FACULTÉ DE MÉDECINE y'/ 4 ÜL ///r^ THÈSE PRÉSENTÉE
À L'ÉCOLE DES GRADUÉS DE L'UNIVERSITÉ LAVAL
POUR L'OBTENTION
DU GRADE DE MAÎTRE ES SCIENCES (M.Sc.)
PAR BRUNO MAINS
BACHELIER ES SCIENCES PURES DE L'UNIVERSITÉ LAVAL
LES PROTÉINES ACIDO-SOLUBLES DU LYMPHOCYTE ACTIVÉ PAR LA PHYTOHAEMAGGLUT1NINE
Il
AVANT-PROPOS
Je desire ici remercier mon directeur de recherche, le Docteur
William I. Waithe pour ses judicieux conseils ainsi que M. Jean Renaud pour son aide technique. Je remercie aussi mon co-directeur, le Docteur Luc Belanger, pour son soutien academique et financier. Mes remer ciements également pour le Dr. Paul Nadeau ainsi que le Dr. Didier Dufour pour l'aide qu'ils m'ont apportée dans la poursuite de mes travaux. Je ne veux pas oublier de remercier les departements de biochimie science et biochimie medicale de l'universite Laval pour leur soutien financier. Finalement, je remercie les responsables de l'abattoir Les Salaisons Brochu Inc. de St-Henri, ainsi que le Dr. Gourde de leur collaboration qui ont rendu possible ce travail.
TABLE DES MATIÈRES
page
AVANT-PROPOS ... ii
TABLE DES MATIÈRES... iv
LISTE DES TABLEAUX ET DES FIGURES ... vii
LES SIGLES, ABRÉVIATIONS ET SYMBOLES ... x
Introduction I Le cycle cellulaire et le lymphocyte ... 2
II La structure chromatinienne et le lymphocyte... 7
Matériel et méthodes I Les méthodes expérimentales... 11
II Isolement des lymphocytes de porc... 11
III Culture, activation à PHA et marquage radioactif des lymphocytes de porc... 13
IV Fractionnement sub-celiulaire ... 13
A) Noyaux ... 13
B) Fractions nucléoplasmiques ... 17
C) Fractions H1 et HMG... 17
D) Fraction acido soluble (As) ... 18
E) Fraction NHCP et ADN... 18
F) Détermination de la radioactivité ... 19
V Dosages colorimétriques ... 19
VI Filtration sur Bio Gel P-100 de la fraction acido-soluble (As) ... 20
VII Électrophorèse ... 20
A) Électrophorèse analytique sur gels SDS... 20
B) Électrophorèse analytique sur gels acide-urée ... 22
C) Électrophorèse préparative sur gels acide-urée ... 22
Résultats I Isolement et conditions de culture des lymphocytes de sang de porc... 25
V
II Le fractionnement sub-cellulaire ... 31
III Enrichissement de As-1 et As-2... 36
A) H1 + HMG... 36
B) Filtration sur Bio Gel P-100 de la fraction As .. 36
G) Électrophorèse préparative pour l'enrichissement de As-1 et As-2 ... 39
Discussion I L'isolation et les conditions optimales de culture du lymphocyte de porc et son fractionnement sub-cellulaire ... 49
II Caractérisation et enrichissement de As-1 et As-2... 50
III Rôle possible de u-H2a chez le lymphocyte ... 51
Conclusion ... 54
LISTE DES TABLEAUX
Page Tableau I: Isolement et culture des lymphocytes de porc 12 Tableau II et III: Fractionnement sub-cellulaire... 15-16
LISTE DES FIGURES
Figure 1 : Influence de l'effet de la concentration de PHA sur
l'incorporation d'isotope ... 27 Figure 2 : Influence de l'effet de la concentration cellulaire
sur l'incorporation d'isotope ... 28 Figure 3 : Influence de la concentration de leucine froide sur
l'incorporation d'isotope ... 29 Figure 4 : Influence de la concentration de leucine froide sur
le rapport d'incorporation de l'isotope chez des
lymphocytes stimulés et non stimulés par PHA... 30 Figure 5 : Gels SDS des fractions Np-3 et As... 32 Figure 6 : Gels acide-urée des fractions As ... 33 Figure 7 : Profil de radioactivité d'un gel SDS de la fraction
As (gel #2 de la figure 5) ... 34 Figure 8 : Effet de la concentration en sel du tampon Np-3 sur
l'extraction des protéines acido-solubles ... 35 Figure 9 : Gels acide-urée des fractions H1 + HMG.et des
y /
fractions As après extraction avec 5% v PCA ... 37 Figure 10 : Profil de radioactivité des gels de la figure 9 ... 38 Figure 11 : Profil d'élution de la fraction As-(H1 + HMG) sur
Bio Gel P-100... 40 Figure 12 : Gels acide-urée de certaines fractions de la
VIII
Figure 13 : Profil de radioactivité des gels 1, 3, 4 de la
figure 12 ... 42 Figure 14 : Profil de radioactivité des gels 5, 6, 7 de la
figure 12 ... 43 Figure 15 : Gel acide-urée des fractions combinées du profil
d'élution sur Bio Gel P-100 ... 44 Figure 16 : Profil de radioactivité des gels de la figure 15 ... 45 Figure 17 : Profils d'élution des électrophorèses
préparatives ... 46 Figure 18 : Gel acide-urée du matériel séparé par électrophorèse
LES SICLES, ABRÉVIATIONS ET SYMBOLES
AA acide aminé
ADN acide déoxyribonucléique ADNase deoxyribonuclease
Arg arginine
ARN acide ribonucléique
ARNm acide ribonucléique messager ARNr acide ribonucléique ribosomique ARNt acide ribonucléique de transfert
As acido-soluble
As (0.10) acido-soluble après lavage de la chromatine avec 0.1 M NaCI
As (0.20) acido-soluble après lavage de la chromatine avec 0.2 M NaCI
As (0.35) acido-soluble après lavage de la chromatine avec 0.35 M NaCI
As —(Hi + HMG) : acido-soluble après extraction de H1 et H MG As-1 protéine acido-soluble 1
As-2 protéine acido-soluble 2 AT P adénosine triphosphaté BSA albumine de sérum de boeuf
G centigrade
Con A concanavaline A
CPM coup par minute
DPM désintégration par minute EDTA éthylène diamine tétraacétate
XI
El F facteur initiateur de traduction
Gly glycine
h heure
HI + HMG histone Hi et le groupe de protéines de grande mobilité HBSS solution Hanks balancée en sel
HMG groupe des protéines de grande mobilité
lieu isoleucine
KD kilodalton
Leu leucine
Lys lysine
Lys+ contenant de la lysine Lys" ne contenant pas de lysine
LA F facteur d'activation lymphocytaire
Met méthionine
min minute
mw poids moléculaire
NCS "Nuclear Chicago Solubilizer"
NHCP protéine chromatinienne non-histone NHP protéine non-histone
Np-1 1^re fraction nucléoplasmique Np-2 2e fraction nucléoplasmique Np-3 3e fraction nucléoplasmique
Np-3 (0.10) 3e fraction nucléoplasmique extraite dans 0.1 M NaCI Np-3 (0.20) 3e fraction nucléoplasmique extraite dans 0.2 M NaCI Np-3 (0.35) 3e fraction nucléoplasmique extraite dans 0.35 M NaCI PBS tampon phosphate salin
PGA acide perchlorique
XII
PHA : phytohaemagg lutinine PH A+ : contenant de la PHA PHA" : ne contenant pas de PHA
Phé phenylalanine
PMSF : phényl méthyl sulfonyl fluoride Pol 1 : ARN polymérase 1
Pol 11 ARN polymérase II
POPOP : p- bis (2 - ( 4 - méthyl - 5 - phényloxazo PPO : 2,5- diphényloxazole
Strep : streptomycine
SDS : sodium dodécyl sulfate TCA : acide trichloroacétique
TCGF : facteur de croissance des lymphocytes T TEMED : N, N, N1, N' - tétraméthyléthylène diamine tp : température de la pièce
T ris : tri hydroxy méthyl aminométhane T ry : tryptophane
U : unité
u-H2a : histone H2a ubiquitiné (complexe) u-H2b : histone H2b ubiquitiné (complexe) Vi : volume de gel dans la colonne
Vo : volume externe du gel dans la colonne Vt : volume total de la colonne
LE CYCLE CELLULAIRE ET LE LYMPHOCYTE
Cette recherche avait pour but d'enrichir et de mieux caractériser deux protéines acido-solubles (As-1 et As-2) afin d'en déterminer éven tuellement les rôles dans le cycle cellulaire et/ou dans la structure chromatinienne des lymphocytes activés. La mise au point d'une technique d'isolement et de culture des lymphocytes de porc a été nécessaire pour partiellement isoler As-1 et As-2 ainsi que pour leur caractérisation par extraction différentielle et par électrophorèse analytique.
Au moment où je me suis joint à l'équipe de chercheurs de mon directeur de thèse (W. I. Waithe), ceux-ci étudiaient le taux de synthèse et le "turnover" de certaines protéines cytoplasmiques et nucléaires. Ils ont démontré que des lymphocytes stimulés par la phytohaemagglutinine (PHA) en absence d'un acide aminé essentiel (sauf lieu et Phé) n'avaient pas d'augmentation majeure de leur taux de synthèse en ARN et en protéines sauf chez trois protéines. Deux de ces protéines (As-1 et As-2) persistaient à être synthétisées même dans un milieu de culture déficient en Lys. L'addition de l'acide aminé essentiel manquant donnait alors une reprise de la synthèse macromoléculaire à un taux très amplement accéléré. Les lymphocytes entraient alors plus rapidement en phase S. La position du bloc (G1 précoce ou tardif) dépendait de l'acide aminé omis dans les cultures (1). Le lymphocyte ne pouvait donc pas entrer en phase S mais restait bloqué dans la phase G1 du cycle cellulaire (1,2).
Le cycle cellulaire est défini par les événements cellulaires qui s'écoulent entre deux mitoses. Pendant cette période, toutes les cellules somatiques suivent un cycle caractéristique de variations morphologiques et biochimiques. La durée d'un cycle complet varie considérablement en fonction du type de cellules. Ce cycle cellulaire se divise en quatre
3
phases. U ADN est synthétisé et la chromatine répliquée pendant la phase S. À la fin de S, les cellules ont deux fois plus d'ADN et entrent dans la phase G2 puis en division cellulaire ou mitose (M). Immédiatement après M les cellules sont alors diploïdes et entrent dans la phase G1 avant de reprendre la synthèse de l'ADN (phase S). G1 et G2 représentent les pauses interphasiques du cycle cellulaire durant lesquelles aucun ADN n'est synthétisé. Les variations de la durée totale du cycle cellulaire sont essentiellement dues à des modifications de la durée de G1 qui est la plus variable de toutes les phases du cycle. Pendant la phase G1 tous les événements nécessaires à la mise en marche de la synthèse de l'ADN se produisent selon un ordre programmé dans le temps. Après la phase M certains types de cellules peuvent entrer dans un état de quiescence Go (3-5) dont on peut les tirer par différents traitements in vivo ou in vitro. Plusieurs études ont démontré que cet état cellulaire est un stade physiologiquement et biochimiquement différent de G1 (3-5).
L'étude des mécanismes qui contrôlent la croissance des cellules normales est d'une importance primordiale pour comprendre la croissance anormale des cellules cancéreuses. Une cellule cancéreuse est une cellule qui a perdu la faculté de contrôler sa croissance, sa division ou plus exactement son cycle cellulaire. Lorsqu'une cellule devient cancéreuse, elle se divise à des périodes et en des sites de l'organisme où normalement on ne devrait pas observer de divisions. L'idéal est de disposer d'un système ou modèle dans lequel l'addition d'un agent externe comme la phytohaemagglutinine (PHA) transforme rapidement des cellules quiescentes en lymphocytes activés. Le lymphocyte du sang constitue un bon exemple d'une cellule en état de quiescence (Go). Lorsque le lymphocyte est exposé in vitro à des antigènes ou à des activateurs non spécifiques comme la phytohaemagg lutine (PHA) et la concanavaline A (Con A), il passe de Go à G1 et entre en S après 24 heures (6,7). L'activation des cellules en Go amène des changements traductionnels, transcriptionnels et post-trans criptionnels ainsi que des transformations ou modifications protéiques. Plusieurs changements résultant de l'activation lymphocytaire sont aussi caractéristiques chez d'autres systèmes (foie en régénération, sérum activant des cultures de fibroblastes, la réponse de certaines cellules aux hormones). Ces systèmes fournissent des modèles pour étudier le contrôle de la croissance des cellules eucaryotiques.
'I
PH A induit la stimulation des lymphocytes et transforme en cellules blastiques jusqu'à 90% des lymphocytes T après 72 heures de culture (6). Cette stimulation des lymphocytes est une réponse complexe impliquant plusieurs sous-populations de lymphocytes T, des macrophages et au moins deux facteurs de croissance. Selon le modèle courant* cette réaction se divise en plusieurs étapes simultanées. L'activateur rend compétent au moins trois types de lymphocytes T. Le premier type de cellules compétentes interagit avec un macrophage pour que ce dernier libère un facteur de croissance LA F (facteur d'activation des lymphocytes : IL-1). LAF à son tour stimule la production de TCCF (facteur de croissance des lymphocytes T: IL-2) par le second type de cellules rendues compétentes ou TCCF productrices. Le dernier type de cellules rendues compétentes, qui représente la majorité des lymphocytes T activés (cytolytiques, suppresseurs, "helpers" ), exprime à sa surface le récepteur du T CCF dès qu'il est stimulé par PH A. Le contact avec TCCF conduit à la stimulation finale de ces cellules et à leur transformation en cellules blastiques (7). Toutes ces réactions se produisent dans les premières 18 heures après l'addition de PHA. Même si la présence de PH A est nécessaire lors des premières 6 à 18 heures pour une réponse maximale, c'est le second signal donné par TCCF qui conduit les cellules (lymphocytes) à la phase S du cycle cellulaire. +
L
ymph
.
»
»
Lymphocyte compétent Lymphocyte TCGF productifLymphocyte ayant récepteur TCGF Macrophage
LAF TCGF
Lymphocyte blastique
* Ce modèle courant est accepté pour des antigènes spécifiques mais pas encore définitivement pour l'activateur non spécifique qu'est PHA.
5
Les petites cellules avec noyau dense et très hétérochromatique, aspect caractéristique des lymphocytes en Go, deviennent de grosses cellules contenant plus de cytoplasme et ayant une augmentation de la proportion d'euchromatine dans le noyau (8). Pendant les premières heures d'activation le nucléole devient visible. Il y a des changements dans les niveaux intracellulaires des nucléotides cycliques, augmentation du transport membranaire, modification des histones ainsi qu'augmentation de la phosphorylation des protéines nucléaires et cytoplasmiques. Les changements dans la maturation des ARNr ont également lieu dans les premières heures d'activation (6,7,9). Dans les lymphocytes quiescents (Go) près de la moitié des ARNr 28S sont détruits dans le noyau ainsi que la plupart des ARNr 18S. Pendant l'activation, la dégradation de ces ARNr diminue par un mécanisme dépendant de la synthèse continue de protéines à courtes demi-vies (10,11). Cette diminution de la dégradation des ARNr est couplée à une augmentation précoce de la synthèse de l'ARNr 45S (12,13). Ceci amène donc une augmentation du nombre de nouveaux ribosomes. Le taux de synthèse protéique augmente aussi dans les deux premières heures. Ceci résulte de l'augmentation de l'initiation de la synthèse protéique grâce à des ARNm préexistant dans le lymphocyte (14,15). Il n'y a pas de changements qualitatifs détectables dans les des protéines nouvellement synthétisées sur gel de polyacrylamide en deux dimensions et en fluorographie pendant la phase G1 du cycle cellulaire des lymphocytes activés (16). Il y a cependant des changements dans le taux différentiel de la synthèse et du "turnover" de certaines protéines spécifiques (16-20). Après 20 heures de stimulation, le taux de synthèse protéique est environ 10 à 20 fois plus élevé qu'à l'état quiescent (Go). L'activité de l'ARN polymérase I (pol I) (transcription de l'ARNr 45S) double 4 heures après l'activation des lymphocytes par PH A et elle est 6 fois plus forte après 20 heures (12,13). L'augmentation de la transcription des ARNr après stimulation des lymphocytes par PHA semble être le résultat d'une augmentation du taux d'élongation des chaînes par des complexes de transcription préalablement initiés (21). Ces résultats sont en accord avec les résultats obtenus dans d'autres systèmes d'activation de croissance. Il semble qu'une grande partie de l'augmentation de l'activité transcriptionnelle des cellules en Go soit due à la transcription d'ARNr nucléolaire (22). Une augmentation précoce de l'activité de l'ARN polymérase II (pol II) (transcription de l'ARNm) du lymphocyte activé par
6
PHA n'a été rapporté que par seulement un auteur (23). Les changements majeurs du passage des phases Go à G1 du cycle cellulaire après activation de croissance cellulaire semblent donc être une augmentation de la trans cription d'ARNr et une augmentation de la synthèse protéique favorisée par des changements au niveau post-transcriptionnel. De plus, il existerait une transcription spécifique d'un ARNm (24) mais aucune autre information n'est disponible sur la transcription d'un nouveau gène pendant l'activation lymphocytaire. Le lymphocyte quiescent a un niveau de synthèse protéique bas, peu de polysomes sont présents, quelques monosomes sont actifs mais la majeure partie des ribosomes sont sous forme de ribosomes libres 80S. L'accumulation de ribosomes libres pourrait possiblement être causée par la faible activité d'un facteur de dissociation (ElF—3) dont le rôle est de séparer les sous-unités 40S et 60S du ribosome qui termine la traduction d'un message (25). Sans cette action du facteur de dissociation le ribosome resterait inapte à reformer le complexe d'initiation de la synthèse protéique. Les quantités de sous-unités natives 40S sont également limitées par une dégradation sélective de l'ARNr 18S nouvel lement synthétisé au niveau du nucléole (26). Malgré ces conditions, des sous-unités 40S et 60S sont disponibles et seraient capables de former le complexe d'initiation 80S mais le processus pourrait être encore consi dérablement ralenti par une quantité limitée d'ARNt initiateur Met (27,28). Cet ARNt initiateur Met serait essentiel à la formation du complexe d'initiation 80S. De plus, les facteurs d'initiation ElF-1 et EIF-2 sont possiblement sous forme inactive et il existerait même des évidences qu'un inhibiteur de la formation du complexe d'initiation soit présent (14) ainsi qu'un inhibiteur de l'élongation (29). Lors de la stimulation du lymphocyte par PHA tous ces mécanismes seraient possiblement renversés. Les facteurs d'initiation deviendraient actifs, les inhibiteurs seraient progressivement inactivés et la dégradation de l'ARNr 18S cesserait avec comme conséquence une augmentation massive de sous-unités 40S provenant de la réserve de ribosomes libres et la formation de polysomes actifs.
7
LA STRUCTURE CHROMATINIENNE ET LE LYMPHOCYTE
La structure des fibres de chromatine est basée sur une unité répétitive de la double hélice de l'ADN qu'on appelle nucléosome (30-34). Le nucléosome peut être isolé après une digestion partielle de la chromatine avec des nucléases. Les nucléases sont plus accessibles à l'ADN non protégé qu'on appelle ADN " Linker". Le nucléosome est ainsi constitué de paires de bases d'ADN enroulées autour du "core" histone. Il est constitué de deux molécules de quatre des cinqs types d'histone H2a, H2b, H3 et H4. La chaîne nucléosomaie qui représente le premier niveau de condensation de la double hélice de l'ADN peut se compacter en un second niveau de condensation pour donner le solénoïde et/ou le supersolénoïde. Le processus d'activation cellulaire requiert probablement des changements importants de l'appareil génique et ainsi de la structure chromatinienne. Il y a des évidences qu'une chromatine transcriptionnellement active soit dans une structure différente de celle d'une région non transcrite. De plus, seulement 10% de l'ADN est généralement transcrit. Il semble que les endonucléases reconnaissent quelque chose de spécial dans les gènes actifs. Il a été démontré que l'augmentation de la sensibilité à l'ADNase I pancréatique des gènes actifs est un phénomène général (35-42). Généralement, les séquences d'ADN dégradées par les nucléases sont complexées à des NHP (43-46), d'autres à des H MG (47-53), des HMG phosphorylées (54), des histones très acétylées (55-58) ou des histones et des NHP ADP-ribosylées (59). La coformation plus ouverte des gènes actifs est conférée par des protéines et/ou leurs modifications.
Les histones sont des protéines qui ont un rôle important à jouer dans la structure chromatinienne (60). Ce sont des molécules relativement
8
petites, fortement basiques, contenant peu d'acide aminés aromatiques et pas de tryptophane. Les histones existent en cinq catégories principales différenciées par leur contenu relatif en Lys et Arg. Ces cinq catégories se divisent en trois groupes. D'abord H1, la plus riche en Lys, a le poids moléculaire le plus élevé soit environ 21 KD. Elle est hétérogène et c'est aussi celle qui a le plus varié au cours de l'évolution. L'histone H1 est localisée sur l'ADN "linker" entre deux nucléosomes. Le deuxième groupe, H2a et H2b, comporte les histones modérément riches en Lys et ont des poids moléculaires respectifs d'environ 12.5 et 14 KD. Finalement, les histones riches en Arg, H3 et H4 ont des poids moléculaires respectifs d'environ 15 et 11 KD. Une caractéristique importante de H3 est qu'elle est la seule histone à posséder de la cystéine ce qui lui permet de former des dimères. Il existerait un niveau de base de synthèse des histones chez le lymphocyte quand il est en phase Go (état de quiescence) de son cycle cellulaire (61). Les mécanismes de contrôle de la synthèse des histones sont d'une grande importance car ils sont liés intimement avec les mécanismes de contrôle de la synthèse de l'ADN et par conséquent avec le contrôle de la prolifération des cellules. Les cinq histones ainsi que les HMG peuvent subir quatre types de modifications covalentes majeures: la méthylation, l'acétylation, la phosphorylation et l'ADP ribosylation (62-67). En plus des quatres modifications majeures précédentes, il peut y avoir ubiquitination des histones H2a et H2b pour donner respectivement u-H2a et u-H2b (69).
Le complexe u-H2a (H2a + ubiquitine) a initialement été désigné par l'appellation A-24 (70-71 ). On retrouve u-H2a dans la chromatine de foie de rat (70), de thymus de veau (72) et de lymphocyte humain (73). L'ubiquitine est une protéine de 8.5 KD qui a été très conservée au cours de l'évolution car on la retrouve dans plusieurs types de cellules animales, les bactéries, les levures et les plantes supérieures (74). La séquence complète d'acides aminés de l'ubiquitine est connue (75). L'ubiquitine est liée à H2a de façon covalente par un lien isopeptidique Gly-Gly entre le 74e acide aminé (Arg) de l'ubiquitine et le 119e acide aminé (Lys) de H2a (33,68,76). Environ 10% des histones H2a de la chromatine sont ubiquitinées (77-80). De plus, il apparaît que u-H2a est en interaction avec H1 (81) et qu'elle est sujette à des modifications covalentes (82-83).
9
L'ubiquitination de l'histone H2a dans les nuclésomes peut jouer un rôle important dans la structure chromatinienne car u-H2a est situé à la partie externe du "core" histone. u-H2a est ainsi disponible pour des interactions avec les fibres de la chromatine (80,84,85). u-H2a peut être hydrolyse en histone H2a et en ubiquitine par une isopeptidase (A-24 lyase) qu'on retrouve au niveau du nucléole (86, 87). Les niveaux de u-H2a et de l'activité A-24 lyase nucléaires pendant une activation de croissance cellulaire suggèrent que tous deux sont impliqués dans les changements d'activité, transcriptionnelle et/ou conformationnelle de la chromatine. Les niveaux de u-H2a diminuent et l'activité A-24 lyase augmente dans les nucléoles de foie en régénération (86). Par ailleurs, des fractions de chromatine active ou inactive ont montré une absence ou une diminution importante de u-H2a dans la chromatine active (51 ,88,89). Sa disparition du nucléole des cellules de foie en régénération (86) et son absence sur des séquences de gène ribosomique hautement actifs (90) suggèrent que l'hydrolyse de u-H2a serait responsable en partie de la modification conformationnelle des nucléosomes de ces sites (31). De plus, des variants d'histones auraient une importance potentielle dans la conformation chromatinienne (62). Les variants de H2a, H2b et H3 ne diffèrent des histones standards que de quelques acides aminés tel que démontré chez plusieurs espèces eucaryotiques (91,92). Les rôles des variants d'histones dans la structure chromatinienne ne sont pas encore éclaircis mais des changements dans leur distribution durant le cycle cellulaire suggèrent qu'ils sont d'une certaine importance dans la condensation chromatinienne (62).
LES MÉTHODES EXPÉRIMENTALES
La plupart des méthodes étaient d'usage courant dans notre laboratoire sauf la technique d'isolement et de culture des lymphocytes de sang de porc ainsi que la méthode d'enrichissement des protéines As-1 et As-2. Notre équipe de recherche avait déjà mis au point des techniques d'isolement et de culture des lymphocytes de sang humain mais une adaptation de ces techniques s'avérait nécessaire pour l'isolement et la culture des lymphocytes de sang de porc dans des conditions optimales. L'enrichissement de As-1 et As-2 par fractionnement sub-cellulaire n'était pas une méthode optimale mais cette approche a été utilisée dans le but de caractériser d'autres protéines nucléaires d'un certain intérêt (résultats non présentés dans cette thèse).
ISOLEMENT DES LYMPHOCYTES DE PORCS
Toutes les manipulations (Tableau I) se font en conditions stériles. Le sang de porc (femelle) est immédiatement défibriné en présence de pénicilline (200 U/ml) et de streptomycine (200 \ig/ml). Une solution de dextran (Pharmacia Fine Chemicals) 6% p/v est ajoutée au sang à concentration finale de 1.5% p/v. La sédimentation des globules rouges se fait à 1 g pendant 35 min à 37° C dans une ampoule à décanter. Le surnageant est ensuite filtré sur une colonne de coton de 40 cm maintenue à 37° C afin de se débarrasser des cellules adhérentes (monocytes + polynucléaires). Les cellules non adhérentes sont récupérées par centrifugation à 400 g pendant 15 min à 20° C. Les lymphocytes sont alors séparés des cellules de densité différente par centrifugation sur un coussin de “Ficoll-Paque" (Flow Laboratories) (densité de 1.077 g/ml)
12
(2,61). Un volume de suspension cellulaire dans une solution de HBSS (Flow Laboratories) est déposé sur deux volumes de "Ficoll-Paque". Les tubes sont centrifugés à 800 g pendant 20 min à 20° C. Les lymphocytes sont récupérés à l'interface et lavés deux fois dans 1 volume de HBSS: la première fois à 200 g pendant 20 min à 20° C, la seconde à 150 g pendant 15 min à 20° C.
TABLEAU I
Isolement et culture des lymphocytes de porc
Sang porc (200 U. Pen.- 200 yg Strep./ml sang
Défibrination Plaquettes
Sang sans
Sédimentation avec dextran (5 x 105 mW) plaquettes Globules rouges Fraction leucocytaire a) Colonne de coton (40 X 2.5 cm), 37° C b) 400g, 15 min, tp. Cellules adhérentes (Monocytes + polynucléaires) Fraction lymphocytaire 1
Ficoll- Isopaque 800 g, 20 min, tp.
Fraction lymphocytaire 2
Lavages dans HBSS a) 200 g, 20 min, tp.
b) 150 g, 15 min, tp.
Culture dans R PMI 1640, 10% v/v sérum de veau foetal sans AA, 100 U/ml de pénicilline et 100 yg /ml de streptomycine.
13
HI CULTURE, ACTIVATION A' PHA ET MARQUAGE RADIOACTIF DES LYMPHOCYTES DE PORC
Les cellules sont resuspendues dans un volume minimal de RPMI 1640 (Flow Laboratories), 10% v/v sérum de veau foetal (Flow Laboratories) sans AA, 100 U/ml de pénicilline, 100 yg/mi de streptomycine. Le décompte cel lulaire et la détermination du pourcentage de polynucléaires des cultures se font sur hémacytomètre dans une solution de Turk (3% v/v acide acétique, 0.01% p/v de bleu de méthylène). Le pourcentage de polynucléaires varie entre 5 et 10%. Les cellules sont incubées pendant 12 h sous atmosphère contrôlée (37 C, 5% CO^ et 95% d'humidité) à une concentration de 10^ cellules/ml dans un erlenmeyer de façon à obtenir un rapport cellules/surface de 5 x 10^ cellules/cm3. Les cellules sont par la suite stimulées avec 10 yg/ml de PHA (Miles). Le marquage d'une partie des cultures se fait avec 3H-Leu (New England Nuclear) (50-65 Ci/mm) à 10 yCi/ml pendant deux heures dans un milieu de marquage contenant 1 mM de Leu. Les suspensions cellulaires sont lavées avec un volume du tampon PBS (140 mM NaCI, 8 mM Na^HPO^ , 1.5 mM KH^PO^ et 3 mM KCI) à 4° C. Les lymphocytes sont centrifugés à 800 g pendant 3 min à 20° C. Le lavage est répété une seconde fois puis le culot de cellules est congelé rapidement dans un mélange éthanol-glace sèche et conservé à -70° C. La détermination de la viabilité cellulaire après culture des lymphocytes se fait sur hémacytomètre avec l'usage d'une solution de bleu Trypan (Flow Laboratories) (0.4% p/v de bleu Trypan dans une solution saline) (93).
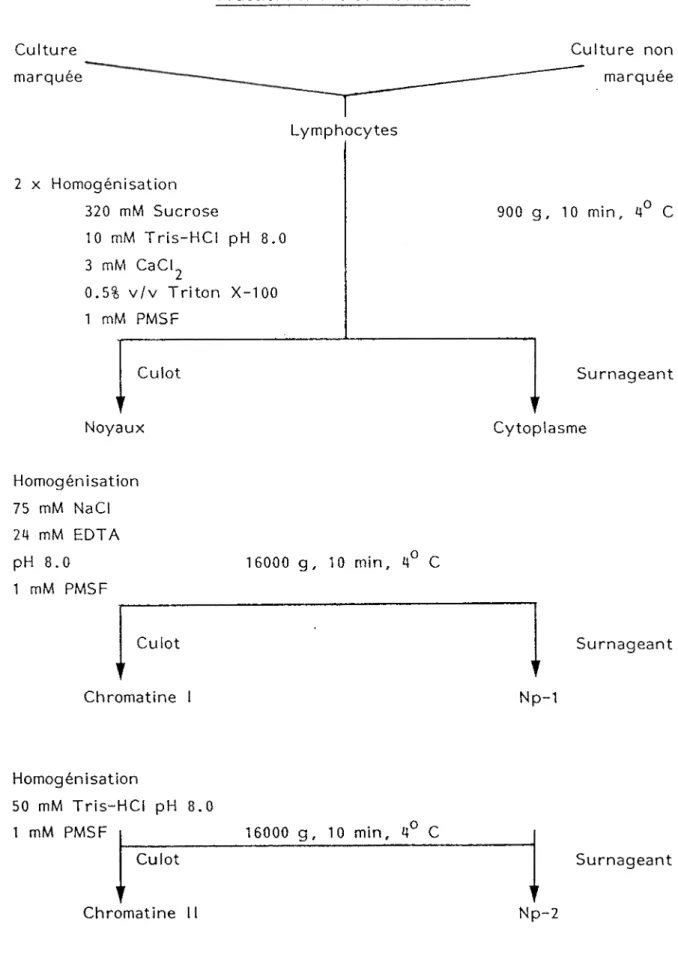
IV FRACTIONNEMENT SUB-CELLULAIRE
Toutes les manipulations (Tableaux II et III) se font à 4°C.
A) Noyaux
Les noyaux sont isolés selon la méthode de Levy R. et colt. (94), laquelle a été modifiée par l'addition de fluorure de phénylméthylsulfonyle (PMSF) pour avoir une concentration de 1 mM dans tous les tampons. Les cellules congelées sont suspendues dans le tampon d'isolement des noyaux (0.32 M sucrose, 10 mM Tris,
14
3 mM CaCI 0,5% v/v triton X-100, 1 mM PMSF, pH 8.0) à une
z 7
concentration finale de 2 à 4 x 10 cellules/ml. Les lymphocytes sont homogénéisés dans un homogénisateur "Bounce" par 10 passages d'un piston à tête serrée. Le relarguage des noyaux est suivi par microscopie en contraste de phase. L'homogénat est transféré dans un tube et centrifugé à 900 g pendant 10 min. Le surnageant est récupéré par inversion du tube et le culot de noyaux est réhomogénisé par cinq passages du piston à tête serrée puis centrifugé à 900 g pendant 10 min. Le surnageant est combiné avec celui de la première centrifugation pour ainsi constituer la fraction cytoplasme.
TABLEAU II Fractionnement sub-cellulaire Culture marquée
T
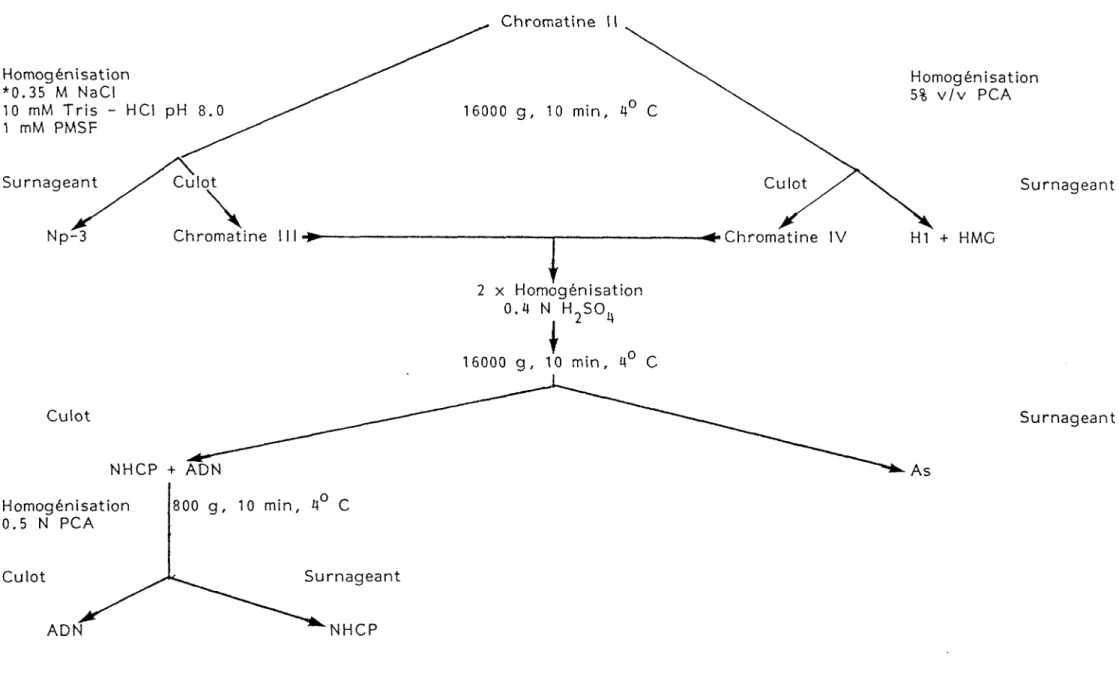
Culture non marquée Lymphocytes 2 x Homogénisation 320 mM Sucrose 10 mM Tris-HCI pH 8.0 3 mM CaCI^ 0.5% v/v Triton X-100 900 g, 10 min, 4° C 1 mM PMSF Culot Noyaux Surnageant Cytoplasme Homogénisation 75 mM NaCI 24 mM EDTA pH 8.0 1 mM PMSF 16000 g, 10 min, 4° C Culot Chromatine I 'r Surnageant Np-1 Homogénisation 50 mM Tris-HCI pH 8.0 1 mM PMSF Culot 16000 g, 10 min, 4° C Chromatine II Surnageant i ' Np-2TABLEAU III
Fractionnement sub-cellulaire (suite) Chromatine Homogénisation *0.35 M NaCI 10 mM Tris - HCl pH 8.0 1 mM PMSF Homogénisation 5% v/v PCA Culot
Surnageant Culot Surnageant
Chromatine 111 * Chromatine IV H1 + HMC 2 x Homogénisation O.if N H„SO„ Homogénisation 0.5 N PCA Surnageant Culot NHCP
17
B) Fractions nucléoplasmiques
Les noyaux isolés sont suspendus dans le tampon d'extraction Np-1 (24 mM EDTA, 75 mM NaCI, 1 mM PMSF, pH 8.0) à raison de
g
l'équivalent de 10 cellules/ml. La suspension est homogénéisée par 20 passages du piston d'un homogénisateur "Potter-Elvejehm" à entraînement motorisé et à vitesse maximale. L'homogénat est centrifugé à 16000 g pendant 10 min. Le surnageant représente la fraction Np-1 et le culot (Chromatine I) sert à l'extraction de la fraction Np-2. Le culot est suspendu dans le tampon d'extraction Np-2 (50 mM Tris, 1 mM PMSF, pH 8.0) à raison de l'équivalent de
7
5x10 noyaux/ml. La suspension est traitée comme la précédente et le surnageant après centrifugation représente la fraction Np-2. Le culot obtenu (chromatine 11) lors de l'isolement de la fraction Np-2 sert pour extraire la fraction Np-3 ou la fraction H1 + HMG. Pour extraire la fraction Np-3 le culot est suspendu dans le tampon d'extraction Np-3 (0.35 M NaCI, 10 mM Tris, 1 mM PMSF, pH 8.0) à raison de l'équivalent de 15 à 20 x 10^ cellules/ml. Deux autres tampons d'extraction Np-3 contenant 0.1 ou 0.2 M NaCI ont été utilisés pour analyser l'effet de la concentration en sel sur l'extraction. Le traitement de la suspension est par la suite identique à celui des fractions Np-1, Np-2 et le surnageant obtenu représente alors la fraction Np-3.
C) Fraction H1 + HMG
H1 et le groupe des protéines de grande mobilité (HMG) sont extraites selon la méthode de Sanders C. & Johns E.W. (95). Le culot de chromatine II est suspendu dans 5% v/v PCA à raison de l'équivalent de 10^ cellules/ml. La suspension est homogénéisée par 20 passages du piston d'un homogénéiseur "Potter-Elvejehm" à entraînement motorisé et à vitesse maximale. L'homogénat est transféré dans un tube et centrifugé à 16000 g pendant 10 minutes. Le surnageant est récupéré par inversion du tube et le culot est suspendu dans le même volume de solution d'extraction. Le tout est
18
homogénéisé une nouvelle fois et de nouveau transféré dans un tube puis centrifugé à 16000 g pendant 10 minutes. Le surnageant est combiné avec le surnageant de la première centrifugation pour ainsi constituer la fraction Kl + HMG. À cette nouvelle fraction on ajoute HCl 12 N pour y obtenir une concentration finale de 0.4 N. On ajoute par la suite 4 à 5 volumes d'acétone froid et le tout est incubé pendant 4 jours à -20° C. Le mélange est par la suite centrifugé à 18000 g pendant 30 min. Le surnageant est aspiré et le culot séché à vide représente les protéines de la fraction H1 + HMG.
D) Fraction acido soluble (As)
Les culots (chromatine III et IV) obtenus lors de l'isolement de la fraction Np-3 et H1 + HMG sont suspendus dans 0.4 N H2S04 (solution d'extraction des protéines acido solubles. As) à raison de l'équivalent de 10^ cellules/ml. La solution est homogénéisée par 20 passages du piston d'un homogénéiseur "Potter-Elvejehm" à entraînement motorisé et à vitesse moyenne. L'homogénat est par la suite lentement agité pendant 30 min à 4° C puis centrifugé à 16000 g pendant 10 min. Le culot est suspendu dans la moitié du volume initial et extrait une nouvelle fois. Le mélange des deux surnageants représente la fraction As auquel on y ajoute 10 volumes d'acétone froid et le tout est incubé pendant 4 jours à -20° C. Le mélange est ensuite centrifugé à 15000 g pendant 30 min. Le surnageant est aspiré et le culot séché à vide représente les protéines de la fraction As.
E) Fraction NHCP et ADN
Le culot obtenu lors de l'isolement de la fraction As est suspendu dans 0.5 N PCA pour précipiter l'ADN à raison de l'équi valent de 5 x 107 cellules/ml. La solution est homogénéisée par 20 passages successifs du piston d'un homogénéisateur dans un "Potter-Elvejehm" à entraînement motorisé et à vitesse maximale. La solution est alors agitée pendant 20 minutes à 70° C puis ramenée à
19
4° C et centrifugée à 800 g pendant 10 minutes. Le surnageant est prélevé à la pipette pasteur et constitue la fraction NHCP tandis que le culot séché à froid représente la fraction ADN.
F) Détermination de la radioactivité.
La technique de détermination de la radioactivité des fractions sub-cellulaires est plutôt standard. On ajoute d'abord 0.3 ml de BSA (2.5 mg/ml) et 3 ml de TCA-a (6% p/v T CA et 65 mg/l Leu) à des échantillons de 100 à 200 yl. Pour les fractions (H1 + HMG) et As on utilise plutôt 3 ml de TCA-b (20% p/v TCA et 65 mg/l Leu). Les fractions sont incubées à 4° C pendant 30 min puis centrifugées à 800 g pendant 10 min à 20° C. On ajoute au culot 0.1 ml de NaOH IN et on incube à 56° C pendant 5 min. On ajoute alors 3 ml de TCA-a aux fractions noyaux, cytoplasme, Np-1, Np-2, Np-3 et 3 ml de TCA-b aux fractions (Kl + HMG) et As. On incube à 4° C, 30 min puis on centrifuge à 800 g, 10 min, 20° C. On ajoute aux culot 0.5 ml de NCS et 1 ml pour la fraction NHCP et ADN. On incube par la suite 5 min à 50° C. La radioactivité est finalement déterminée par comptage à scintillation après l'addition de 10 ml de toluène 0.5% p/v PPO et 0.05% p/v PO PO P.
V DOSAGE COLORI MÉTRIQUE
Le dosage des protéines se fait selon la méthode de Lowry O.H. modifiée par Hartree E.F. (96). L'albumine de sérum de boeuf (BSA) est utilisé comme standard. Le dosage de l'ADN se fait selon la méthode de Burton K. (97). De l'ADN de thymus de veau est utilisé comme standard.
20
VI FILTRATION SUR BIO-GEL P-100 DE LA FRACTION ACIDO SOLUBLE (As)
La technique de filtration sur Bio Gel P-100 utilisée est celle de Nadeau P. et colt. (98). Les dimensions de la colonne sont de 115 cm de
3 hauteur et 2.5 cm de diamètre ce qui fait un volume d'environ 565 cm . Le Bio Gel P-100 est mis en suspension dans le solvant de colonne (10 mM HCl, 0.02% p/v NaNg) pendant 24 heures puis coulé après avoir lavé et dégazé la suspension de gel. La colonne est équilibrée et l'élution se fait à 10 ml/h avec une pompe péristaltique. Le Vo, V et le Vi sont évalués respectivement à 1 35, 525 et 390 ml après avoir élué une solution de 5 ml de ferritine (mw: 440 KD) à 4 mg/ml dans le solvant d'élution de la colonne. Environ 17 mg des protéines de la fraction As dans 5.4 ml de solvant d'élution sont chromatographiés. La collection de l'éluat est faite à raison d'un ml/tube et un échantillonnage est réalisé à tous les cinq tubes afin de localiser les pics de matériels radioactifs. Les échantillons sont alors solubilisés dans 1 ml de solvant NCS. La radioactivité est déterminée par comptage à scintillation après avoir ajouté 10 ml de toluène contenant 0.5% P/v PPG et 0.05% P/v PO PO P. Les tubes d'un pic de matériel radio actif sont combinés puis lyophilisés.
Vil ÉLECTROPHORÈSE
A) Électrophorèse analytique sur gels SDS
La technique d'électrophorèse analytique sur gels SDS utilisé est celle de Thomas J.O. & Kornberg R.D. (99). Les fractions Np-3 sont ajustées à 1% V/v de bêta-mercaptoéthanol et 1% P/v de SDS. La dialyse des échantillons est nécessaire afin d'éviter d'éventuelles précipitations de matériel pendant l'électrophorèse et afin d'obtenir ainsi des conditions optimales de migration élec trophorétique. Les fractions sont dialysées pendant environ 15 h contre le tampon de dialyse I (tampon phosphate 10 mM pH 7.0-7.4, 5 M urée, 1% P/v SDS et l% V/v de bêta-mercaptoéthanol). Le tampon de dialyse l est changé pour le tampon de dialyse II
21
(tampon de dialyse I contenant 0.1% P/v SDS plutôt que 1% p/v) et la dialyse se poursuit pendant environ 10 h. Les fractions Np-3 sont concentrées contre du séphadex G-200 et dialysées pendant environ 15 h contre le tampon dialyse III (tampon de dialyse II constitué d'urée ultra pure). À chacune des fraction Np-3 il y a addition de 2 à 5 yl d'une solution de bleu de bromophénol (0.05% p/v) comme indicateur du front de migration lors de l'électro phorèse. Les fractions As lyophylisées sont remises en solution avec un volume de 40 à 50 yl de tampon d'échantillon SDS (tampon phosphate 10 mM pH 7.0-7.4, 5 M urée ultra pure, 0.1% P/v SDS et 1% V/v de bêta-mercaptoéthanol) auquel il y a aussi addition de 2 à 5 yl d'une solution de bleu de bromophénol (0.05% p/v). Un gel de séparation (11.7% p/v acrylamide, 0.3% P/v bis-acrylamide, 0.375 M tris, 0.1% pyv SDS, 0.025% P/v persulfate d'ammonium, 0.1% V/v TEMED, pH 8.8) de 10 cm de longeur et d'un diamètre de 0.55 cm est utilisé. Le gel de tamisage (4.38% P/v acrylamide, 0.12% P/v bis-acrylamide, 0.125 M tris, 0.1% P/v SDS, 0.03 P/v persulfate d'ammonium, 0.1% V/v TEMED, pH 6.8) est coulé sur le gel de séparation sur une hauteur de 1 cm. Le tampon d'électrophorèse I (192 mM glycine, 25 mM Tris et 0.1% P/v SDS) est maintenu à 18° C pour l'électrophorèse. Celle-ci est à I mA/gel jusqu'à ce que le front de migration pénètre dans le gel de séparation puis de 2 mA/gel jusqu'à ce que le bleu de bromophénol du front de migra tion commence à sortir du gel de séparation. Les gels sont placés dans une solution de fixation (50% V/v CH^OH et 10% V/v CH^COOH pendant environ 15 h. Les gels sont colorés pendant 2 h dans une solution de coomassie R-250 (0.25% P/v de bleu de coomassie R-250, 50% V/v CHgOH et 10% V/v CH„COOH). Les gels sont décolorés avec 20% v/v CH^OH et 7.5% /v CH^COOH jusqu'à ce que les bandes protéiques soient bien identifiables. Les gels sont photo graphiés et tranchés à tous les millimètres. Le matériel protéique des tranches est extrait pendant environ 15 h à 40° C dans 1 ml de solvant NCS contenant 10% V/v d'eau. La radioactivité est déter minée par comptage à scintillation dans 10 ml de toluène contenant 0.5% P/v P PO et 0.05% P/v POPOP.
22
B) Électrophorèse analytique sur gels acide-urée
La technique d'électrophorèse analytique sur gels acide-urée utilisée est celle de Panyim S. & Chalkley R. (100). Toutes les
fractions histoniques lyophilisées sont remises en solution avec 40 à 50 ni de tampon d'échantillon acide-urée (9.5 M urée ultra pure dans 0.9 N CH^COOH). On y ajoute 2 à 5 yl d'une solution de pyronnine (0.05% P/v de pyronnine, 9.5 M urée et 0.9 N CH^COOH) comme indicateur du front de migration. Un gel (10% P/v acrylamide, 0.1% P/v bis-acrylamide, 6.25 M urée, 5.4% V/v CH^COOH, 0.125%
P/v
persulfate d'ammonium, 0.5% v/v TEMED) de 10 cm de longeur et d'un diamètre de 0.55 cm est utilisé. Le tampon d'électrophorèse II (0.9 N CH^COOH) est maintenu à 18° C pour l'électrophorèse. Une pré-électrophorèse à 0.5 mA/gel pendant environ 15 h est nécessaire. L'électrophorèse est à 1 mA/gel jusqu'à ce que la pyronnine du front de migration sorte du gel. Les gels sont directement colorés pendant une heure dans 1% P/v "d'Amido black", 50% v/v CH^OH et 10% v/v CH^COOH. Les gels acide-urée sont par la suite traités de la même façon que les gels SDS.C) Electrophorèse préparative sur gels acide-urée
La technique d'électrophorèse préparative sur gels acide-urée utilisée est la même que celle de l'électrophorèse analytique sur gels acide-urée (100). Le système d'électrophorèse de gel préparatif utilisé est celui de la compagnie BRL (modèle 1100 PC). Les principales fractions de la filtration sur Bio Gel P-100 de la fraction As lyophilisées sont remises en solution dans 40 à 50 yl de tampon d'échantillon acide-urée (9.5 M urée ultra pure dans 0.9 N CHgCOOH). Le traitement des échantillons est identique à celui de l'électrophorèse analytique sur gels acide-urée. Le gel utilisé est le même que celui de l'électrophorèse analytique sur gels acide-urée. Seulement 7 cm de gel est utilisé dans le tube communiquant avec la chambre d'élution. Il y a pré-électrophorèse à 125 volts sans réfrigération pendant environ 5 h dans un tampon 0.9 N CH^COOH.
23
L'électrophorèse est réalisée dans les mêmes conditions que la pré-électrophorèse. L'élution de la chambre d'élution du système d'électrophorèse de gel préparatif est à un débit de 7 ml/h et la collection du matériel protéique à I ml/tube. L'échantillonnage est réalisé à tous les tubes afin de localiser les pics de matériels radioactifs qui auraient été séparés. Un volume de 100 -pi de chacun des tubes de l'élution de gel acide-urée préparatif est solubilisé dans un ml de solvant NCS. La radioactivité est déterminée par comptage à scintillation dans 10 ml de toluène contenant 0.5% ^/v PPO et 0.05% ^/v POPOP.
25
ISOLEMENT ET CONDITIONS DE CULTURE DES LYMPHOCYTES DE SANG DE PORC
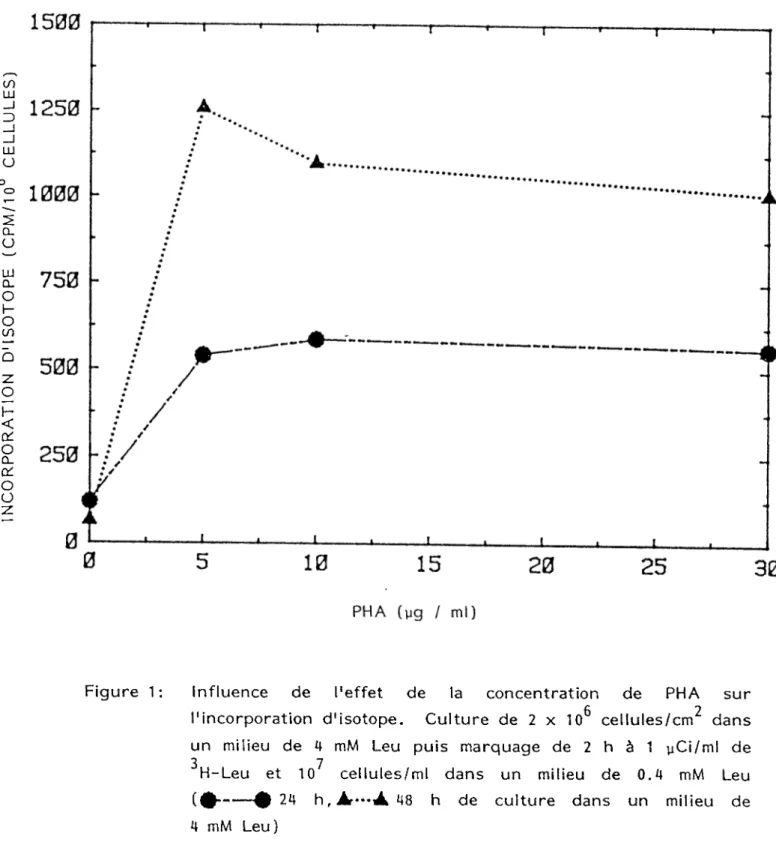
La mise au point des techniques d'isolement et de culture des lymphocytes de porc était prérequises pour l'enrichissement en quantité suffisante de As-1 et As-2. L'isolement des lymphocytes de sang de porc est généralement réalisé avec un rendement final supérieur à 65% et un pourcentage de polynucléaires ne variant qu'entre 5 et 10% avant la culture. La viabilité des lymphocytes de sang de porc stimulés ou non avec PHA après une journée ou deux de culture s'avère satisfaisantes. Cette viabilité cellulaire est de 92% avant la mise en culture à une concentration quelconque de 2 X 106 cellules/ml et avec un rapport cellules/surface de 3 X 10^ cellules/cm^. La viabilité cellulaire des lymphocytes de sang de porc non stimulés avec PHA est respectivement 75% et 65% après 24 heures et 48 heures de culture. La viabilité cellulaire des lymphocytes de sang de porc stimulés à une concentration quelconque de 10 yg PH A/ml est respectivement 81% et 74% après 24 heures et 48 heures de culture. L'isolement de lymphocytes de rate ou de ganglions de porc ont donné un rendement final et une viabilité cellulaire très inférieure à ceux obtenus à partir du sang de porc (résultats non-présentés ici car non-reproductibles). Le choix sJ avéra it donc le sang de porc comme source éventuelle de lymphocytes en grande quantité. Le maximum de stimulation des lymphocytes de porc par PHA Miles est obtenu avec une dose entre 5 et 10 yg de PHA/ml. PHA augmente de 5 à 6 fois la synthèse protéique des lymphocytes après 24 h de culture et de 10 à 12 fois après 48 h (figure 1). L'augmentation de la synthèse protéique va jusqu'à tripler après 12 h de cultures à des concentrations de 2, 5 et 10 x 10 cellules/ml (figure 2). La culture de 10 cellules/ml semble donc possible pendant de courte période de temps. La détermination de la concentration minimale de Leu qui n'interrompt pas la séquence des changements biochimiques déclenchée par le stimulus permet de diminuer la participation de Leu froide intracellulaire pendant le marquage radioactif avec dH-Leu et d'augmenter ainsi l'activité spécifique des protéines
26
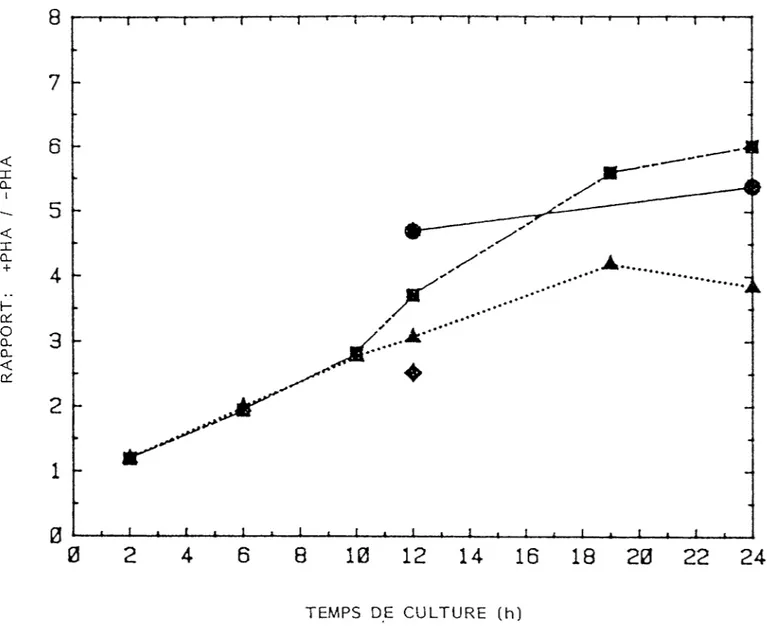
marquées. L'incorporation d'isotope chez les lymphocytes stimulés à PH A est sensiblement similaire pour des cultures en milieu 4 mM Leu et I mM Leu (figure 3). Le rapport + PHA/-PHA est beaucoup plus important pour des cultures en milieu 0.04 mM Leu, ce qui démontre un milieu déficient en Leu froide pour la culture optimale des lymphocytes. Le rapport de la synthèse protéique + PHA/-PH A en milieu de 4 mM Leu augmente linéairement jusqu'à 20 h et par la suite il commence à diminuer (figure 4). Dans un milieu de 1 mM Leu le rapport de la synthèse protéique + PHA/-PHA est comparable à celui d'un milieu de 4 mM jusqu'à la dixième heure mais il est subséquemment plus grand. Le marquage des cellules dans un milieu de 0.4 mM Leu après une culture dans un milieu de 4 mM
3
Leu donne un incorporation de H-Leu 2 fois plus grande qu'un marquage dans un milieu de 4 mM Leu (figure 4). La stimulation des cellules directement après isolement donne aussi des résultats comparables à ceux des cellules incubées pendant 12 h avant stimulation avec PH A (figure 4). Avec ces résultats (figure 1-4) il apparaît justifiable de faire des cultures de lymphocytes de porc pendant 12 h dans les conditions suivantes:
1) stimulation immédiatement après l'isolement avec 10 yg de PH A Mi les/ml,
2) concentration cellulaire à 10^ cellules/ml,
3) rapport cellules/surfaces de 5 x 10^ cellules/cm^, 4) culture dans un milieu de 1 mM Leu et
5) marquage radioactif pendant 2 h dans le même milieu avec 10 yCi/ml 3
IN
C
O
R
P
O
R
A
T
IO
N
D
'I
S
O
T
O
P
E
(C
P
M
/1
0
C E L L U L E S ) 271250
1000
PHA (yg / ml)Figure 1 : Influence de l'effet de la concentration de PHA sur l'incorporation d'isotope. Culture de 2 x 106 cellules/cm2 dans un milieu de 4 mM Leu puis marquage de 2 h à 1 yCi/ml de
3 7
H-Leu et 10 cellules/ml dans un milieu de 0.4 mM Leu
(
0
---0
24 h, Af-A 48 h de culture dans un milieu deIN
C
O
R
P
O
R
A
T
IO
N
D
'I
S
O
T
O
P
E
(C P M x 1 0 C E L L U L E S )Figure 2: influence de l'effet de la concentration cellulaire sur l'incorporation d'isotope. Culture pendant 12 h avec 10 yg PHA/ml et 5 X 106 cellules/cm2 dans un milieu de 4 mM Leu. Marquage de 2 h à 1 yCi/ml de 3H-Leu et 107 cellules/ml dans un milieu de 0.4 mM Leu ( culture sans PHA, 1111 culture avec PHA, Rapport +PH,Al/PHA.
IN
C
O
R
P
O
R
A
T
IO
N
D
'I
S
O
T
O
P
E
(DPM x 1 0 C E L L U L E S ) 29 0I . ■ * -I—rr.i 1 . 1 .T ~I l___l___ I___1___,___i___I___i___I—l—.—i—I—i___ ,___l0
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
TEMPS DE MARQUAGE (min)
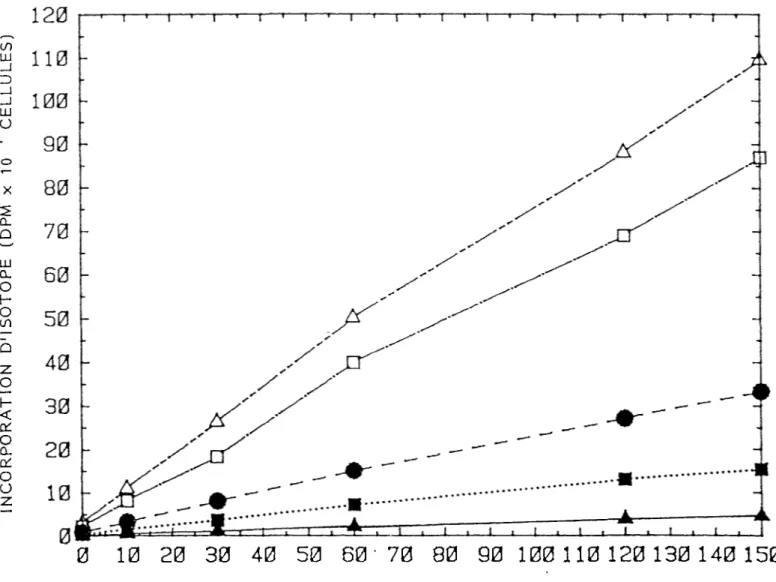
Figure 3: Influence de la concentration de leucine froide sur l'incorporation d'isotope. Culture pendant 16 h dans un milieu de 4 mM Leu avec 10 yg PHA/ml à 2 x 10^ cellules/ml et 5 x 10^
2 3
cellules/cm . Le marquage est à 2 y Ci/ml de H-Leu dans un milieu de:
À—: 4 mM Leu; *•••: 1 mM Leu; 0— : 0.4 mM Leu;
R
A
P
P
O
R
T
:
+ P H A /-P
H
A
3020
22
24
TEMPS DE CULTURE (h)Figure 4: Influence de la concentration de leucine froide sur le rapport d'incorporation de l'isotope chez des lymphocytes stimulés et non stimulés par PHA. Lymphocytes non-stimulés ou simulés avec 10 vg PHA/ml à 107 cellules/ml et 5 x 106 cellules/cm2. Le marquage se déroule pendant 2 h à 1 yCi/ml de 3H-Leu sans en changer le milieu de culture. A : milieu de culture de 4 mM Leu, Bh—: 1 mM Leu, @—: milieu de culture de 4 mM Leu puis marquage dans un milieu de 0.4 mM Leu, ^— : lymphocytes stimulés directement après isolation et cultivés dans un milieu de 4 mM Leu.
31
LE FRACTIONNEMENT SUB-CELLULAIRE
La radioactivité des fractions cytoplasmiques et des noyaux est respectivement de 20 et 80% de la radioactivité totale de l'homogénat cellulaire car le lymphocyte est justement une cellule contenant déjà peu de cytoplasme. La radioactivité des fractions Np-1 et Np-2 sont respectivement de 15% et 10% de la radioactivité totale de l'homogénat cellulaire. La radioactivité de la fractions Np-3 après extraction dans 0.1, 0.2 et 0.35 M NaCI est respectivement de 55, 60 et 190 DPM/yg ADN. La grande majorité de la radioactivité de l'homogénat cellulaire se retrouve dans la fraction As. L'électro phorèse des fractions nucléoplasmiques extraites dans 0.1, 0.2 et 0.35 M NaCI ainsi que la fraction acido-soluble permet d'identifier spécifiquement si As-1 et As-2 sont extraites avec une solution saline contenant entre 0.1 à 0.35 M de NaCI (figure 5 et 6). Les profils de radioactivité de ces gels se retrouvent aux figures 7 et 8. Le profil de radioactivité du gel SDS de la fraction As (figure 7) permet d'affirmer que le poids moléculaire de As-2 est de 21.5 KD. Sur les gels acide-urée, il apparaît que As-2 n'est pas extraite avec des tampons salins Np-3 de 0.1, 0.2 et 0.35 M NaCI (figure 8). On ne peut en dire autant sur Âs-1 car celle-ci a une activité spécifique trop basse et se retrouve en quantité trop faible pour être identifiée positivement sur les gels SDS et acide-urée.
32
1
2
3 4 5
Figure 5: Gels SDS des fractions Np-3 et As (coloration au bleu de coomassie)
1) Marqueur de P. M. (par ordre de poids moléculaire décroissant: albumine humaine, chaînes lourdes de gamma-globuline humaine, ovalbumine, chaînes légères de gamma-globuline humaine, ribonucléase de pancréas de boeuf et cytochrome C de coeur de cheval.
2) Fraction As 3) Fraction Np-3^ 1 M NaC|) 41 Fraction NP"3(0.2 M NaCl) 51 Fraction NP"3(0.35 M NaCI)
N.B.: POUR CETTE THÈSE, LA PLUPART DES GELS ONT ÉTÉ INTEN TIONNELLEMENT SURCHARGÉS POUR AVOIR SUFFISAMMENT DE COMPTE DE RADIOACTIVITÉ CAR LA PLUPART DES PRO TÉINES ONT UNE FAIBLE ACTIVITÉ SPÉCIFIQUE. C'EST CE QUI EXPLIQUE LA MOINS BONNE QUALITÉ DE CERTAINS GELS.
33
As-2 —►
1
2
3
#
#
H2A+H2B +H3-»
H 4 —*-
!
Figure 6: Gels acide-urée des fractions As (coloration à l'~ 1) Fraction As 2) Fraction As^ 1 3) Fraction As|Q 2 M NaC|) S) Fraction As(Q 35
Amido black" ). M NaCI )
R
A
D
IO
A
C
T
IV
IT
E
( % D P M 3H 43 117 H2a + H2b + H3 + H4 As-2 MIGRATION (mm)Figure 7: Profil de radioactivité d'un gel SDS de la fraction As (gel # 2 de la figure 5). La migration des protéines s'est faite de gauche à droite. La position des protéines étalons est indiquée à la partie supérieure de la figure. Le % DPM représente la radioactivité totale qui est récupérée du gel (36000 DPM)
ami'll iiiiai i 31,000 .1,
113 • 113a • MOI,
/ ^ ut
A.-?
COMME IIIIAI , jOOUU U|
113 « ... ..
MIGRATION ( mm ) MIGRATION ( mm )
•--- « CUMME IIIIAI 1.39000 tip
113 • 113a < ll.'l. MIGRATION ( mm ) CL a OP UJ I— > I— u
<
o Q êCOMI’ll' UllAt., 1.0000 tip.
MIGRATION ( mm )
Figure 8: Effet de la concentration en sel du tampon Np-3 sur l'extraction des protéines As-1 et As2. Profil de radioactivité des gels de la figure 6. A) Gel #1 (As), B) Gel #2 (AsQ^ M NaCI^
Gel #2 (ASq 2 ^ NaC|) (^0.35 ^ NaCI^" ^ migration des protéines s'est faite de gauche à droite. Le % DPM représente la radioactivité d'une tranche de gel comparativement à la radioactivité totale du gel.
36
ENRICHISSEMENT DE As-1 ET As-2
A) H1 + H MG
L'extraction de la fraction H1 + H MG se fait efficacement (figure 9) et les profils de radioactivité des gels acide-urée radioactifs confirment davantage l'efficacité d'extraction de Ht (figure 10). Ces profils de radioactivité permettent aussi d'affirmer que As-1 et As-2 ne sont pas des HMG car la plupart sinon toutes les HMG sont extraites avec 5% v/v PCA.
B) Filtration sur Bio Gel P-100 de la fraction As - (H1 + HMG)
La raison pour laquelle j'utilise une fraction As- (H1 + HMG) plutôt que As pour la filtration sur Bio Gel P-100 est qu'il y a une possibilité d'élution simultanée de H1 et As-2 à cause de leur poids moléculaire sensiblement voisin. Son profil d'élution sur Bio Gel P-100 se retrouve à la figure 11. Il y a jusqu'à 82% de récupération de la radioactivité du matériel après élution. J'anticipais cinq pics de matériels (As-2, As-1, H2a + H2b, H3 et H4) mais le profil d'élution obtenu possède plutôt trois pics majeurs de matériel. Une électrophorèse sur gels acide-urée des fractions 300, 320, 350, 380, 420, et 470 (Fractions 2 à 7 des figures 11 et 12) ainsi que les profils de radioactivité de ces fractions (figures 13, 14) permettent d'identifier le matériel de chacun des pics de ce profil d'élution. Il semble que l'élution sur Bio Gel P-100 de As-1 se fait avec H3
(figure 11, pic II d'élution) et que celle de As-2 se fait avec H2a + H2b (figure 11, pic I d'élution).
37 1 2
■i
As-1 —
As ~2
—*-H 2 A + —*-H 2 B + —*-
H3—*-H4 —
:
•
Figure 9: Gels acide-urée de la fraction H1 + H MG (2) et de la fraction As-(H1 + HMG) (1) après extraction avec 5% v/v RCA (colora tion à I1 "Amido black").
R
A
D
IO
A
C
T
IV
IT
É
(% DPM)RA
DI
OAC
T
IVI
T
É
(% D P M ) 38 COMPTE TOTAL: 13 00 0 10 20 30 40 50 60 70 80 90 100 MIGRATION ( mm ) COMPTE TOTAL: 9100 dp, MIGRATION ( mm )Figure 10: Profil de radioactivité des gels de la figure 9 A) Gel #2, B) Gel #1. La migration des protéines s'est faite de gauche à droite. Le % DPM représente la radioactivité d'une tranche de gel comparativement à la radioactivité totale du gel.
39
Afin de poursuivre la purification de As-1 et As-2 les fractions de chacun des 3 pics de matériel radioactif du profil d'élution (figure 11) sont combinées. Une électrophorèse de ces trois fractions (figure 15) ainsi que leurs profils de radioactivité (figure 16) permettent d'identifier As-1 et H3 avec la fraction 11, As-2 et H2a + H2b avec la fraction I et H4 avec la fraction III.
C) Electrophorèse préparative pour l'enrichissement de As-1 et As-2
Une électrophorèse préparative des trois fractions combinées du profil d'élution sur Bio Gel P-100 (figure 11) permet l'enrichissement finale de As-1 et As-2 et des histones H2a + H2b, H3 et H4 (figure 17). La récupération du matériel radioactif après l'électrophorèse préparative est supérieure à 90% ce qui en fait une technique d'isolement simple et efficace. De plus, les profils d'élutions des gels acide-urée d'électrophorèses préparatives sont comparables aux profils des gels acide-urée d'électrophorèses analytiques. Une électrophorèse analytique sur gel acide-urée des cinq pics de matériels radioactifs séparés par électrophorèse préparatives (figure 17) permet d'estimer la pureté du matériel des fractions combinées du profil d'élution sur Bio Gel P-100 (figure 18). Il y a ainsi enrichissement de H2a + H2b, H3, H4, As-1, et As-2.
R
A
D
IO
A
C
T
IV
IT
E
( D P M 40 + H2b 1001** FRACTIONSFigure 11: Profil d'élution de la fraction As - (H1 + HMG) sur Bio Gel 9
P-100 (18 mg de protéines provenant de 7 x 10 cellules ont été déposés sur la colonne). Les chiffres arabes du profil d'élution correspondent aux fractions disposées sur les gels acide-urée de la figure 12. Les chiffres romains du profil d'élution correspondent aux fractions combinées et disposées sur les gels acide-urée de la figure 15).
41
1 2 3 4 5 6 7
Figure 12: Gels acide-urée de certaines fractions de la figure 11 (coloration à l'"Amido black"). 1) Fraction As - (H1 + HMG) déposé sur la colonne de Bio Gel P-100, 2-7) Fractions (2,3,4,5,6,7) de la figure 11. (Ces gels acide-urée sont les fractions identifiées en chiffres arabes de la figure 11).
42 CL Q OP LU H > h-O
<
O a<
ai 5 CL a OP ai H>
V-U<
O Q<
Où CL û OP LU t>
H U<
O a<
al MIGRATION ( mm ) 35 30 25 15 A.-2 V COMI’Tf ÎUTAL. 4 «*00 H* h;\. • k>i> U 4 A 10 20 30 <0 50 OU VH HH 90 48 4 C 10 ?0 30 40 5U 041 .’H 00 90 MIGRATION ( mm )Figure 13: Profil de radioactivité des gels 1, 3 et 4 de la figure 12. A) Gel #1, B) Get #3, C) Gel #4. La migration des protéines s'est faite de gauche à droite. Le % DPM représente la radioactivité d'une tranche de gel comparativement à la radioactivité totale du gel.
- 43 -UJ o
<
o û<
q: uj t-> t-u<
O Q<
ex Z a. Q X UJ h-> t-L) < O û<
ex MIGRATION (""i 4B 10 20 30 40 511 fin 70 80 90 MIGRATION («h)Figure 14: Profil de radioactivité des gels 5, 6 et 7 de la figure 12. A) Gel #5, B) Gel #6, G) Gel #7. La migration des protéines s'est faite de gauche à droite. Le % DPM représente la radioactivité d'une tranche de gel comparativement à la radioactivité totale du gel.
44
S
td
. 1
2
3
H3 DIM.
As-1
—*-As ~2 ——
HI —
H2A+H2B +H3 ^
H 4
—*-Figure 15: Gels acide-urée des fractions combinées du profil d'élution sur Bio Gel P-100 (figure 11) (coloration à l'"Amido black"). Std) histone standard type ll-A de Sigma, 1) Fraction I, 2) Fraction II, 3) Fraction III. (Ces gels acide-urée sont les fractions identifiées en chiffre romains de la figure 11).
45 Z O K LU t-> U
<
O Û<
ce ï a. a & LU h-> U<
O Û<
ce X a. Q LU H > 1— U < O û<
£XUIMM K KIT AL. 4000 MIGRATION ( ) te ?a 30 40 su nu 70- eu 90 MIGRATION ( "" ) 14 13 . - -• COMPTE TOTAL. 260 0 DPM 12 T, 11 <i 10 h 9 ' 1 8 1 1 7 i » 1 l 6 . ' 1 /11 5 i> 1 i 4 i\ i ' 3 1, / \ i \1 2 1 V L * 1 AA ■ . » ■ 1- - ■ • 1 ■ » ■ ---‘-- -—‘ *■ 10 20 30 40 60 60 70 60 90 MIGRATION (mh)
Figure 16: Profil de radioactivité des gels de la figure 15. A) Gel #1, B) Gel #2, G) Gel #3. La migration des protéines s'est faite de gauche à droite. Le % DPM représente la radioactivité d'une tranche de gel comparativement à la radioactivité totale du gel.
46 2 Û. a ULI
>
H U<
O Q<
DC FRACTIONS CL Û LU H>
F-U<
O Q<
DC FRACTIONS CL Û LU>
H U<
O Û<
DC FRACTIONSFigure 17: Profil d'élution des électrophorèses préparatives. La direction de l'élution des gels préparatifs est de la gauche vers la droite. A) Fraction I, B) Fraction II, C) Fraction III.
H3
H2A+H2B
As-1
^—As-2
— HI
•^H2A+H2B+H3
H4
Figure 18: Gel acide-urée du matériel séparé par électrophorèse prépa rative. (Coloration au bleu de Coomassie) 1) H2a + H2b, 2) H3, 3) H4, 4) As-1, 5) As-2, 6) Histone standard de type 11—A de Sigma. Des quantités équivalentes d'échantillons ont été déposées sur les gels.
149
L'ISOLATION ET LES CONDITIONS OPTIMALES DE CULTURE DU LYMPHOCYTE DE PORC ET SON FRACTIONNEMENT SUB- CELLULAIRE
Les résultats démontrent que le porc est l'un des animaux de choix pour l'obtention d'une grande quantité de lymphocyte (4). Un meilleur rendement d'isolation lymphocytaire, une moins grande contamination cellulaire et une meilleure viabilité des lymphocytes du sang de porc expliquent la raison du délaissement des ganglions et de la rate de porc comme source éventuelle de matériel lymphocytaire (résultats non présentés car non-reproductibles). Néanmoins, le grand avantage à utiliser le sang de porc comme source lymphocytaire c'est que nous sommes moins limité par la quantité de matériel cellulaire. La culture des lymphocytes s'est faite dans R PMI 1640 10% sérum de veau foetal sans AA afin d'avoir une quantité assez bien déterminée de Leu froide dans le milieu de culture. Il est important de déterminer la concentration optimale de PH A pour stimuler les lymphocytes de porc afin d'obtenir le maximum d'incorporation isotopique. Une concentration de PH A inférieure ou supérieure à la concentration optimale de PH A diminue l'incorporation isotopique et ainsi l'activité spécifique des protéines qui nous intéresse. La concentration cellulaire peut aussi influencer cette incorporation isotopique mais ce ne fut pas le cas ici car les cultures se sont faites sur une courte période (12h) et sans que les cellules atteignent la période S du cycle cellulaire. Le temps de marquage ainsi que la concentration de Leu froide peut aussi influencer l'activité spécifique des protéines qui nous intéresse. Le fractionnement cellulaire des lymphocytes de porc après la mise au point des conditions optimales de culture et de marquage radioactif donne suffisamment de matériel radioactif pour des électrophorèses. La plupart de la radioactivité se retrouve dans la fraction noyau car le lymphocyte est une cellule qui contient déjà peu de cytoplasme (8). La fraction As extraite des noyaux et plus particulièrement la protéine As-2 sont très riches en radioactivité. En partant de l'hypothèse qu'As-2 est u-H2a ou A-24, cette incorporation de radioactivité serait possiblement conséquente du "turnover" important de u-H2a chez le lymphocyte en G1 (61). Le fractionnement sub-cellulaire
50
-utilisé dans ce travail n'est pas optimal pour l'enrichissement de As-1 et As-2 car il a plutôt été utilisé dans le but de caractériser d'autres protéines nucléaires d'un certain intérêt (résultats non-présentés). Néanmoins, l'enrichissement de As-1 et As2 a été réalisé. L'isolation des nucléosomes par digestion enzymatique partielle par des nucléases puis une extraction acide des protéines acido-solubles auraient permis l'isolation directe de As-1 et As-2 ainsi que des histones.
CARACTÉRISATION ET ENRICHISSEMENT DE As-1 ET As-2
Le poids moléculaire de As-2 est sensiblement voisin de celui de u-H2a. Il n'a pas été possible de déterminer celui de As-1 car celle-ci a une activité spécifique trop basse et/ou se retrouve en quantité trop faible pour être identifiée positivement. Néanmoins, je peux affirmer ici qu'As-1 et As-2 sont des protéines acido-solubles comme les histones car elles sont toutes deux extraites avec 0.4 N ^SO^. Je peux aussi affirmer qu'As-1 et As-2 ne sont pas des HMG ou des NHCP car elles ne sont pas extraites avec 0.35 M NaCI. Je peux donc affirmer qu'As-1 et As-2 possèdent une grande affinité pour la chromatine. As-1 est associé avec H3 dans toutes conditions mais les résultats démontrent qu'As-1 n'est pas un dimère de H3 (figure 15, gel #2) ni un produit de dégradation car en culture cellulaire l'incorporation de radioactivité chez As-1 est généralement supérieur à celle des histones (résultats non publiés de Waithe W. I. et coll. ). Avec toutes ces affirmations, il est possible qu'As-1 soit un variant de H3. Ceci expliquerait la raison de la présence de H3 dans toutes conditions mais afin de résoudre cette question il serait nécessaire de purifier assez de As-1 pour la caractériser davantage (poids moléculaire, composition en acides aminés, etc... Néanmoins, un protocole d'enrichissement de As-1 a maintenant été établi dans cette thèse. Pour As-2 il s'agirait possiblement de u-H2a car As-2 semble avoir le même poids moléculaire et le même com portement que A-24 isolé antérieurement par Busch H. et coll (72). La séquence en acide aminé de As-2 n'a pas pu être déterminé mais d'autres propriétés de As-2 laissent croire qu'il s'agirait possiblement de u-H2a. La technique d'enrichissement de As-2 est sensiblement la même que celle qu'a utilisé l'équipe de Busch H. et coll. dans son isolation de u-H2a. La
51
mobilité de As-2 sur gel acide-urée et sur gel acide-urée triton (résultats non-publiés de Waithe W.l. et coll. ), son absence d'incorporation de Try radioactif (résultats non-publiés de Waithe W.l. et coll.), son taux de "turnover" (résultats non publiés de Waithe W.l. et coll.) et son patron d'élution sur Bio-Gel P-100 démontrent tous qu'il s'agirait fortement de u-H2a.
RÔLE POSSIBLE DE u-H2a CHEZ LE LYMPHOCYTE
La modification de H2a dans le nucléosome joue un rôle important dans la structure superhélice de la chromatine. L'extrémité carboxylique de H2a de même que l'ubiquitine de u-H2a sont situées à la partie externe du "core" nucléosome et cela permet des interactions avec les fibres de chromatine (80, 84, 85, 89). L'addition d'ubiquitine à H2a ou son retrait de u-H2a peut affecter la conformation de la chromatine (80, 90). Avec ce qui est connu de u-H2a ainsi que de la structure chromatinienne, certaines fonctions de la formation ou du retrait de u-H2a dans le nucléosome peuvent être envisagées (80, 88-90). L'ubiquitination de H2a pourrait être un signal pour la fermeture de certains sites spécifiques de gênes potentiellement actifs. u-H2a peut servir d'inhibiteur d'activité dans les gènes par une interaction internucléosomale avec l'ADN "linker" ou elle pourrait maintenir la chromatine inactive en accroissant l'enroulement de la superhélice des fibres de chromatine. La nature des changements dans la quantité de u-H2a de la chromatine de cellules activés ou quiescentes n'est pas encore claire de même que la distribution de u-H2 dans les nucléosomes des fibres de chromatine. La formation ou le retrait de u-H2a est peut être associé à une activité transcriptionnel et/ou à un changement dans la configuration chromatinienne pendant l'activation de croissance. Cette étude des protéines acido-solubles du lymphocyte de sang de porc activé par PHA a donc permis d'élaborer de nouvelles étapes dans l'étude du rôle de ces protéines dans l'activation lymphocytaire. Il serait souhaitable à
l'avenir de: