Caractérisation d’un modèle murin déplété en la protéine FANCI:
phénotypes méiotiques et comportementaux, et résistance aux
aldéhydes
Mémoire
Mariline Béliveau Lapointe
Maîtrise en biologie cellulaire et moléculaire
Maître ès sciences (M. Sc.)
Québec, Canada
Caractérisation d’un modèle murin déplété en la
protéine FANCI : phénotypes méiotiques et
comportementaux, et résistance aux aldéhydes
Mémoire
Mariline Béliveau Lapointe
Sous la direction de :
R
ESUMEL’anémie de Fanconi (AF) est une maladie récessive rare associée à un défaut dans la voie de réparation des cassures double-brin de l’ADN causées par des agents pontants (voie de l’AF). Ces agents pontant l’ADN créent un lien covalent entre les deux brins et bloquent les polymérases lors de la réplication ou de la transcription. Mon projet s’intéresse à une protéine importante de la voie de l’AF : FANCI. Celle-ci est décrite comme agissant de pair avec la protéine FANCD2 pour recruter la machinerie de réparation de l’ADN. Nous posons l’hypothèse que FANCI a un rôle important lors du développement, lors de la méiose et est impliquée dans la résistance des cellules aux aldéhydes.
Suite à la création d’un modèle murin déplété en FANCI, nous avons effectué des essais comportementaux de mémoire ou d’anxiété pour caractériser le modèle plus en profondeur. Ensuite, des résultats de coupes histologiques de gonades ont mené à la confirmation de la stérilité des souris KO. Plus spécifiquement, un étalage de chromosomes méiotiques et une immunofluorescence ont été effectués pour vérifier la localisation de FANCI sur ces derniers. De plus, le traitement à la mitomycine C (agent pontant) de cellules embryoniques conférant une instabilité génomique, nous avons voulu tester l’effet d’une source endogène, les aldéhydes, sur des cellules déplétées en FANCI. Nous avons observé les foyers des protéines 53BP1 et γ-H2AX pour vérifier l’accumulation de cassures double-brin.
Les essais comportementaux montrent une tendance non significative. Une co-localisation de FANCI avec le marqueur de résection RPA est observée. De plus, la déplétion de FANCI rend les cellules résistantes aux aldéhydes. Les foyers 53BP1 et γ-H2AX montrent qu’une petite population de ces mêmes cellules a un nombre très élevé de dommages à l’ADN alors que d’autres ont un niveau de dommages similaire au contrôle.
A
BSTRACTFanconi anemia (FA) is a rare recessive disease associated with a defect in the pathway of DNA double strand breaks repair (FA pathway). These double stranded breaks are caused by crosslinking agents by creating a covalent bond between the two opposite strands and blocking polymerases during DNA replication or transcription. My project’s principal interest is a major protein it this pathway : the FANCI protein. It is described as acting together with FANCD2 protein to recruit DNA repair machinery. We hypothesised that FANCI has an important role in development, in meiosis and that it is implicated in cells’ aldehydes resistance.
After the creation of a mouse model depleted in FANCI, with did anxiety and memory behavior tests to deeply characterize our mouse model. Also, results of gonad histologic cuts confirmed mouse sterility. More specifically, we did a meiotic spread and immunofluorescence to see FANCI on meiotic chromosomes. We also treated embryonic fibroblasts with mitomycin C, an exogenous crosslinking agent, and then wanted to check with an endogenous source, like aldehydes, on FANCI depleted human cells. We also observed 53BP1 and γ-H2AX foci formation to check for double strand break accumulation.
Behavior tests do not show any significative tendancy. We observe a colocalization between FANCI and the resection marker RPA. Moreover, FANCI depletion gives an aldehyde resistance to cells. 53BP1 and γ-H2AX foci show a population of cells with a very high level of foci and others that have the same amount of foci as the control cells.
T
ABLE DES MATIERESRésumé ... III Abstract ... IV Table des matières ... V Liste des figures ... VIII Liste des abréviations ... X Remerciements ... XIV
Introduction ... 1
1. Les bases génétiques du cancer ... 1
2. Le cycle cellulaire ... 5
3. Les mécanismes de réparation de l’ADN ... 7
3.1 La recombinaison homologue ... 10
4. L’anémie de Fanconi ... 13
4.1 La voie FA/BRCA ... 16
5. Les protéines FANCI et FANCD2 ... 20
5.1 La protéine FANCD2 et ses rôles indépendants ... 20
5.2 La protéine FANCI et ses rôles indépendants ... 24
5.3 Les rôles dépendants des deux protéines associées et non associées à la voie canonique ... 26
6. Les aldéhydes ... 28
6.1 L’acétaldéhyde ... 28
6.2 Le formaldéhyde ... 31
6.3 Autres aldéhydes ... 34
7. La méiose ... 36
8. Objectifs des travaux de maîtrise ... 38
Matériel et méthodes ... 40
1. Modèle murin Fanci conditionnel ... 40
1.2 Génotypage ... 41
1.2.1 Extraction d’ADN au phénol-chloroforme ... 41
1.2.2 Précipitation de l’ADN ... 41
1.2.3 Amplification et migration de l’ADN ... 42
2. Essais comportementaux ... 42
2.1 Openfield ... 43
2.2 Novel object recognition ... 43
2.3 Spatial object recognition ... 43
3. Immunofluorescence sur étalage de chromosomes méiotiques ... 44
3.1 Étalage de chromosomes méiotiques ... 44
3.2.1 Purification de l’anticorps FANCD2 ... 45
3.3 Immunofluorescence avec la protéine RPA ... 46
4. Culture cellulaire ... 46
5. Essais de survie cellulaire avec aldéhydes ... 46
5.1 Transfection des cellules ... 47
5.2 Acétaldéhyde ... 47
5.3 Formaldéhyde ... 47
5.4 Dénombrement des cellules ... 48
5.5 Immunobuvardage pour vérification de l’efficacité des siARNs ... 48
5.5.1 Lyse des cellules ... 49
5.5.2 Immunobuvardage ... 49
6. Immunofluorescence sur cellules humaines ... 50
6.1 Immunofluorescence ... 50
6.2 Immunobuvardage ... 51
7. Essai de monoubiquitination ... 51
Résultats ... 53
1. Phénotypes observés chez la souris Fanci KO ... 53
1.1 Ratio Mendélien faible ... 53
1.2 Problèmes oculaires ... 53
1.3 Stérilité des souris Fanci KO ... 54
1.4 Les souris Fanci KO ne présentent pas de problème d’anxiété ou de mémoire ... 55
2. Les protéines FANCI et FANCD2 ne présentent pas le même niveau de co-localisation avec le marqueur de resection RPA ... 56
2.1 Co-localisation entre les protéines FANCI et FANCD2 et le marqueur de méiose SCP3 .... 56
2.2 Co-localisation entre les protéines FANCI et FANCD2 et le marqueur de résection RPA . 58 3. Les cellules sont résistantes au formaldéhyde et à l’acétaldéhyde lors de la déplétion de FANCI ... 59
4. La déplétion de FANCI montre parfois un niveau très élevé de dommages à l’ADN, parfois un niveau similaire à celui de la déplétion de FANCD2 ... 63
5. FANCI et FANCD2 ne montrent pas un niveau plus élevé de monoubiquitination lorsque les cellules sont traitées au formaldéhyde ... 66
Discussion ... 68
1. FANCI a un rôle important lors du développement ... 68
2. FANCI à un rôle différentiel de celui de FANCD2 lors de la méiose ... 70
3. FANCI à un rôle différentiel de celui de FANCD2 dans la résistance des cellules aux aldéhydes ... 71
Conclusion ... 74
Liste des tableaux
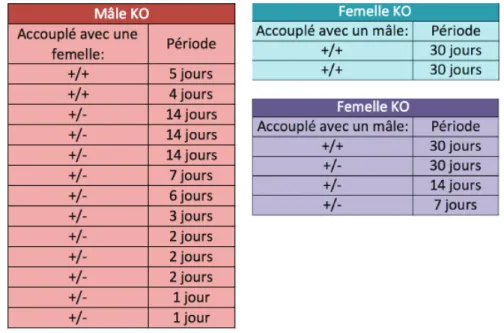
Tableau 1 : Nombre de souris générées pour chaque phénotype et ratio Mendélien ... 53 Tableau 2 : Fiche des partenaires d’accouplement de trois souris Fanci KO ... 55
L
ISTE DES FIGURESFigure 1 : Les six propriétés caractéristiques du cancer ... 2
Figure 2 : Les nouvelles propriétés caractéristiques du cancer ... 4
Figure 3 : Le cycle cellulaire et ses points de contrôle ... 6
Figure 4 : Résumé des différents mécanismes de réponse aux dommages à l’ADN ... 8
Figure 5 : Les voies de réparation qui emploient la recombinaison homologue pour corriger les cassures double-brin ... 11
Figure 6 : La voie FA/BRCA et la recombinaison homologue ... 18
Figure 7 : Formation et réparation des cassures double-brin dans un contexte de réorganisation des chromosomes ... 38
Figure 8 : Problèmes oculaires observés chez les souris Fanci knock-out ... 54
Figure 9 : Différentes phases de la prophase I de la méiose observées à l’aide du marqueur de méiose, la protéine SCP3 ... 56
Figure 10 : Co-localisation entre les protéine FANCI ou FANCD2 et le marqueur de méiose, la protéine SCP3. ... 57
Figure 11 : Co-localisation entre les protéines FANCI ou FANCD2 et le marqueur de résection, la protéine RPA. ... 58
Figure 12 : Quantification des foyers observés pour les protéines FANCI ou FANCD2 et le marqueur de résection, la protéine RPA. ... 59
Figure 13 : Les cellules déplétées en la protéine FANCI montrent une résistance au formaldéhyde et à l’acétaldéhyde ... 61
Figure 14 : Les cellules déplétées en FANCI montrent une mortalité accrue ... 63
Figure 15 : Immunofluorescence de marqueurs de dommages à l’ADN sur des cellules déplétées en FANCD2 ou FANCI ... 64
Figure 16 : Quantification des foyers 53BP1 ... 66
Figure 17 : La monoubiquitination ne semble pas être plus importante lorsque les cellules sont traitées au formaldéhyde ... 67
Figure A1 : Délétion ciblée du locus Fanci murin ... 91
Figure A2 : Les souris Fanci KO ont une fertilité réduite ... 92
Figure A4 : Immunobuvardages afin de valider la déplétion des protéines par siARN 96 Figure A5 : Souris présentant une microcéphalie ... 97
L
ISTE DES ABREVIATIONS°C Degré Celsius
% Pourcent
53BP1 Tumor protein p53 binding protein 1 (TP53BP1)*
ADH5 Alcohol dehydrogenase 5 (class III), chi polypeptide
ADN Acide désoxyribonucléique AKT AKT sérine/thréonine kinase ALDH2 Aldehyde dehydrogenase 2
ARN Acide ribonucléique
ATM Ataxia telangiectasia mutated ou ATM serine/threonine kinase*
ATP Adénosine triphosphate
ATR Ataxia telangiectasia and Rad3 related ou ATR serine/threonine kinase*
ATRIP ATR interacting protein
BIR Break-induced replication (mécanisme de réparation de l’ADN)
BL100 Complexe formé des protéines FANCB, FANCL et FAAP 100 BRCA1 Breast cancer 1 ou BRCA1, DNA repair associated* (FANCS)
BRCA2 Breast cancer 2 ou BRCA2, DNA repair associated* (FANCD1)
BRIP1 BRCA1 interacting protein C-terminal helicase 1 (FANCJ) BSA Bovine serum albumine
BLM Bloom syndrome RecQ like helicase*
Cdks Kinases dépendantes des cyclines CHK1 Checkpoint kinase 1
CHUL Centre hospitalier de l’Université Laval cm Centimètre
cm3 Centimètre cube
CO2 Dioxyde de carbone
CTiP RB binding protein 8, endonuclease (RBBP8)*
CUE Coupling of ubiquitin conjugaison to endoplasmic reticulum degradation
CYP2E1 Cytochrome P450 family 2 subfamily E member 1
DAPI 4’, 6’-diamidino-2-phénylindole DDR Réponse aux dommages à l’ADN DNA2 DNA replication helicase/nuclease 2
DNAPK Protein kinase, DNA-activated, catalytic polypeptide (PRKDC)*
Dr(e)(s) Docteur(e)(s)
DSBR Voie canonique de réparation des cassures double-brin
DT40 Cellules de poulet provenant du lymphome de la bourse, induit par le virus de la leucose aviaire
DTT Dithiothreitol
EDGE Motif protéique nommé d’après sa séquence d’acides aminés EDTA Acide éthylène-diamine-tétra-acétique
EME1 Essential meiotic structure-specific endonuclease 1
ERCC1 ERCC excision repair 1, endonuclease non-catalytic subunit EXO1 Exonuclease 1
FA/BRCA Fanconi anemia/Breast Cancer
FAN1 FANCD2 and FANCI associated nuclease 1 FANC Fanconi anemia complementation group
FBS Fetal bovine serum
FRT Flippase recognition target
γ-H2AX Forme phosphorylée de H2A histone family X H2AX H2A histone family X*
HCl Acide chlorhydrique
HEPES 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
ID2 Complexe formé des protéines FANCI et FANCD2 KCl Chlorure de potassium
kDa Kilodalton
KO Knock-Out
Ku70 X-ray repair cross complementing 6 (XRCC6)*
Ku80 X-ray repair cross complementing 5 (XRCC5)*
Lox P Locus of X-over P1
M Molaire ou mole par litre
MCM Minichromosome maintenance complex component
MDC1 Mediator of DNA damage checkpoint 1
MHF1 Centromere protein S (CENPS)*
MHF2 Centromere protein X (CENPX)*
MiTF Melanogenesis associated transcription factor*
µL Microlitre mL Millilitre µM Micromolaire µmol/L Micromole par litre mM Millimolaire
MRE11 MRE11 homolog, double strand break repair nuclease*
MRN Complexe formé des protéines MRE11, RAD50 et NBS1 MUS81 MUS81 structure-specific endonuclease subunit
NaCl Chlorure de sodium NADH Ubiquinone oxidoreductase
NaF Fluorure de sodium NaOH Hydroxyde de sodium
Na2P2O7 Pyrophosphate de tetrasodium
Na3VO4 Orthovanadate de sodium
NBS1 Nijmegen breakage syndrome 1 ou Nibrin (NBN)*
ng Nanogramme
NHEJ Réparation par jonction des extrémités non-homologues
nm Nanomètre
P32 Isotope radioactif du phosphore
P53 Tumor protein p53 (TP53)*
P63 Tumor protein p63 (TP63)*
PAF Complexe formé des protéines PHLPP 1 et 2, FANCI et D2, USP1 et UAF1 PALB2 Partner and localizer of BRCA2 (FANCN)
PBS Phosphate saline buffer
PCNA PCNA clamp associated factor (PCLAF)* PCR Réaction de polymérisation en chaîne
PFA Paraformaldéhyde pH Potentiel hydrogène
PHLPP PH domain and leucine rich repeat protein phosphatase PIP PCNA-interacting protein motif
PMSF Phenylmethylsulfonyl fluoride
pPDA p-Phenylenediamine
RAD17 RAD17 checkpoint clamp loader component* RAD50 RAD50 double strand break repair protein* RAD51 RAD51 recombinase (FANCR)
RAD51C RAD51 paralog C (FANCO)
RAD52 RAD52 homolog, DNA repair protein*
RB Rétinoblastome ou RB transcriptional corepressor 1 (RB1)* REV1 REV1, DNA directed polymerase
RIF1 Replication timing regulatory factor 1*
ROS Réactifs de l’oxygène RPA Protéine de réplication A RPM Révolution par minute
SCP3 Synaptonemal complex protein 3 (SYCP3)*
SDS Sodium dodecyl sulfate
SDSA Réparation par synthesis-dependant strand annealing siARN ARN interférent (Silencing RNA)
SLX1 SLX1 structure-specific endonuclease subunit
SLX4 SLX4 structure-specific endonuclease subunit (FANCP) SMC1 Structural maintenance of chromosomes 1
SOX2 SRY-box 2*
SPO11 Initiator of meiotic double stranded breaks
SPO11β Un des deux isoformes de SPO11 SSA Réparation par single-strand annealing
S/TQ Motifs protéiques étant la cible des protéines ATM et ATR pour la phosphorylation
SYCP1 Synaptonemal complex protein 1
SYCP2 Synaptonemal complex protein 2
SYCP3 Synaptonemal complex protein 3 (ou parfois SCP3 dans le texte)
TBS Tris-buffered saline
TE Tris-EDTA
Tris 2-Amino-2-(hydroxyméthyl)propane-1,3-diol
U2OS Cellules humaines provenant d’un ostéosarcome de tibia UAF1 WD repeat domain 48 (WDR48)*
UBE2T Ubiquitin conjugating enzyme E2 T (FANCT)
USP1 Ubiquitin specific peptidase 1
UV Ultraviolet
VACTERL Vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal abnormalities and limb anormalities
XPA XPA, DNA damage recognition and repair factor
XPF ERCC excision repair 4, endonuclease catalytic subunit (ERCC4)* (FANCQ) XRCC2 X-ray repair cross complementing 2 (FANCU)
*Afin de respecter le gène cité dans les sources, le nom d’un gène (qui réfère à une protéine) qui n’est pas celui cité dans ce mémoire, mais qui est le nouveau nom approuvé selon le HUGO Gene Nomenclature Committee (http://www.genenames.org/) et par Uniprot (http://www.uniprot.org/) est aussi inscrit mais identifié par un astérisque.
R
EMERCIEMENTSLa réussite de ce projet de recherche repose sur plusieurs personnes sans qui je n’aurais jamais réussi à traverser cette épreuve passionnante qu’est la maîtrise. Je dois premièrement remercier mon Directeur de recherche, chez qui j’avais d’abord effectué un stage en 2014 et qui m’a fait assez confiance pour me donner un projet de recherche dans son laboratoire. Je dois d’abord le remercier pour toutes ses excellentes idées et suggestions scientifiques, mais aussi pour tous les encouragements et conseils dans les moments difficiles, autant sur le plan scientifique que personnel. Jean-Yves, l’année 2016 a été très haute en émotions pour moi, mais tu as toujours su trouver les bons mots pour me redonner le moral et je t’en remercie. Tu as pris le temps d’écouter mes idées, de m’amener à me questionner et à pousser plus loin mes expériences scientifiques. Nous avons de la chance d’avoir un Directeur de recherche aussi patient et compréhensif ! Je dois ensuite remercier Émilie Dubois, qui m’a transféré son projet avec une confiance aveugle, après m’avoir transmis sa passion pour celui-ci. Merci de toujours avoir été là, d’avoir répondu à tous mes messages aussi rapidement, et de m’avoir consacré autant de temps. Je dois aussi remercier tous les membres de mon laboratoire, Marie-Christine Caron qui m’a aidée avec toutes les manipulations en rapport avec les souris, Amélie Rodrigue qui m’a aidée avec tout ce qui était culture cellulaire et Yan Coulombe, pour avoir répondu à mes nombreuses questions et incompréhensions pendant toute la durée de mon passage au laboratoire Masson. Je vous l’ai déjà dit, tous les trois êtes des personnes en or et les étudiants de Jean-Yves ont de la chance de vous avoir. J’aimerais ensuite remercier Niraj Joshi, pour tous ses conseils et son aide avec mon projet, en plus de son amitié, des pas de danse, de la musique et de la nourriture indienne ! J’aimerais remercier Maripier Hainse pour son amitié, son support et son amour pour la gastronomie, nos dîners et soupers au restaurant étaient toujours extrêmement plaisants en ta compagnie. Merci à Anthony Couturier pour son aide avec les logiciels et pour son humour. Merci à Julia O’sullivan, Hemantha Adhikary, Denis Velic, Kenny Dubois et Marie-Michelle Genois, qui m’ont tous aidée à un moment ou à un autre avec mon projet de recherche. Un merci spécial à Stéphanie Bérubé pour son mémoire, qui m’est d’une très grande aide présentement, et à la Dre Isabelle Brodeur pour la correction de mon mémoire.
J’aimerais ensuite remercier mes amies, premièrement Anne-Marie Doyon, ma complice au laboratoire et dans tellement d’autres sphères de ma vie. Myriam Sévigny, Justine Létourneau, Blandine Bulot, Caroline V.G. Scallion, Julie Dubuc et Frédéric Droulers, mes amis depuis plusieurs années déjà pour leur support immense lorsque j’en avais besoin. J’aimerais aussi remercier mes colocataires et amis, Noémie Leduc, ma première amie officielle à Québec, pour ta grande écoute et ton support depuis tout ce temps. Jean-François Meilleur pour ton amitié et tes blagues, la façon dont tu me permets d’être moi-même dans la vie de tous les jours. Je remercie aussi mon copain, Maxime Bourelle-Langlois, pour son support incroyable à l’approche de mon séminaire et pour m’avoir poussé à toujours travailler fort et à donner le meilleur de moi-même lors de l’écriture de mon séminaire ; pour son amour, ses gestes et ses attentions, qui remplissent ma vie de bonheur.
Pour terminer, Je dois remercier ma mère, qui m’encourage dans tout ce que j’entreprends dans la vie et qui est toujours là pour moi. Même lorsque j’ai décidé de partir à Québec pour mes études graduées, tu m’a toujours supportée et encouragée. Merci pour ton écoute et tes conseils. Je dois aussi remercier Marc-André Filion, qui m’a toujours encouragée à aller à l’Université, et qui même dans la maladie, s’informait toujours de la progression de mon projet de maîtrise. C’est en grande partie grâce à toi que je suis rendue ou je suis maintenant.
I
NTRODUCTION1. LES BASES GENETIQUES DU CANCER
Le cancer touche des milliers de personnes à travers le monde depuis déjà plusieurs siècles. Néanmoins, la recherche sur cette maladie n’a débuté officiellement qu’en 1971 lorsqu’un montant d’argent énorme a été déversé pour la recherche sur le cancer par monsieur Nixon, 37ème Président des États-Unis d’Amérique. Par contre, qui dit financement ne dit pas
nécessairement réussite immédiate ; il aura fallu jusqu’aux années 80 et 90 pour se détacher de l’idée que les cancers sont causés uniquement par des virus, puis ensuite de découvrir qu’ils dépendent de six propriétés caractéristiques particulières (figure 1). Dix ans plus tard, deux nouvelles propriétés caractéristiques émergent, en plus de deux autres permettant l’activation de ces huit propriétés caractéristiques (figure 2) (Weinberg 2014).
La première propriété caractéristique est la signalisation proliférative constante. En effet, les tissus sains régulent très finement les facteurs de croissance qui dictent l’entrée et la progression des cellules dans le cycle cellulaire, permettant leur croissance ainsi que la division cellulaire. Lorsque cette régulation est défectueuse ou inhibée, les facteurs de croissance restent constamment liés aux cellules, conférant à celles-ci une division cellulaire constante (Hanahan and Weinberg 2011). Les cellules cancéreuses peuvent elles-mêmes produire leurs propres facteurs de croissance, ou utiliser les cellules saines environnantes afin qu’ils leurs soient fournis (Cheng, Chytil et al. 2008). Les cellules cancéreuses auront aussi acquis une insensibilité aux signaux antiprolifératifs. Ce sont ces signaux qui régulent la prolifération cellulaire (Hanahan and Weinberg 2011). En effet, il existe deux différents types de protéines prenant action en tant que signaux antiprolifératifs, aussi appelés suppresseurs de tumeur. Ce sont les
Gatekeepers et les Caretakers. Lorsque les deux allèles d’un Gatekeeper sont inactivés, la cellule ne
peut plus inactiver les facteurs de croissance ; un bon exemple de ce type de suppresseur de tumeur est le gène du rétinoblastome (RB). Les individus ayant une mutation sur un des allèles de ce gène auront des cellules saines jusqu’à l’inactivation du gène sur le deuxième allèle. Dès lors, la cellule ayant subi cette inactivation peut proliférer et éventuellement former une tumeur. Alors que cette mutation est très rare, elle doit arriver à deux reprises chez les
individus sains pour abolir l’action du suppresseur de tumeur ; les personnes ayant à la naissance une mutation sur ce gène ont une prédisposition très élevée au cancer. Un exemple de Caretaker est le gène P53, menant à la production d’une protéine impliquée dans l’arrêt du cycle cellulaire lorsque la cellule rencontre des dommages à l’ADN, mais aussi impliquée dans la voie de l’apoptose, ou mort cellulaire programmée. Une mutation dans ce gène rend la cellule moins apte à se soucier des dommages à l’ADN, pouvant augmenter le taux de mutation et mener à des défauts dans d’autres gènes importants. Puisque ce gène est aussi impliqué dans l’apoptose, la résistance à cette dernière étant la troisième propriété caractéristique du cancer, la cellule qui serait normalement tuée dû à sont haut taux de mutation restera en vie, pouvant ensuite mener à la formation d’une tumeur (Deininger 1999, Hanahan and Weinberg 2011).
Figure 1 : Les six propriétés caractéristiques du cancer
Figure tirée de (Hanahan and Weinberg 2011)
Ensuite vient la capacité de réplication illimitée ou perte de la sénescence. Cette dernière est l’entrée dans un état non prolifératif mais viable. La cellule saine contient un nombre limité de divisions cellulaires. Une fois ce nombre atteint, elle se dirigera soit vers l’état sénescent ou vers un état critique : la mort cellulaire. Lorsqu’une cellule provenant d’une population de
cellules en état critique acquiert la capacité de prolifération illimitée, on dit que cette cellule est immortalisée (Hanahan and Weinberg 2011). C’est la télomérase, une enzyme ajoutant des répétitions de nucléotides aux extrémités de l’ADN, lui conférant une protection contre la dégradation et éventuellement la mort cellulaire, qui permettrait cette immortalité (Blasco 2005). En effet, celle-ci est retrouvée en quantité significative dans la grande majorité des cellules immortalisées spontanément (incluant celles des cancers humains), alors qu’elle n’est presque pas présente dans des cellules saines. Ensuite viens la capacité d’angiogenèse des cellules tumorales. La vascularisation est présente normalement à l’embryogénèse, où de nouveaux vaisseaux sanguins seront produits, créés à partir de ceux préexistants, par la formation de cellules endothéliales. Chez l’adulte, l’angiogenèse est active de façon transitoire lors de la guérison de blessures ou lors du cycle menstruel chez la femme. Comme les cellules tumorales ont besoin elles aussi d’être oxygénées et d’évacuer leur dioxyde de carbone, elles parviendront à maintenir l’angiogenèse active afin de toujours construire de la nouvelle vascularisation allant avec leur prolifération continue. C’est ce qui mène à la sixième propriété caractéristique, c’est-à-dire la capacité d’invasion et de diffusion métastatique. Les cellules acquièrent lentement des changements biologiques leur permettant une invasion locale, puis une intravasation, c’est-à-dire une invasion des vaisseaux sanguins. Les cellules procèdent ensuite à l’extravasation, la sortie des vaisseaux sanguins, pour aller loger dans un nouveau tissu, puis formeront une micro-métastase, qui deviendra par la suite une tumeur macroscopique.
Récemment, deux nouvelles propriétés caractéristiques ont émergé des recherches sur le cancer, mais restent à être étudiées plus en profondeur. La première est la reprogrammation du métabolisme énergétique. Les cellules cancéreuses préconiseront la glycolyse, bien plus faible en production d’ATP plutôt que la phosphorylation oxydative, utilisée en condition aérobie par les cellules saines. Néanmoins, une glycolyse élevée permettrait aux intermédiaires de cette dernière de former des nucléotides et acides aminés, utiles pour former des macromolécules et organelles pour de nouvelles cellules puisqu’elles sont en prolifération constante. La deuxième propriété émergente est l’évitement de la destruction immunitaire. En effet, certains cancers seraient capables de détourner le système immunitaire en désactivant des composés qui ont été relâchés afin d’éliminer les cellules cancéreuses, comme en sécrétant des immunosuppresseurs.
Figure 2 : Les nouvelles propriétés caractéristiques du cancer
Les deux nouvelles propriétés caractéristiques émergentes, ainsi que les deux permettant l’activation des huit propriétés caractéristiques. Figure tirée de (Hanahan and Weinberg 2011) Il existe aussi deux propriétés permettant l’activation des huit propriétés caractéristiques du cancer, dites propriétés caractéristiques émergentes. Il existe le phénomène de l’inflammation cancéreuse, c’est-à-dire que les cellules néoplasiques vont imiter la réaction inflammatoire. Cette réaction amènera aux cellules plusieurs facteurs utiles à chacune des propriétés caractéristiques du cancer. La deuxième propriété est celle de l’instabilité génomique et des mutations. Comme mentionné au départ, une mutation acquise par une cellule peut lui conférer un avantage par rapport aux autres cellules dans son environnement. Les Caretakers doivent être capables soit de reconnaître les dommages à l’ADN et recruter les protéines de réparation, soit de réparer l’ADN, ou encore d’intercepter les molécules qui peuvent endommager l’ADN avant qu’elles ne se lient à ce dernier (Hanahan and Weinberg 2011). Le bon fonctionnement des mécanismes de réparation de l’ADN est donc essentiel pour prévenir l’apparition de cellules cancéreuses. Il existe plusieurs mécanismes de réparation tous spécifiques à un type de lésion en particulier. Par contre, avant de voir les différents
mécanismes de réparation de l’ADN, il est important de faire un survol du cycle cellulaire puisque ces mécanismes ont lieu à différentes étapes de ce dernier.
2. LE CYCLE CELLULAIRE
Le cycle cellulaire sert à transmettre les informations génétiques d’une cellule à une autre en effectuant une division cellulaire. Il existe plusieurs étapes à ce cycle : premièrement, si les cellules ne sont pas en division cellulaire, elles seront dans un état appelé quiescent, qui constitue la phase G0 (ou Gap 0) du cycle cellulaire. Ensuite, lorsqu’elles se retrouveront dans un moment propice à la division, elles seront stimulées par des facteurs de croissance et passeront à la phase G1 (ou Gap 1), étape précédant la phase S, étant la phase de réplication de
l’ADN. Ensuite, la cellule passera en phase G2 (ou Gap 2) puis en phase M, la phase de la mitose (figure 3) (Nigg 2001). Le cycle est régulé majoritairement par des cyclines et des kinases dépendantes des cyclines (ou Cdks pour Cyclin-dependant kinases) (Malumbres and Barbacid 2005). En effet, afin que le processus de division cellulaire se déroule correctement, la cellule utilise des points de contrôle au travers du cycle, chacun ayant leurs propres rôles. Le premier point de contrôle est celui de la phase G1/S. À la phase G1, certaines cyclines et Cdks ont comme rôle la phosphorylation de la protéine RB, afin de permettre la poursuite des événements du cycle cellulaire. En effet, RB est un répresseur des facteurs de transcription de la famille E2F, des histones déacétylases ainsi que des complexes de remodelage de la chromatine (Cobrinik 2005). Sa phosphorylation est donc essentielle afin de permettre à la cellule d’atteindre le point de restriction, c’est-à-dire le moment irréversible pendant la phase G1 ou celle-ci ne nécessitera plus de stimuli mitogéniques afin de continuer sa progression dans
Figure 3 : Le cycle cellulaire et ses points de contrôle
On observe les différentes phases du cycle cellulaire ainsi que les points de contrôle qui régulent chacune des phases. Ce cycle finement régulé permet la division cellulaire et donc la transmission de l’information génétique à la nouvelle cellule formée. Image tirée et adaptée de (Chin and Yeong 2010)
Néanmoins, si la cellule possède des dommages à l’ADN, la phase G1 sera arrêtée par les voies
impliquant les protéines ATM, ATR et P53 (Kastan and Bartek 2004). La protéine ATM est une protéine qui est en faible concentration dans la cellule normalement. Lors de cassures double-brin de l’ADN, elle sera activée et pourra aller phosphoryler ses substrats au noyau ou aux sites de cassures, entre autres les protéines P53, NBS1, BRCA1 et SMC1 (Bakkenist and Kastan 2003). La protéine ATM possède aussi des médiateurs, les protéines MDC1, 53BP1 et BRCA1 qui s’accumulent aux cassures de l’ADN. Ces dernières modulent ATM suite à la phosphorylation de l’histone H2AX (ou γ-H2AX dans sa forme phosphorylée), une modification de la chromatine marquant les dommages à l’ADN. Les médiateurs pourront par la suite former des foyers aux dommages à l’ADN de ces régions, entre autres pour les substrats d’ATM (Kastan and Bartek 2004). La protéine ATR existe en complexe constant avec la protéine ATRIP. Lors de la formation d’ADN simple-brin dû à une cassure simple-brin,
double-brin ou d’une fourche de réplication bloquée, la protéine de réplication A (RPA) s’y liera, permettant à ATR de se localiser sur l’ADN aussi grâce à l’interaction entre ATRIP et RPA. Cette dernière peut alors phosphoryler ses substrats, entre autres les protéines RAD17, CHK1 et P53 (Zou and Elledge 2003, Kastan and Bartek 2004). Lors de la phase G1, ATM et
ATR phosphoryleront P53 amenant une cascade d’inhibition de différentes protéines, menant finalement à l’inhibition de la phosphorylation de RB, et empêchant la progression à la phase S du cycle cellulaire, afin que l’ADN soit réparé avant sa réplication (Kastan and Bartek 2004).
Lors de l’entrée en phase S, le cycle rencontre le deuxième point de contrôle, c’est-à-dire le point intra-S, utilisé pour empêcher la progression de la réplication de l’ADN (Visconti, Della Monica et al. 2016). Celui-ci est aussi régulé par ATM et ATR, activées suite à des dommages provenant entre autres de sources endogènes causées à l’ADN. Ce point de contrôle comporte deux rôles principaux : premièrement, les fourches de réplication qui étaient sur le point d’être initiées seront inhibées temporairement. Deuxièmement, les fourches de réplication bloquées doivent être protégées (Bartek, Lukas et al. 2004, Kastan and Bartek 2004). Le dernier point de contrôle est celui à la phase G2 du cycle cellulaire, appelé G2/M. Celui-ci est utile pour
empêcher la cellule d’entrer en mitose si elle acquiert des dommages lors de la phase G2, ou si
l’ADN n’a pas été correctement ou pas du tout réparé lors des phases précédentes (Nyberg, Michelson et al. 2002). Les protéines ATM et ATR sont encore une fois impliquées lors de ce point de contrôle, ainsi que les protéines 53BP1 et BRCA1 (Kastan and Bartek 2004). Par la suite, les cellules ayant passé tous ces points de contrôle avec succès peuvent procéder à la mitose, puis à la cytocinèse, et le cycle peut recommencer si celles-ci n’entrent pas en phase G0
(Malumbres and Barbacid 2005).
3. LES MECANISMES DE REPARATION DE L’ADN
Chaque cellule du corps humain est exposée chaque jour à des dizaines de milliers d’événements endommageant l’ADN (Lindahl and Barnes 2000). L’organisme a donc mis en place plusieurs mécanismes de réparation spécifiques aux différentes lésions causées par des sources endogènes ou exogènes aux cellules et à l’individu. Globalement, on appelle ces mécanismes la réponse aux dommages à l’ADN (ou DDR pour DNA Damage Response)
(figure 4). Cette dernière perçoit les dommages, signale leur présence et arrête temporairement le cycle cellulaire afin de réparer l’ADN. Si les dommages sont irréparables ou trop nombreux, la DDR peut aussi envoyer des signaux à la cellule afin qu’elle active l’apoptose. Un premier mécanisme est la réparation par excision de base. Celui-ci est en charge d’éliminer les dommages oxydatifs, les sites abasiques, la formation d’uraciles et les cassures simple-brin causés soit par des réactions spontanées dans la cellule, soit par des réactifs de l’oxygène (ou ROS pour Reactive Oxygen Species), soit par des agents alkyants, ou encore par des rayons-X. Ce mécanisme est caractérisé par le clivage de la base endommagée par une ADN glycosylase (Dexheimer 2013). De plus, il existe deux types de réparation par excision de base : l’insertion d’un petit morceau d’ADN, représentant de 80 à 90 % du type choisi ou l’insertion d’un grand morceau d’ADN plus spécifique aux lésions résistant à l’ADN polymérase β (Fortini and Dogliotti 2007).
Figure 4 : Résumé des différents mécanismes de réponse aux dommages à l’ADN
Synthèse des différents mécanismes de réparation de l’ADN. Ces mécanismes sont spécialisés selon le dommage ou la cassure générée à l’ADN. Légende : BER réparation par excision de bases, MMR correction des mésappariements, GG- réparation par excision de nucléotides globale, TC-NER réparation par excision de nucléotides couplée à la transcription, NHEJ
réparation par jonction des extrémités non-homologues, HR recombinaison homologue. Image tirée de (Dexheimer 2013).
La réparation par excision de nucléotides, un autre mécanisme de réparation de l’ADN, est très semblable au mécanisme précédent, mais est beaucoup plus complexe et nécessite une certaine distorsion de l’hélice d’ADN, ce que la réparation par excision de bases ne requiert pas. Ce deuxième mécanisme répare les composés plus volumineux comme les dimères de pyrimidine et les pontages intra-brin (un lien covalent entre deux bases sur un même brin d’ADN) causés par la lumière UV ou par des hydrocarbures aromatiques polycycliques. Les étapes de ce mécanisme sont la reconnaissance, l’ouverture de l’hélice entourant la lésion, l’excision d’un court segment d’ADN simple-brin et la réparation de la séquence. Il existe aussi deux différents types de réparation pour ce mécanisme: La réparation par excision de nucléotides globale et celle couplée à la transcription. Ces deux types de mécanisme sont semblables, mis-à-part pour l’étape de reconnaissance du dommage à l’ADN qui n’est pas reconnu par le même complexe de protéines (Dexheimer 2013).
Un troisième mécanisme est la correction des mésappariements. Ce dernier répare les erreurs de la polymérase lors de la réplication causant parfois des mésappariements, des insertions ou des délétions de bases. Il comporte trois étapes importantes chez l’humain : la reconnaissance du mésappariement par l’hétérodimère MutSα, l’excision de l’erreur par l’exonucléase 1 et la synthèse de l’ADN afin de réparer le brin qui contenait le mésappariement. Ces trois mécanismes de réparation de l’ADN sont tous affectés à la réparation des cassures simple-brin de l’ADN. Il existe par contre des cassures encore plus dommageables pour les cellules : les cassures double-brin (Dexheimer 2013).
Il existe un premier mécanisme de réparation des cassures double-brin appelé réparation par jonction des extrémités non-homologues (ou NHEJ pour Non-Homologous End Joining). Ce dernier à lieu principalement aux phases G0 et G1 du cycle cellulaire (mais est observé dans
toutes les phases du cycle cellulaire (Dexheimer 2013)), lorsqu’un gabarit complémentaire n’est pas disponible puisque l’ADN n’est pas encore en phase de réplication. C’est pourquoi on dit que ce mécanisme n’est pas fidèle et est sujet à l’erreur (Bhattacharjee and Nandi 2016). En
plus de réparer les cassures double-brin, celui-ci répare les pontages inter-brin (un lien covalent entre deux bases sur des brins complémentaires de l’ADN) causés par des rayons-X, des radiations ionisantes ou des agents anti-tumoraux. Ce mécanisme consiste à la reconnaissance de la cassure par l’hétérodimère formé des protéines Ku70 et 80. Celles-ci encercleront les extrémités de l’ADN brisé puis recruteront des kinases dépendantes de l’ADN nommées DNAPK qui sont capables d’autophosphorylation pour stabiliser et protéger l’ADN, puis contrôler la ligation des deux extrémités de l’ADN (Meek, Dang et al. 2008). Si la cassure double-brin a formé des bouts cohésifs, ceux-ci devront subir des modifications avant qu’ait lieu la ligation et ainsi que la cassure soit réparée (Dexheimer 2013). Il existe un autre mécanisme appelé réparation par jonction des extrémités non-homologues alternatif ou NHEJ-alternatif. Il comporte quelques différences majeures avec le NHEJ classique : premièrement, il utilise des séquences comportant des micro-homologies de 10 à 25 paires de bases pour lier les deux bouts d’ADN. Deuxièmement, la réparation par ce mécanisme est indépendante des protéines Ku et DNAPK. Il est utilisé pour réparer les cassures double-brin causées par des réarrangements, des inversions, des translocations ainsi que des délétions majoritairement lors de la phase S du cycle cellulaire (Bhattacharjee and Nandi 2016). Un deuxième mécanisme important impliqué dans la réparation des cassures double-brin est la recombinaison homologue. Puisqu’il est le mécanisme le plus important en lien avec ce projet, il sera vu plus en détail dans la prochaine section.
3.1 La recombinaison homologue
La recombinaison homologue est un mécanisme de réparation qui a lieu lors des phases S et G2 du cycle cellulaire. À ce moment, l’ADN est répliqué et un gabarit − une chromatide sœur −
est disponible pour réparer la cassure sans faire d’erreur. C’est pourquoi la recombinaison homologue est le mécanisme de réparation le plus fidèle chez la cellule Eucaryote (San Filippo, Sung et al. 2008). Celle-ci est impliquée dans la prévention et la réparation des fourches de réplication bloquées, a un rôle dans le maintien des télomères et orchestre la ségrégation des chromosomes homologues lors de la méiose I (Sung and Klein 2006). Il existe six voies de réparation qui emploient la recombinaison homologue pour corriger les cassures double-brin : La voie canonique de réparation des cassures double-brin (ou DSBR pour Double Strand Break Repair), la réparation par synthesis-dependant strand annealing (SDSA), la réparation par
single-strand annealing (SSA), le break-induced replication (BIR), le NHEJ-alternatif, décrit un peu plus
haut, et la réparation des cassures double-brin à l’aide d’un gabarit d’ARN (figure 5) (San Filippo, Sung et al. 2008, Bhattacharjee and Nandi 2016).
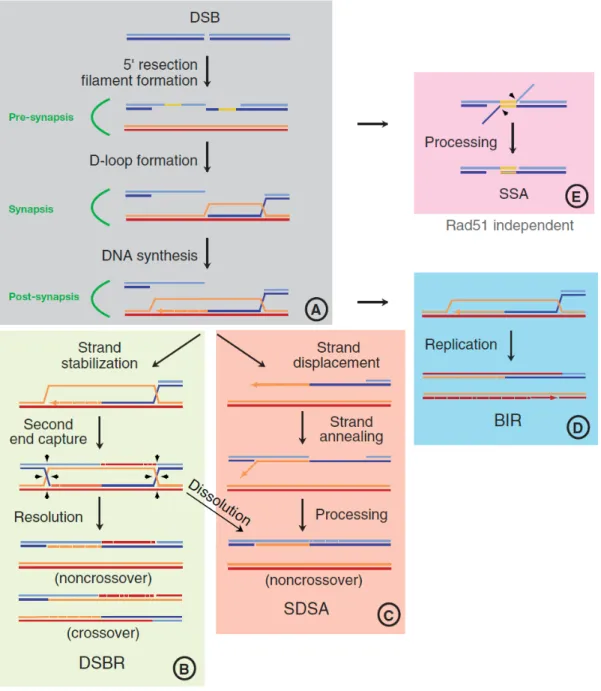
Figure 5 : Les voies de réparation qui emploient la recombinaison homologue pour corriger les cassures double-brin
A Lors d’une cassure double-brin, cette dernière subira la résection afin de créer des extrémités
simple-brin qui seront prises en charge par RAD51, puis l’invasion aura lieu. Des jonctions de Holliday sont formées. Ces jonctions sont résolues soit par B la voie canonique de réparation
de cassures double-brin, par C la réparation par synthesis-dependant strand annealing ou par D le
break-induced replication. E la réparation par single-strand annealing ne requiert pas l’invasion d’un
brin. Le NHEJ-alternatif et la réparation des cassures double-brin à l’aide d’un gabarit d’ARN ne sont pas montrés sur cette figure. Image tirée et adaptée de (Krejci, Altmannova et al. 2012) La voie canonique de réparation des cassures double-brin emploie un mécanisme qui consiste en trois étapes principales : Premièrement, la pré-synapse, où la résection aura lieu. Ensuite, la synapse, suite à l’invasion d’un brin de la chromatide brisée dans la chromatide intacte, puis la post-synapse, où les jonctions de Holliday seront résolues (Tacconi and Tarsounas 2015). Avant même la première étape, la cellule doit choisir quel mécanisme de réparation elle emploiera afin de réparer la cassure double-brin. En effet, comme mentionné plus tôt, le NHEJ peut être exploité à n’importe quelle étape du cycle cellulaire. Certaines protéines vont alors entrer en compétition pour le choix du mécanisme de réparation à adopter. Par exemple, les protéines BRCA1 et CtIP vont agir comme promoteur de la résection, menant alors à la recombinaison homologue, alors que des protéines comme 53PB1 et RIF1 l’inhiberont, favorisant le NHEJ (Jasin and Rothstein 2013). Une fois la recombinaison homologue choisie, la résection sera effectuée par le complexe MRN, composé des protéines MRE11, RAD50 et NBS1, ce qui créera des extrémités 3’ puisque les extrémités 5’ seront réduites par le complexe (Sartori, Lukas et al. 2007). Ensuite, la résection est poursuivie par deux nouvelles protéines, l’hélicase BLM ainsi que l’exonucléase EXO1 (Nimonkar, Ozsoy et al. 2008). L’apparition des extrémités simple-brin attire la recombinase RAD51 ; par contre, ce processus est lent, alors RAD51 entre en compétition avec RPA qui se liera aux extrémités 3’ en premier, afin de les protéger (Wang and Haber 2004). Cette étape permet néanmoins de retirer les structures secondaires pouvant empêcher la liaison efficace de la recombinase à l’ADN simple-brin. Ce sont la protéine RAD52 ainsi que les nombreux paralogues de RAD51 qui aideront ce dernier à remplacer la protéine RPA sur l’ADN (Forget and Kowalczykowski 2010). Par la suite, RAD51 formera un filament autour de l’ADN simple-brin, appelé le filament pré-synaptique qui recherchera la séquence homologue sur l’autre chromatide sœur (Sung and Robberson 1995). Lorsque la séquence homologue sera trouvée, le brin envahira l’autre chromatide sœur, formant alors un ensemble de trois brins d’ADN avec le filament de RAD51, appelé le complexe synaptique (Bianco, Tracy et al. 1998). La synthèse de l’ADN à l’aide du brin complémentaire est effectuée par la polymérase η, puis subit ensuite une ligation, formant une
double structure intermédiaire appelée jonction de Holliday (McIlwraith, Vaisman et al. 2005). Lors de la résolution des jonctions, deux possibilités sont offertes : les endonucléases spécifiques, appelées résolvases, peuvent couper une différente paire de brins pour chaque jonction ; cela mènera à un échange de matériel génétique (Hunter and Kleckner 2001). Cette approche est favorisée lors de la méiose. Puisqu’il ne doit pas y avoir d’échange de matériel génétique lors de la mitose, pouvant mener à une perte d’hétérozygotie, les jonctions subissent une migration afin de n’en former qu’une seule et celle-ci est résolue à l’aide de l’hélicase BLM et de la topoisomérase III. L’ADN est alors coupé sur le même brin, évitant l’échange de matériel génétique (Sung and Klein 2006).
Un autre mécanisme employant la recombinaison homologue est le SDSA. Il emploie exactement les mêmes étapes que pour le DSBR jusqu’à l’étape du complexe synaptique. En effet, il n’y aura pas de doubles jonctions de Holliday formées. Dû à la méthode de résolution des brins de ce mécanisme, il n’y a pas d’échange de matériel génétique (Sun, Nandi et al. 2008). Le mécanisme de réparation par SSA est surtout utilisé lorsqu’une cassure double-brin survient dans des séquences répétées d’ADN. Ce processus ne requiert pas d’invasion des brins alors ce n’est pas toute la machinerie de la recombinaison homologue qui est impliquée. L’ADN subira la résection afin de former des extrémités simple-brin qui pourront être appariées ensembles. L’ADN qui n’est pas homologue entre les répétitions appariées sera clivé, ce qui place le SSA dans les mécanismes de réparation étant mutagènes (Paques and Haber 1999). Le BIR est plutôt employé lorsque la cassure double-brin n’a qu’une seule extrémité, par exemple à une fourche de réplication brisée dont l’autre moitié a été perdue. Ce mécanisme mutagène, employé pour redémarrer les fourches de réplications bloquées, utilisera la méthode de migration du brin jusqu’à ce que celui-ci retrouve le gabarit ou la fin de l’ADN (Michel 2000). La dernière méthode, celle de la réparation des cassures double-brin à l’aide d’un gabarit d’ARN, utilise ce dernier de la même façon que pour le DSBR et utilise aussi les protéines RAD52 et RAD51 (Wahba, Gore et al. 2013).
L’anémie de Fanconi est une maladie rare, principalement à transmission autosomique récessive, qui a été décrite pour la première fois par le pédiatre suisse Guido Fanconi en 1927, lorsqu’il a observé des défauts à la naissance ainsi qu’une pancytopénie (une réduction accrue des globules rouges, blancs et des plaquettes) chez trois frères âgés entre cinq et sept ans (Fanconi 1967, Auerbach 2009). La prévalence de la maladie est d’environ un individu sur 100 000 (Duxin and Walter 2015). Par contre, il existe deux ethnies où la fréquence est plus élevée : les juifs ashkénazes dont la fréquence d’hétérozygotes est d’un sur 89 et les afrikaners où la fréquence d’hétérozygotes est d’environ un individu sur 77 (Lanneaux, Poidvin et al. 2012). On estime qu’au moins 0,5 % de la population en général est hétérozygote sur un locus de l’anémie de Fanconi et que plus de mille individus dans le monde sont atteints de la maladie (Auerbach 2009).
Cette dernière présente une très grande hétérogénéité phénotypique. En effet, 75 % des personnes atteintes ont des malformations congénitales et le ⅔ des ces patients ont des malformations squelettiques, qui sont les plus fréquentes (Giri, Batista et al. 2007). Environ 40 % des patients seront atteints d’anomalies cutanées, 20 % auront des malformations rénales et de l’appareil urinaire, 10 % ont des anomalies oto-rhino-laryngologiques, puis 7 % des patients auront des anomalies du tube digestif. Il existe aussi chez certains patients des anomalies cardiaques et endocriniennes (Kutler, Singh et al. 2003). Les hommes atteints de l’anémie de Fanconi sont très souvent infertiles. En effet, leur sperme comporte un nombre très bas ou tout simplement une absence de spermatozoïde. Chez les femmes, la moitié d’entre elles sont infertiles ; une grossesse peut être possible chez l’autre moitié. Néanmoins, celle-ci est souvent associée à de graves complications (Alter, Frissora et al. 1991). Presque tous les patients seront atteints d’anormalités hématologiques au cours de leur vie. En effet, entre la naissance et 40 ans, l’âge médian pour contracter une pancytopénie est de 7 ans (Dokal and Vulliamy 2008). De plus, la défaillance de la moelle osseuse est de 90 % à l’âge de 40 ans (Auerbach 2009). Les patients sont aussi très à risque de développer une leucémie aiguë myéloïde. L’incidence est approximativement 500 fois supérieure à celle de la population en général et l’âge médian des patients qui contractent cette leucémie est de 13 ans (Auerbach and Allen 1991). Les individus atteints de l’anémie de Fanconi ont aussi une fréquence élevée de certains cancers spécifiques ou de formation de tumeurs associées à la mutation d’un des gènes mentionnés plus bas, comme des cancers de la tête, du cou, de la peau, de l’œsophage, du foie, du cerveau et des
cancers gynécologiques, souvent associés au papillomavirus humain (Joenje and Patel 2001, Kutler, Wreesmann et al. 2003, Rosenberg, Socie et al. 2005).
Jusqu’à maintenant, 20 gènes ont été identifiés comme causant l’anémie de Fanconi. Ces gènes sont FANC A, B, C, D1 (BRCA2), D2, E, F, G, I, J (BRIP1), L, M, N (PALB2), O (RAD51C),
P (SLX4), Q (XPF), R (RAD51), S (BRCA1), T (UBE2T) et U (XRCC2) (Castella, Jacquemont
et al. 2015, Park, Virts et al. 2016). Chacun de ces gènes classe les patients atteints dans différents groupes de complémentation. En effet, les cellules déplétées en l’une de ces protéines seront sensibles aux agents pontant l’ADN, comme la mitomycine C. Ces agents causent des pontages interbrins, c’est-à-dire un lien covalent entre les brins complémentaires de l’ADN, qui sera discuté plus en détail à la section 4.1, pouvant éventuellement mener à des cassures double-brin, néfastes pour le génome. La sensibilité des cellules à ces agents causera une instabilité génétique, causant par exemple des chromosomes quadriradiaux, qui sont caractéristiques à l’anémie de Fanconi. Un patient sera diagnostiqué comme atteint de la maladie si ses cellules présentent cette instabilité génomique lorsque soumises à la mitomycine C, au diépoxybutane ou à la cisplatine (Michl, Zimmer et al. 2016). Il sera ensuite classé dans un des groupes de complémentation, celui dans lequel ses cellules ne retrouveront pas une résistance à l’agent pontant lorsque fusionné avec une cellule de chacun des différents groupes (Auerbach 2009, de Winter and Joenje 2009). Environ 85 % des patients atteints ont une mutation bi-allélique soit sur FANCA, FANCC ou FANCG (Auerbach 2009). De plus, comme mentionné plus tôt, la maladie est à transmission autosomique récessive, à l’exception des patients atteint sur le gène FANCB, qui se situe sur le chromosome X, et le gène FANCR, dont les mutations sont dominantes négatives (Michl, Zimmer et al. 2016). De plus, ce dernier étant un nouveau gène associé à l’anémie de Fanconi découvert chez deux patients, est pour l’instant classé comme conférant un syndrome ressemblant à l’anémie de Fanconi (ou FA-like
syndrome), puisque malgré l’instabilité génomique observée, ces patients ne présentent pas de
défaillance de la moelle osseuse (Ameziane, May et al. 2015, Wang, Kim et al. 2015). Il en est de même pour deux protéines de la famille des paralogues de RAD51 : RAD51C (FANCO) et XRCC2 (FANCU). Ces deux protéines forment un complexe avec les protéines RAD51B et D. Dans les deux cas, les patients présentant une mutation bi-allélique sur un de ces gènes ont des malformations congénitales mais ne présentent pas de défaillance de la moelle osseuse (Kottemann and Smogorzewska 2013, Park, Virts et al. 2016).
Finalement, les patientes ayant une mutation dans un des gènes du complexe cœur de la voie FA/BRCA (voir section 4.1) ne seront pratiquement pas plus à risque de contracter un cancer ovarien ou du sein, probablement parce qu’elles sont pour la majorité infertiles et qu’elles ont un niveau très bas d’œstrogène (Alter 1996, Berwick, Satagopan et al. 2007). Néanmoins, les femmes ayant une mutation mono-allélique sur les gènes suppresseurs de tumeur BRCA1 (FANCS) et BRCA2 (FANCD1) ont un risque de 82 % de contracter un cancer du sein au cours de leur vie, et de 54 et 23 % respectivement de chances de contracter un cancer ovarien (King, Marks et al. 2003). Par contre, l’hétérozygotie ne confère pas l’instabilité génomique observée chez les personnes ayant des mutations bi-alléliques sur ces gènes. Ces patients sont cependant très rares puisque ces gènes sont essentiels à la survie. Ils possèdent plutôt des mutations hypomorphes, c’est-à-dire que le produit du gène est moins actif que la protéine sauvage, permettant la survie de l’individu (Michl, Zimmer et al. 2016). Ce sont les interactions étroites entre BRCA1 et BRCA2 avec la protéine FANCD2 qui ont mis la puce à l’oreille des chercheurs afin qu’ils découvrent le lien entre l’anémie de Fanconi et la réparation de l’ADN (Verlinsky, Rechitsky et al. 2001). C’est pourquoi la voie de l’anémie de Fanconi a été renommée la voie FA/BRCA (de Winter and Joenje 2009), dont le mécanisme moléculaire sera expliqué en détail dans la prochaine sous-section.
4.1 La voie FA/BRCA
Les pontages interbrins sont très dommageables pour l’ADN. Ceux-ci bloquent les polymérases lors de la réplication ou de la transcription. Comme mentionné plus tôt, il existe plusieurs sources d’agents pontant l’ADN. Ces agents peuvent être exogènes comme la mitomycine C, le diépoxybutane et la cisplatine, qui sont utilisés comme agents chimiothérapeutiques (Kee and D'Andrea 2010). Il existe aussi des sources endogènes comme les aldéhydes, qui seront discutés plus en détail à la section 6. Lorsqu’un gène de la voie FA/BRCA est inactif dans la cellule, les défauts du mécanisme de réparation mèneront à une accumulation de pontages inter-brins, pouvant ensuite générer une cascade vers un nombre élevé de mutations ainsi que vers une instabilité génomique. Ce sont ces défauts dans le mécanisme qui causent l’instabilité génomique dans chacune des cellules des patients atteints de l’anémie de Fanconi (Michl, Zimmer et al. 2016).
Toutes les protéines impliquées dans la maladie de l’anémie de Fanconi mentionnées ci-haut font partie soit de la voie de réparation spécifique des pontages inter-brins appelée voie FA/BRCA ou dans des réactions en aval afin de compléter la réparation de l’ADN et sont classées en trois groupes distincts. Neuf de ces gènes codent pour des protéines qui formeront le complexe cœur de la voie de l’anémie de Fanconi, ce qui constitue le groupe I. Ces protéines dont le rôle est la monoubiquitination des protéines FANCD2 et FANCI sont FANCA, B, C, E, F, G, L, M et T. Cette étape de monoubiquitination des protéines FANCD2 et FANCI ou complexe ID2, formant le groupe II, est cruciale pour la suite du mécanisme. Les neuf protéines restantes, formant le groupe III, composé de FANCD1, J, N, O, P, Q, R, S et U vont orchestrer les mécanismes en aval, comme la recombinaison et les réactions nucléolytiques (Zhang and Walter 2014).
Plus en détail (voir la figure 6), le modèle le plus récent de la voie FA/BRCA débute lorsqu’une ou deux fourches de réplication convergentes sont bloquées à cause d’un pontage inter-brin. C’est cet événement qui activera le mécanisme, puisque l’hélicase FANCM et ses partenaires FAAP24, MHF1 et MHF2 seront recrutés au dommage suite à l’éviction de l’hélicase MCM, démantelant partiellement le réplisome (Ciccia, Ling et al. 2007, Collis, Ciccia et al. 2008). FANCM se liera à l’ADN tout près de la lésion et recrutera le complexe cœur. À ce moment le point de contrôle dépendant d’ATR sera activé afin que le cycle cellulaire soit arrêté le temps que la lésion soit réparée. C’est la protéine FANCL, une E3 ubiquitine ligase, qui monoubiquitinera le complexe ID2, à l’aide de FANCT, l’enzyme de conjugaison d’ubiquitine (E2), de FANCB et FAAP100, formant un sous-complexe dont le rôle consiste en la sous-unité catalytique (Wang and Smogorzewska 2015). Les autres protéines du complexe cœur forment aussi des sous-complexes dont les fonctions sont plus ou moins connues. Les protéines FANCA, FANCG et FAAP20 forment un sous-complexe utile pour la rétention du complexe cœur à la chromatine, favorisant la monoubiquitination d’ID2, mais n’étant pas essentielle à celle-ci (Hodson, Purkiss et al. 2014, Huang, Leung et al. 2014). Les protéines restantes forment le dernier sous-complexe : les protéines FANCC, FANCE et FANCF. Ce dernier guide la sous-unité catalytique afin qu’elle soit localisée au site du dommage sur la chromatine (Huang, Leung et al. 2014).
Figure 6 : La voie FA/BRCA et la recombinaison homologue
Une fois le complexe ID2 monoubiquitiné, dont les caractéristiques et les rôles seront vus en détails dans la section 5, ce sont les protéines du groupe III qui prennent le relais afin de terminer la réparation de l’ADN en procédant à l’incision endonucléolytique, à la synthèse translésionnelle et à la réparation de la cassure double-brin (Michl, Zimmer et al. 2016). L’incision endonucléolytique consiste à couper le brin d’ADN de part et d’autre de la lésion. Il existe plusieurs nucléases impliquées à cette étape comme SLX1, MUS81 et EME1 puis FAN1, mais le modèle le plus récent semble être que FANCP recrute les protéines FANCQ et ERCC1 afin qu’elles effectuent les incisions (Kim, Spitz et al. 2013, Klein Douwel, Boonen et al. 2014). Suite à ces dernières, une cassure double-brin est formée sur le brin complémentaire qui a reçu les incisions avec sa nouvelle copie partiellement répliquée, et de l’autre côté, la molécule qui formait le pontage reste accrochée à l’autre brin complémentaire ayant aussi une nouvelle copie d’ADN partiellement répliquée (Michl, Zimmer et al. 2016). C’est sur ce dernier que sera effectuée la synthèse translésionnelle dépendante de REV1 (une désoxycytidyle transférase, qui a pour rôle le transfert d’un nucléotide désoxycitidine sur un brin d’ADN en face d’une désoxyguanosine ou en face d’un site abasique) et de la polymérase ζ (Budzowska, Graham et al. 2015). Cette dernière a un site catalytique permissif, qui peut s’adapter à plusieurs structures d’ADN, permettant la synthèse par-delà la lésion mais selon un mécanisme de synthèse qui est sujet à des erreurs (Kunkel 2009). La molécule pontante sera ensuite retirée grâce au mécanisme de réparation par excision de nucléotides, décrit à la section 3. La cassure double-brin sera elle réparée la plupart du temps par recombinaison homologue (section 3.1). Toutefois, comme mentionné à la section 3, le mécanisme de NHEJ a lieu tout au long du cycle cellulaire, et un choix doit parfois être fait entre ces deux mécanismes de réparation des cassures double-brin (Kottemann and Smogorzewska 2013).
Finalement, FANCJ est une hélicase ayant la capacité de défaire les structures quadruplexes G tertiaires présentes dans les séquences riches en guanosines pouvant bloquer la réplication. Elle comporte aussi une interaction avec la protéine BRCA1, et ses autres rôles sont peu connus (Wu, Shin-ya et al. 2008). La protéine FANCN (PALB2) est quant à elle impliquée dans la recombinaison homologue et est essentielle au recrutement de BRCA2 et de RAD51 au site de dommage à l’ADN, sans quoi la réparation ne pourrait être effectuée. Elle assure aussi la
connexion entre BRCA1 et BRCA2, augmentant leur efficacité pendant la réparation (Xia, Sheng et al. 2006).
5. LES PROTEINES FANCI ET FANCD2
5.1 La protéine FANCD2 et ses rôles indépendants
La protéine FANCD2 a été découverte en 2001 grâce au travail des laboratoires entre autres de Grompe, Taniguchi et d’Andrea. Ce sont ces recherches qui ont connecté l’anémie de Fanconi aux mécanismes de réparation de l’ADN puisque la protéine FANCD2 co-localisait avec BRCA1 non seulement en formant des foyers dans les noyaux de cellules humaines suite aux irradiations γ mais aussi sur des immunofluorescences effectuées sur des étalages de chromosomes méiotiques. De plus, la monoubiquitination de la protéine a aussi été démontrée ainsi que sa conservation dans plusieurs organismes utilisés en recherche biomoléculaire (Garcia-Higuera, Taniguchi et al. 2001, Timmers, Taniguchi et al. 2001). Près de deux ans plus tard, un modèle murin déplété en la protéine FANCD2 a été créé par le groupe Grompe. À ce moment, les modèles murins knock-out (KO) des gènes Fanca, Fancc et Fancg avaient déjà été créés et ont pu servir de comparaison à ce nouveau modèle effectué chez des souris C57BL/6J. Ce dernier montre des phénotypes similaires à celui des modèles murins précédents comme une sensibilité des fibroblastes embryoniques de souris à la mitomycine C, un retard de croissance post-natal, des tubules de testicules normaux et anormaux chez les mâles, un niveau de follicules ovariens réduit chez les femelles ainsi que des anormalités à la phase zygotène de la prophase I de la méiose. Le modèle montre cependant des phénotypes plus marqués comme la microphtalmie présente chez 78 % des souris KO et un hypogonadisme plus sévère. De plus, un ratio Mendelien faible de 16,5 % au lieu du 25 % attendu, observé aussi chez les souris KO Brca2 et un taux élevé de formation de tumeurs, inexistant chez les modèles Fanc précédents, ont poussé l’équipe à élaborer deux théories pour expliquer ces phénotypes plus sévères. Premièrement, la protéine FANCD2 serait sous forme non monoubiquitinée dans les modèles KO de Fanca, Fancc et Fancg. Elle agirait donc de façon hypomorphique dans ceux-ci ; la déplétion complète de la protéine rendrait donc le phénotype plus sévère. Le deuxième modèle est que la protéine serait multifonctionnelle et aurait des fonctions non reliées à la voie
FA/BRCA ; cette perte de multifonctionnalité rendrait le phénotype plus prononcé (Houghtaling, Timmers et al. 2003).
Plus de 10 ans plus tard, la littérature regorge de nouvelles fonctions pour cette protéine qui est maintenant beaucoup mieux caractérisée. Seulement 6 % des patients atteints de l’anémie de Fanconi font partie de ce groupe de complémentation ; néanmoins, le niveau de pénétrance de la maladie est de 100 %, alors que les autres groupes montrent seulement 30 % de pénétrance (De Kerviler, Guermazi et al. 2000). La protéine quant à elle fait 1451 acides aminés et a une masse moléculaire de 164 kDa (Joo, Xu et al. 2011). Cette protéine peut être monoubiquitinée sur la lysine 561, lui permettant d’être ciblée à un endroit particulier de la cellule, la chromatine en l’occurrence (Sigismund, Polo et al. 2004). Elle possède aussi plusieurs domaines protéiques, comme un motif EDGE en N-terminal, requis pour la résistance aux pontages inter-brin (Montes de Oca, Andreassen et al. 2005). Son domaine CUE (pour coupling of ubiquitin
conjugaison to endoplasmic reticulum degradation), situé dans la même région, est important pour son
association avec FANCI ainsi que pour leur rétention sur la chromatine (Rego, Kolling et al. 2012). Un troisième domaine est le motif PIP, étant très conservé et ayant comme rôle l’interaction avec la protéine PCNA. Ce domaine est nécessaire pour la formation des foyers FANCD2 aux sites de dommages à l’ADN ainsi que pour la monoubiquitination (Howlett, Harney et al. 2009). Un domaine plus récemment découvert est le domaine Tower, utile pour la monoubiquitination ainsi que pour la résistance aux pontages inter-brin (Liang, Li et al. 2016). Finalement, la protéine contient aussi un domaine de localisation nucléaire (ou NLS) dans la région N-terminale lui permettant de se rendre au noyau (Boisvert, Rego et al. 2013). FANCD2 monoubiquitinée co-localise aux foyers induits par des dommages à l’ADN avec les protéines BRCA1, BRCA2, RAD51, FANCJ, FANCN et γ-H2AX durant la phase S du cycle cellulaire (Garcia-Higuera, Taniguchi et al. 2001, Folias, Matkovic et al. 2002, Taniguchi and D'Andrea 2002, Hussain, Wilson et al. 2004, Wang, Andreassen et al. 2004, Bogliolo, Lyakhovich et al. 2007, Reid, Schindler et al. 2007, Xia, Dorsman et al. 2007). Elle peut aussi se lier aux jonctions de Holliday et doit être phosphorylée pour que son ubiquitination et celle de FANCI soit efficace (Park, Margossian et al. 2005, Smogorzewska, Matsuoka et al. 2007).