Accumulative Charge Separation Inspired by Photosynthesis
Texte intégral
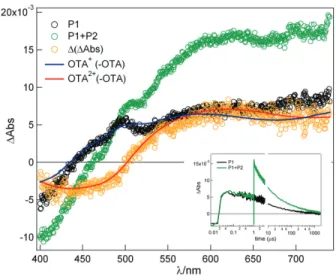
Figure

Documents relatifs
A control problem was introduced first in [25] for a Penrose−Fife type model with Robin-type boundary conditions for the temperature and a heat flux proportional to the gradient of
These results were consis- tent with the existence on the low temperature state Fermi surface (T < Tp2 of small electron and hole pockets with a size of the order of lo~~ the
However, Johnson et a1./9/ showed that if the individual excitations are uniformlv distributed along the track at the time of decav and acted at their point of decav then the
The mechanism of d-electron transfer suggested to partly account for the change in hyperfine field is supported by the observed concentration dependence of
It appears that the weak amplitude rising component of the NDI-centered fluorescence can be ascribed to an OPE-LES f NDI-LES transition, most probably through energy transfer, with
FIG. Schematic level structure corresponding to various combination modes. Explanation as in Fig.. in each triplet is based on energy only and the numbering of the triplets
The variation of the width as a function of Z is in good agreement with a model derived from the analysis of the 1/Z perturbation series, suggesting that the state acquires a
9.In 16---, the Pilgrim fathers , the Puritans established the
