biais (comparaison d’étude prospective vs rétrospective, hétérogénéité des patients entre les différents bras, différents critères d’inclusion), une méta-analyse récente retrouvait de manière significative une meilleure survie globale (p=0.02) et sans récidive (p=0.02) à 2 ans pour la potentialisation de la radiothérapie par Cisplatine plutôt que par Cetuximab85.
5.2.2 Association à une poly-chimiothérapie
La seconde utilisation du Cetuximab, la plus fréquente, est son association à une polychimiothérapie cytotoxique. Avant l’introduction du Cetuximab, le standard de traitement de première ligne à visée palliative était l’association sel de platine - 5 Fluoro-uracile (PFU). Avec l’apparition des anticorps anti monoclonaux et leur efficacité en association avec la radiothérapie, plusieurs études se sont intéressées à l’ajout du Cetuximab dans les protocoles standards de chimiothérapie (Platine - 5FU). La plus concluante, étude comparative randomisée réalisée par Vermorken et al.17 (étude EXTREME), comparait le PFU +/- Cx. Les auteurs retrouvaient un bénéfice significatif de survie globale (médiane de survie de 7,4 mois pour le bras PFU vs 10,1 pour le bras PFU - Cx (HR 0,80 ; IC95% (0.64 - 0.99) ; p=0,04). Ce bénéfice était également retrouvé en termes de contrôle locorégional et de survie sans progression (Figure 11)17.
Figure 11 : Réponse au traitement et survie selon les groupes Cx + ou C- dans l'étude EXTREME, d'après Vermorken et al./
L’association Sel de platine – 5 Fluoro-Uracile – Cetuximab est, depuis lors, devenue le standard de première intention de chimiothérapie à visée palliative dans les carcinomes épidermoïdes des VADS, avec obtention de l’AMM en France pour cette indication en 2009.
A noter que ce protocole propose, au-delà des 6 cures de PFUCx, une poursuite du Cx en entretien en monothérapie, jusqu’à progression tumorale.
Chez certains patients extrêmement fragiles, le Cetuximab peut être utilisé en monothérapie lorsque l’ensemble des autres traitements semble déraisonnables, mais le bénéfice est faible.
Plus récemment, l’étude de phase II du GORTEC 2008-03, a démontré l’efficacité d’une chimiothérapie associant Docetaxel - Cisplatine - Cetuximab (TPEx) dans cette même indication86. Ces résultats encourageants doivent encore être comparés à ceux du PFU-Cx. C’est ce que vise l’étude de phase IIb du GORTEC TPExtreme (GORTEC 2014-01) devant inclure 416 patients et comparant, de manière randomisée, les protocoles TPEx et Extreme (Figure 12).
Figure 12 : Protocole de l'étude TPExtreme (GORTEC 2014-01)
Il faut noter que l’ensemble de ces études a été réalisé sur des patients sélectionnés (OMS Performance Status 1, Age < 70 ans, sans comorbidité significative) et bien différents de la réalité clinique. A notre connaissance, il n’existe pas à l’heure actuelle d’étude comparative sur patients non-sélectionnés, aussi serait-il intéressant de confirmer ces résultats dans une large cohorte de cette population.
5.2.3 Effets secondaires
Bien que relativement bien toléré, le Cetuximab ajoute des effets secondaires propres à ceux des chimiothérapies. Le risque principal lié à l’utilisation du Cetuximab est le choc anaphylactique allergique qui peut survenir dès le début de l’induction. Bien que rares (1 à 5% de réactions de grade 3-4), les réactions anaphylactiques sévères peuvent engager le pronostic vital (0.1% des patients)87,88.
Beaucoup plus fréquente mais généralement moins sévère, la toxicité dermatologique constitue le principal effet secondaire observé sous Cetuximab. Elle surviendrait chez plus de 80% des patients dont 20 à 30% de toxicité de grade 3-4. Elle se caractérise par un érythème associé à un rash acnéiforme du tronc et du visage, une xérose cutanée et une atteinte unguéale à type de fissure ou de péri-onyxis. Cette atteinte s’explique par la présence de nombreux récepteur EGFR au sein des cellules de la couche basale de l’épiderme87. L’association de Doxycicline, d’émollients et de corticoïdes locaux permet le plus souvent de contrôler ces réactions. L’apparition de ces lésions serait un facteur prédictif de bonne réponse au traitement. En effet, dans leur étude comparant Cisplatine - Cetuximab vs Cisplatine – Placebo, Herbst et al.89 retrouvaient une médiane de survie globale de 2, 2 mois pour les patients ne présentant pas de réaction cutanée, pour une médiane de 7,1 mois pour les patients ayant une réaction de grade 2 ou 3 (Figure 13).
Les hypomagnésémies par inhibition de la réabsorption de magnésium au niveau de l’anse de Henlé surviennent dans environ 10% des cas. Elles sont souvent rapidement résolutives mais peuvent nécessiter une supplémentation au long cours87. D’autres effets
secondaires plus rares sont également cités : diarrhée, déshydratation, atteinte oculaire (kératite, conjonctivite), cytolyse hépatique.
Dans l’étude de Vermorken et al.17, les patients traités par Cetuximab présentaient plus de réaction cutanée (p<0.001), d’anorexie (p=0.05), de sepsis (p=0.02) et d’hypomagnésémie (p=0.05) que sous PFU. Néanmoins il n’existait pas plus d’arrêt de traitement (20%) dû aux effets secondaires dans le groupe Cetuximab par rapport au groupe chimiothérapie seul.
Il importe également de considérer l’augmentation conséquente des coûts de santé qu’impliquent ces traitements pour seulement une partie de bons répondeurs90.
Ces éléments imposent de trouver des facteurs pronostics de réponse à ces traitements afin de sélectionner les patients pouvant en tirer réellement un bénéfice et ceux pour lesquels l’apport du Cetuximab n’implique qu’une augmentation des effets secondaires et des risques de complications et de coût de santé.
5.2.4 Facteurs prédictifs de réponse ou de résistance au Cetuximab
Plusieurs facteurs ont déjà été testés en ORL, principalement orientés sur l’expression de l’EGFR et de ses voies, sans pour autant avoir montré à l’heure actuelle de significativité suffisante pour être intégrés à la pratique courante.
5.2.4.1 Expression EGFR (IHC / copie de gènes)
L’une des premières hypothèses était que l’expression de l’EGFR pouvait être corrélée à la réponse au Cetuximab. Celle-ci a donc été évaluée sur les tumeurs des patients de l’étude EXTREME. Il existait une tendance à une meilleure PFS et OS chez les patients ayant reçu du Cetuximab et présentant des scores élevés d’expression de l’EGFR en immunohistochimie mais sans résultat significatif et avec un bénéfice médiocre. Les auteurs concluaient que l’expression de l’EGFR ne pouvait être utilisée comme facteur prédictif de réponse au Cetuximab91.
anomalies vis-à-vis de l’EGFR pourraient donc être plus sensibles aux anti-EGFR. Licitra et al.92 ont ainsi étudié, sur la même population, la corrélation entre le bénéfice de survie apporté
par le Cetuximab et nombre de copies de gène de l’EGFR en FISH. Là encore, aucune différence significative n’a pu être retrouvée entre les groupes.
5.2.4.2 Mutation du récepteur EGFR
Dans les cancers du poumon non à petites cellules les mutations du récepteur EGFR ont un rôle central dans l’efficacité ou la résistance aux traitements. Les mutations activatrices au niveau des codons 19 ou 21 sont les plus fréquentes et sont responsables de l’activation permanente de leur domaine TK ; ainsi les tumeurs arborant ces récepteurs présentent une très grande sensibilité aux inhibiteurs de tyrosine kinase93. Leur présence constituerait une contre-indication théorique au Cetuximab, le blocage du récepteur survenant en amont de ces domaines. Dans les cancers des VADS ces mutations ne sont retrouvées que de manière sporadique93,94. Les analyses exhaustives de codons 18 à 20 montrent d’ailleurs dans les cancers
ORL une grande conservation de ces domaines et leurs mutations sont trop rares pour que leur recherche soit intégrée à la pratique courante94,95.
Par ailleurs, un polymorphisme de la partie extracellulaire du récepteur EGF, EGFRvIII, principalement secondaire à des délétions acquises des exons 2-7, a également été rapporté dans 30 à 40% des glioblastomes. Ils aboutissent à une version tronquée du récepteur avec perte du domaine extracellulaire. La fixation des anticorps monoclonaux anti-EGFR serait alors impossible ce qui entraînerait une résistance au Cetuximab. Dans une vaste étude sur 638 carcinomes épidermoïdes des VADS, Katthri et al.96 n’ont pu mettre évidence que 2 cas de mutation EGFRvIII (0,31%), excluant son intérêt prédictif de sensibilité au traitement.
5.2.4.3 Mutation KRAS
Les mutations activatrices de RAS, tel que KRAS codon 12 et 13, sont l’un des facteurs majeurs de résistance au Cetuximab dans les cancers du côlon. 30 à 40% de ces tumeurs présentent ces mutations rendant la recherche de ces anomalies systématiques avant d’initier un traitement anti-EGFR.57 En ORL cette mutation intéresserait moins de 5% des tumeurs27,28. Ainsi, dans une série étudiant la prévalence de ces mutations sur 197 patients traités par radio chimiothérapie, seuls 3,5% présentaient une mutation KRAS codon 12 et aucun ne présentait de