La caractérisation de deux nouvelles mutations du gène
SCN5A liées au syndrome de QT long révèle une
déstabilisation de l’état inacti
vé de Nav1.5
Mémoire
Quentin Plumereau
Maîtrise en sciences cliniques et biomédicales - avec mémoire
Maître ès sciences (M. Sc.)
La caractérisation de deux nouvelles mutations du gène SCN5A liées
au syndrome du QT long révèle une déstabilisation de l’état inactivé
de Nav1.5
Mémoire de Maîtrise en Sciences Cliniques
et Biomédicales
Quentin Plumereau
Faculté de Médecine
Université Laval
Sous la direction de : Professeur Mohamed Chahine
Résumé
Les canaux sodiques sont des protéines membranaires d’une importance primordiale. Ils sont impliqués dans le déclenchement des potentiels d’action et sont nécessaires à l’excitabilité des cellules. Ils participent à la contraction des muscles striés squelettiques, cardiaques et lisses. Des dysfonctionnements de ces canaux, dûs à des mutations, peuvent engendrer de graves pathologies et atteintes cardiaques telles que le syndrome du QT long, le syndrome de Brugada, ou la cardiomyopathie dilatée.
L’étude de ces mutations constitue un enjeu essentiel dans la compréhension des mécanismes de ces canaux et du développement des pathologies associées. Plus les connaissances avancent et plus il devient alors possible d’envisager des études précises sur le développement des traitements appropriés.
Cette étude se concentre sur l’analyse de deux nouvelles mutations du gène SCN5A codant pour le canal Nav1.5, et a permis de mettre en évidence les caractéristiques biophysiques dysfonctionnelles de ces mutants et de confirmer le diagnostic médical.
Abstract
Sodium channels are important transmembrane proteins. They are involved in the upstroke of action potential and they are necessary for the cardiac cell excitability. Their dysfunctions caused by mutations can lead to serious pathologies and cardiac impairments such as long QT syndrome, Brugada syndrome, dilated cardiomyopathy and even sudden death.
The study of these mutations is a key step toward the comprehension of the pathophysiological mechanisms involved. The knowledge progress allows to lead accurate studies on drug development. This study focused on the analysis of two novel SCN5A mutations and allowed to highlight the dysfunctional biophysic characteristics of these mutants and to confirm the medical diagnosis.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... vi
Liste des tableaux ... vii
Liste des abréviations ... viii
Remerciements ... ix
Avant-propos ... x
Introduction ... 1
Physiologie et fonctionnement cardiaque ... 1
Anatomie du cœur ... 1
Valves cardiaques ... 3
Tissu conducteur ... 4
Cardiomyocytes ... 6
Les cardiomyocytes ventriculaires ... 6
Électrocardiogramme et potentiel d’action ... 7
Patch Clamp ... 10
Canaux sodiques ... 11
Les canaux sodiques voltages-dépendants ... 11
Syndrome du QT long ... 20
Historique ... 20
Prévalence ... 21
Étiologie ... 21
Physiopathologie et manifestations cliniques ... 23
Prise en charge ... 24
Sujet d’étude ... 24
Hypothèses et objectifs ... 25
Chapitre 1 : Novel G1481V and Q1491H SCN5A mutations linked to long QT syndrome destabilize the Nav1.5 inactivation state ... 26
1.1 Résumé ... 26
Novel G1481V and Q1491H SCN5A mutations linked to long QT syndrome destabilize the Nav1.5 inactivation state ... 28 1.3 Abbreviations ... 29 1.5 Introduction: ... 31 1.6 Methods ... 32 1.7 Results ... 34 1.8 Discussion ... 36 1.9 Conclusion ... 39 1.10 References ... 41 Discussion et Conclusion ... 55 Perspectives ... 57 Bibliographie ... 58
Liste des figures
Introduction
Figure 1 : Schéma d’une coupe d’un cœur………...………1
Figure 2 : Schéma de la circulation sanguine dans le corps………2
Figure 3 : Schémas du réseau des artères et veines coronaires…...………..…………..3
Figure 4 : Schéma d’une coupe de l’oreillette gauche et du ventricule gauche, mettant en évidence les structures anatomiques des valvules, cordages et muscles papillaires………..………..4
Figure 5 : Schéma représentatif du tissu conducteur………...………..5
Figure 6 : Cardiomyocyte ventriculaire.………..………...………7
Figure 7 : Représentation graphique d’un ECG normal……….……….8
Figure 8 : Représentation d’un potentiel d’action ventriculaire……….….……….9
Figure 9 : Schéma représentant les 3 étapes de la configuration cellule entière……….10
Figure 10 : Schéma des différents états de canaux voltage-dépendants……….14
Figuré 11 : Portes d’activation et d’inactivation rapides………....15
Figure 12 : Structure cristalline par Cryo-EM de rNav1.5C……….………17
Figure 13 : La structure cristalline Cryo-EM du canal rNav1.5C et le complexe de la flécaïnide…….…19
Novel G1481V and Q1491H SCN5A mutations linked to long QT syndrome destabilize the Nav1.5 inactivation state Figure 1 : Standard ECG recorded soon after birth………43
Figure 2 : Analysis of whole-cell Na+ currents recorded from HEK 293 cells expressing Nav1.5 WT, Q1491H, and G1481V………44
Figure 3 : Gating properties of steady-state activation and inactivation and window currents………….45
Figure 4 : The gating properties of slow inactivation, recovery from slow inactivation, and closed-state inactivation………..46
Figure 5 : Frequency dependence of WT, Q1491H, and G1481V………48
Figure 6 : Persistent Na+ currents in WT, Q1491H, and G1481V inhibited by TTX………...……49
Figure 8 : Open state structure of the human Nav1.5 sodium channel………51
Liste des tableaux
Introduction
Table 1 : Liste des différents membres de la famille des Nav ainsi que de leur localisation………..13
Table 2 : Critères de diagnostic du LQTS………..23
Novel G1481V and Q1491H SCN5A mutations linked to long QT syndrome destabilize the Nav1.5 inactivation state
Table 1 : Biophysical properties of the Nav1.5 WT and mutant channels………...52
Supplement Table S1 : List of cardiomyopathy (CM) and arrhythmia syndrome (AS) causative genes tested………54
Liste des abréviations
D : Domaine transmembranaire E : GlutamateECG : Électrocardiogramme
EGFP : Enhance Green Fluorescence Protein G : Glycine
H : Histidine
HCS : Site de restriction hydrophobe HEK : Human Embryonic Kidney
IFM : Isoleucine-Phénylalanine-Méthionine LQTS : Syndrome du QT long
Na+ : Ion sodium
Nav : Canaux sodiques voltage-dépendants
NCX : Échangeur Na+/Ca2+
PA : Potentiel d’action
QTc : Intervalle QT corrigé selon la fréquence cardiaque RS : Réticulum sarcoplasmique
RyR : Récepteur à la ryanodine S : Segment transmembranaire
SERCA : Sarco/endoplasmic reticulum Ca2+- ATPase
SF : Filtre sélectif TTX : Tétrodotoxine V : Valine
Remerciements
Je tiens à remercier le Professeur Mohamed Chahine pour m’avoir offert l’opportunité de faire une maîtrise au sein de son laboratoire. Il m’a accordé sa confiance, a su me motiver et a été à l’écoute dans les moments difficiles. Ça a été un immense plaisir que de travailler sous sa supervision et de l’avoir comme mentor lors de cette maîtrise. Je tiens également à remercier Olivier Thériault qui m’a aidé à la mise en place de mes expériences et à l’analyse de mes résultats. Sans lui, je me serais heurté à de nombreux problèmes et il a su avoir la patience de m’expliquer comment résoudre ces problèmes afin que je sois capable de m’en sortir seul. Mes remerciements d’adressent aussi à Valérie Pouliot pour les constructions des mutants qu’elle a réalisés et à Hugo Poulin pour l’aide et les conseils qu’il m’a apportés. Leur bonne humeur et leur disponibilité ont contribué grandement à ce que ma maîtrise se déroule de façon idéale. Enfin, mes remerciements s’adressent également à Marion Pierre et Mohammed Djemai, mes deux camarades étudiants avec qui j’ai partagé de bons moments, des fous rires, de l’entraide et qui ont su être là dans les bons comme les mauvais moments. Je ne serais pas là où j’en suis sans l’aide de tous les membres de ce laboratoire.
Avant-propos
Ce mémoire contient l’article ayant été rédigé au cours de la maîtrise. Il s’intitule Novel G1481V and Q1491H SCN5A mutations linked to long QT syndrome destabilize the Nav1.5 inactivation state (Q.
Plumereau, O. Thériault, V. Pouliot, A. Moreau, E. Morel, V. Fressart, I. Denjoy, A. Delinière, F. Bessière, P. Chevalier, TM. Gamal El-Din et M. Chahine) sous presse dans Canadian Journal of Cardiology et constitue le chapitre 1. Mon rôle dans cet article a été de réaliser les expériences, d’analyser et d’interpréter les résultats. J’ai également réalisé les figures et participé à la rédaction de l’article.
Cette étude a été réalisée au Centre de recherche CERVO, dans l’équipe du Professeur Mohamed Chahine. Cette équipe se concentre sur l’étude de la structure et des fonctions des canaux sodiques dans le système nerveux et dans le cœur afin d’identifier de nouvelles cibles thérapeutiques. L’étude de nouvelles mutations de gènes codant pour des canaux sodiques entre donc dans la thématique de ce laboratoire. En effet, les nombreux contacts du Professeur Mohamed Chahine à travers le monde, permettent au laboratoire de bénéficier de nombreuses collaborations et de pouvoir être en première ligne sur l’étude des canaux sodiques.
Introduction
Physiologie et fonctionnement cardiaque
Anatomie du cœur
Le cœur est un organe fibro-musculaire (muscle strié) d’environ 250 g qui a pour rôle d’assurer la circulation sanguine, une activité vitale dans le fonctionnement du corps humain. Il est situé dans la cage thoracique (2/3 de son volume étant du côté gauche), dans une cavité péricardique et il est entouré par les poumons, le diaphragme et l’œsophage. Le myocarde, tissu musculaire du cœur, est dans un sac à double paroi, le péricarde, constitué d’une membrane fibreuse (paroi externe et rattachée aux autres organes) et d’une membrane séreuse (double feuillet coulissant l’un contre l’autre). Le cœur permet de faire circuler le sang, chargé en oxygène et nutriments, de façon à pouvoir alimenter tous les organes et tissus. Le cœur fonctionne comme une pompe, faisant circuler le sang dans un sens unique, grâce à des structures bien particulières.
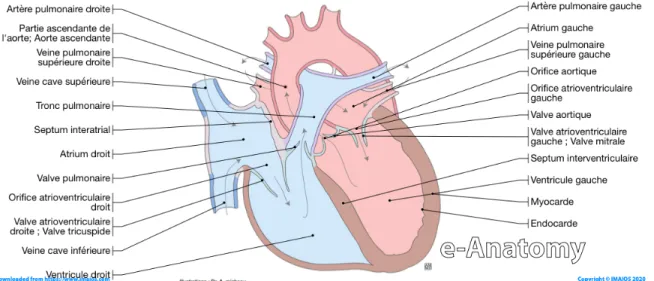
Figure 1 : Schéma d’une coupe d’un cœur. La partie sur la gauche représente les veines caves, l’oreillette droite, le ventricule droit et les veines pulmonaires. La partie de droite représente l’artère pulmonaire, l’oreillette gauche, le ventricule gauche et l’aorte. Le bleu indique le sang pauvre en oxygène et la partie rouge, le sang riche en oxygène. Micheau A, Hoa D, eAnatomy, www.imaios.com, DOI: 10.37019/e-anatomy
Le myocarde est un muscle creux, composé de 4 compartiments : deux oreillettes (atrium), séparées par le septum interauriculaire et deux ventricules, séparés par le septum interventriculaire, dont la
partie interne est recouverte par l’endocarde (épithélium monocouche en contact avec le sang). Le cœur a un rythme au repos de 60 à 80 battements par minute, soit 100 000 battements par jour et se contracte plus de 3 milliards de fois au cours d’une vie. Un cycle de battement cardiaque commence lorsque l’oreillette droite se remplit de sang pauvre en oxygène lors de sa diastole (décontraction) via les veines caves supérieure et inférieure (Figure 1) mais également via le sinus coronaire (Figure 3). Lors de sa systole (contraction), le sang va passer via l’orifice tricuspidien et venir remplir le ventricule droit qui est en phase diastolique. Le ventricule droit va ensuite se contracter pour expulser le sang vers le tronc pulmonaire qui va se séparer en artères pulmonaires droite et gauche. Le sang expulsé ne peut revenir dans l’oreillette droite grâce à la valve tricuspide qui va venir obstruer l’orifice tricuspidien lors de la systole ventriculaire. Le sang alors chargé en oxygène après son passage dans les capillaires pulmonaires, arrive dans l’oreillette gauche. Celle-ci, en se contractant, va expulser le sang vers le ventricule gauche, qui à son tour va expulser le sang vers l’aorte afin de pouvoir alimenter l’ensemble du corps en nutriments et oxygène (Figure 2).
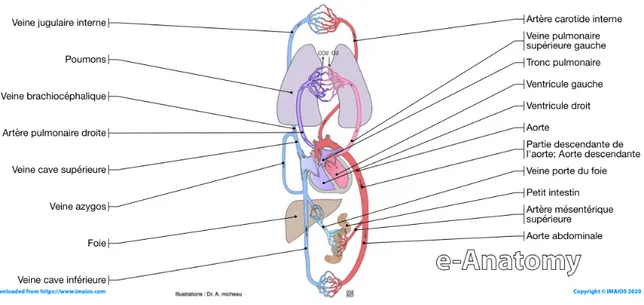
Figure 2 : Schéma de la circulation sanguine dans le corps. Le coté bleu correspond au sang pauvre en oxygène et le coté rouge correspond au sang riche en oxygène. Micheau A, Hoa D, eAnatomy, www.imaios.com, DOI: 10.37019/e-anatomy
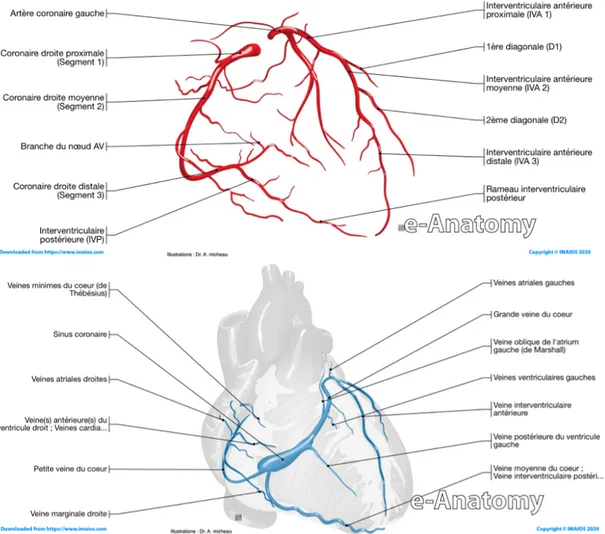
Figure 3 : Schémas du réseau des artères coronaires haut en rouge) et des veines coronaires (en-bas en bleu). Micheau A, Hoa D, eAnatomy, www.imaios.com, DOI: 10.37019/e-anatomy
Le sang irrigue également le cœur via les artères coronaires qui prennent leur source au niveau de l’aorte, juste après la valve aortique. Les artères principales gauches et droites se subdivisent en rameaux qui vascularisent les ventricules et forment une couronne (figure 3).
Valves cardiaques
Les valves cardiaques sont des structures qui séparent les cavités du cœur en évitant le reflux du sang pour qu’il ne circule pas dans le mauvais sens. Il y a 4 valves principales : la valve mitrale, la valve tricuspide, la valve pulmonaire et la valve aortique.
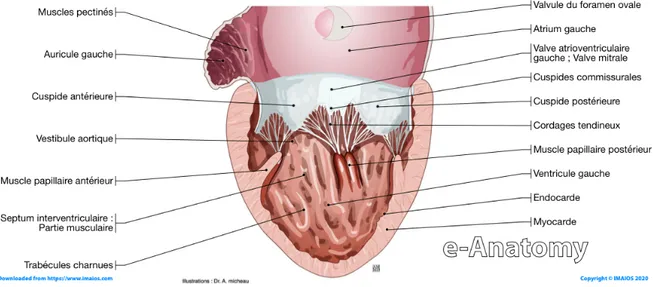
Figure 4 : Schéma d’une coupe de l’oreillette gauche et du ventricule gauche mettant en évidence les structures anatomiques des valvules, cordages et muscles papillaires. Micheau A, Hoa D, eAnatomy, www.imaios.com, DOI: 10.37019/e-anatomy
Elles sont toutes rattachées aux ventricules par des cordages (tendons) et des piliers (muscles papillaires) qui aident à éviter le reflux sanguin (Figure 4). La valve tricuspide est composée de 3 feuillets (valvules) et sépare l’oreillette droite du ventricule droit. La valve pulmonaire est composée de 3 valvules et sépare le ventricule droit de l’artère pulmonaire. La valve mitrale est composée de 2 valvules et sépare l’oreillette gauche du ventricule gauche. Enfin la valve aortique est composée de 3 valvules et sépare le ventricule gauche de l’aorte.
Tissu conducteur
L’activité contractile automatique du cœur est due à la présence du tissu cardionecteur ou tissu nodal, qui est composé de 2 nœuds (nœud sinusal et nœud auriculoventriculaire) et d’un filament qui se ramifie dans les ventricules (Figure 5). Cet automatisme est spécifique au tissu nodal cardiaque et permet au cœur de se contracter de façon rythmique sans l’influence des autres organes, y compris du système nerveux. Il peut par exemple continuer à battre hors du corps humain (ou de celui de tout autre mammifère) pendant plusieurs heures, s’il est perfusé. Il est cependant régulé par le système nerveux sympathique et parasympathique. Le système nerveux sympathique va permettre l’accélération du rythme cardiaque (augmentation de la fréquence cardiaque) grâce à la libération d’un agent tachycardisant (adrénaline) alors que le système nerveux parasympathique va faire baisser la fréquence cardiaque grâce à la libération d’un agent bradycardisant (acétylcholine).
Figure 5 : Schéma représentatif du tissu conducteur. Micheau A, Hoa D, eAnatomy, www.imaios.com, DOI: 10.37019/e-anatomy
Les nœuds sinusal et auriculoventriculaire sont des amas cellulaires indiscernables à l’œil nu. Le premier nœud est le nœud sinusal (nœud de Keith et Flack) qui se situe au niveau de l’oreillette droite, entre les deux embouchures des veines caves. Il génère l’impulsion permettant au cœur de battre à 90 battements par minute. Ceci est possible grâce aux cellules autorythmiques qui produisent des potentiels d’action (PA). Cependant, le système nerveux parasympathique permet de réguler la fréquence à 70-80 battements par minute, notamment grâce au nerf vague qui libère de l’acétylcholine. Elle peut également être accélérée par le système nerveux sympathique par libération d’adrénaline et de noradrénaline. Lors de la dépolarisation du PA, l’influx électrique est transmis de proche en proche dans les oreillettes avant d’arriver au nœud auriculoventriculaire (nœud d’Aschoff-Tawara) situé dans l’oreillette droite, proche de la valve tricuspide. Ce nœud transmet alors la dépolarisation au faisceau de His situé dans le septum interventriculaire, se séparant en 2 branches et se terminant en fibres de Purkinje, situées au niveau interne des ventricules, sous l’endocarde.
Afin de permettre une contraction optimale des ventricules, le nœud auriculoventriculaire impose un retard de conduction de 40 ms pour que le sang soit éjecté efficacement des oreillettes, procurant du même coup un temps de remplissage ventriculaire adéquat, permettant par la suite une contraction des ventricules et une éjection du sang ventriculaire tout aussi efficaces.
Cardiomyocytes
Le myocarde est un muscle strié. La contraction de ce muscle est assurée par les cellules musculaires appelées cardiomyocytes. Ces cellules mesurent 120 𝜇m de long et de 20 à 30 𝜇m de diamètre chez l’adulte et possèdent un ou deux noyaux, de nombreuses mitochondries et des myofibrilles disposées de façon linéaire. Ce tissu se différencie des muscles lisses de par sa structure, car les cardiomyocytes sont reliés entre eux par des disques intercalaires qui contiennent des desmosomes (zone de jonction cellule-cellule) et des jonctions communicantes (jonction gap) qui permettent la propagation du PA. Il existe 3 types de cardiomyocytes. Les cardiomyocytes contractiles permettent la contraction du myocarde, les cellules cardionectrices permettent l’initiation et la conduction du PA et les cellules myoendocrines localisées au niveau des oreillettes permettent de diminuer la pression artérielle par libération du facteur natriurétique auriculaire qui joue un rôle sur les concentrations de sodium, de potassium et sur l’excrétion de l’eau via la fonction rénale.
Les cardiomyocytes ventriculaires
Au repos, les cardiomyocytes ventriculaires ont un potentiel de repos transmembranaire de -90 mV. La dépolarisation de la membrane constitue la première étape de la contraction. Cette dépolarisation est due à l’entrée d’ions via les connexines, qui permet l’ouverture des canaux sodiques voltage-dépendants. L’entrée massive d’ions sodium (Na+) au sein de la cellule engendre une dépolarisation
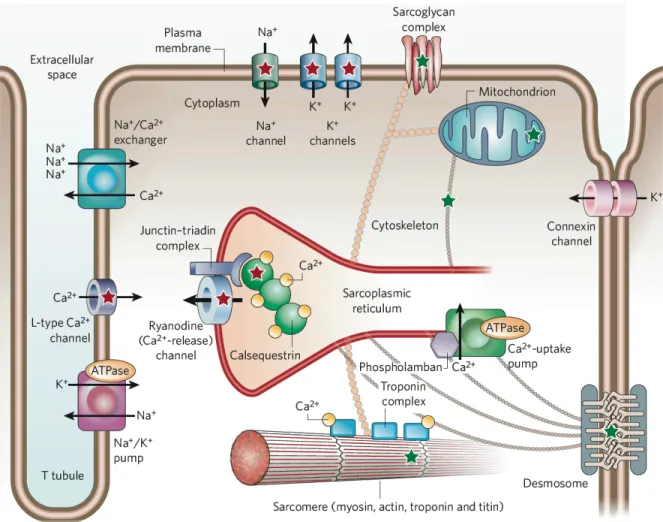
rapide de la membrane, le déclenchement du PA et ultimement l’inactivation des canaux sodiques. Le PA se propage le long de la membrane jusqu’aux tubules T d’une part, en activant successivement les canaux calciques et potassiques. L’augmentation du calcium intracellulaire permet l’activation des récepteurs à la ryanodine (RyR) présents sur le réticulum sarcoplasmique (RS). L’activation des RyR permet le relargage massif du calcium présent dans la lumière du RS, qui vient alors se lier à la troponine-C (Figure 6). Cette protéine inhibe habituellement l’interaction entre l’actine et la myosine, mais en présence de calcium, elle change de conformation et permet aux têtes de myosine de se lier aux filaments d’actine, entraînant le raccourcissement du sarcomère. Ceci représentant la contraction musculaire.
Figure 6 : Cardiomyocyte ventriculaire. Les structures et les protéines impliquées dans le couplage excitation-contraction sont illustrées. Le déplacement des ions via des canaux ioniques et des transporteurs, au niveau de la membrane plasmique et du réticulum sarcoplasmique fait suite à la dépolarisation membranaire et permet la contraction musculaire1.
La relaxation suit la contraction. Elle résulte du restockage du calcium dans le RS grâce à la pompe SERCA (sarco/endoplasmic reticulum Ca2+- ATPase). La troponine-C libre inhibe alors l’interaction
actine-myosine.
Électrocardiogramme et potentiel d’action
Les PA sont des variations du potentiel électrique d’une cellule en fonction du temps. L’activité électrique du cœur peut être mesurée à la surface du corps à l’aide d’un électrocardiogramme (ECG). Le premier ECG humain publié date de 1887 et a été réalisé par Augustus D. Waller.
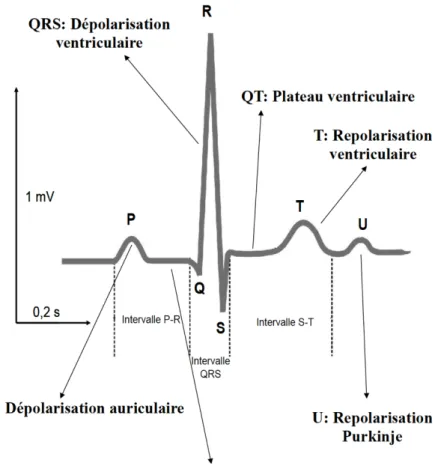
Figure 7 : Représentation graphique d’un ECG normal. Tiré du Pr. Jean-Yves Le Guennec, Inserm U1046 - CNRS UMR 9214 UMontpellier, France.
C’est en 1906 que les premiers ECG pathologiques sont alors mis en évidence. Il faut attendre 1942 pour que l’ECG à 12 dérivations, toujours utilisé de nos jours, soit mis au point par Emanuel Goldberger2. Les 12 dérivations correspondent à 12 enregistrements, 6 dérivations frontales et 6
dérivations précordiales. Ces enregistrements sont obtenus grâce à des électrodes présentes à la surface de la peau (bras, jambe gauche et torse). Cet examen est interprété par un cardiologue et permet de déceler les anomalies électriques cardiaques.
Sur un ECG, un cycle cardiaque (contraction et relaxation) correspond à un ensemble de 6 déflexions P, Q, R, S, T et U. L’onde P correspond à la dépolarisation (et contraction) des oreillettes. Le segment QRS correspond à la dépolarisation (et contraction des ventricules), l’onde T correspond à la repolarisation (relaxation) des ventricules et l’onde U correspond à la repolarisation des fibres de Purkinje (Figure 7). L’intervalle QT d’un ECG est donc l’expression (en fonction du temps) de la
contraction et de la relaxation ventriculaire. Un intervalle QT raccourcit lorsque la fréquence cardiaque augmente et augmente lorsqu’elle ralentit. Plusieurs formules peuvent alors être utilisées afin de déterminer le QT corrigé (QTc) par la fréquence ; la plus commune étant la formule de Bazett : QTc = QT/Racine carrée de l’intervalle RR. L’intervalle RR, représentant en fait la durée d’un cycle cardiaque complet, permet donc de déterminer la fréquence cardiaque.
Le PA ventriculaire peut également être enregistré. Il traduit l’expression de l’intervalle QT au niveau cellulaire et est composé de 5 phases.
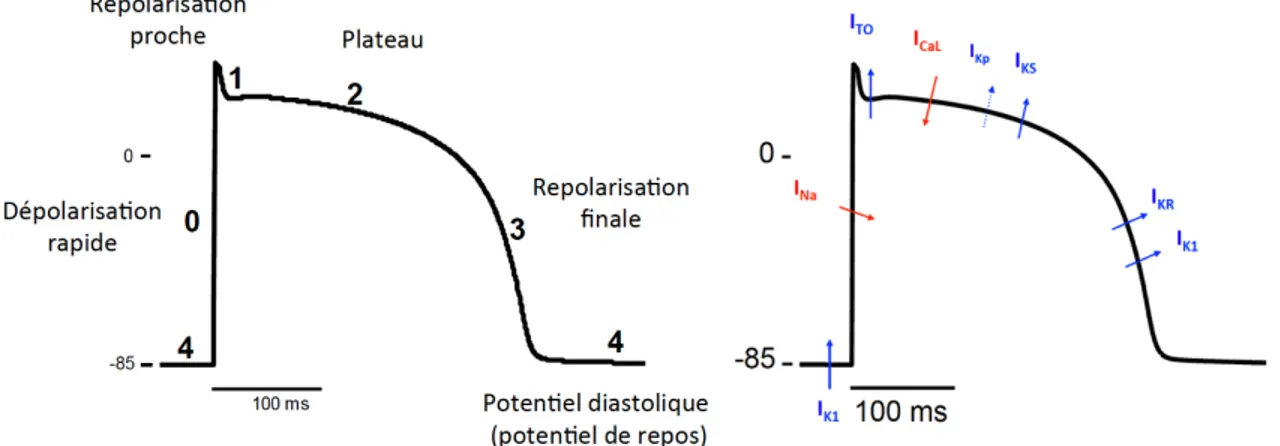
Figure 8 : Représentation d’un potentiel d’action ventriculaire. À gauche, le potentiel d’action ventriculaire présentant les 5 phases (0 à 4) de l’ensemble contraction et relaxation d’un cardiomyocyte ventriculaire. À droite, le potentiel d’action ventriculaire présentant l’activation des différents canaux aux différentes phases. Tiré du Pr. Jean-Yves Le Guennec, Inserm U1046 - CNRS UMR 9214 UMontpellier, France
La phase 0 correspond à la dépolarisation de la cellule par entrée massive de sodium au sein de la cellule. Une légère repolarisation se produit ensuite due à l’inactivation rapide des canaux sodiques voltage-dépendants et à l’activation d’un courant potassique transitoire Ito. C’est la phase 1
(repolarisation proche). L’activation concomitante des canaux calciques (entrants) et potassiques (sortants) permet ensuite de maintenir le voltage transmembranaire relativement constant pendant un certain temps, c’est la phase 2 (plateau). L’inactivation progressive des canaux calciques et le maintien de l’activation des canaux potassiques, en particulier les canaux du courant IK1, entraînent finalement la repolarisation du PA. La phase 4 est la phase finale et correspond au retour au potentiel de repos (Figure 8). Le potentiel d’action comprend une période réfractaire pendant laquelle un deuxième stimulus ne parviendrait pas à déclencher un second potentiel d’action.
Patch Clamp
La technique de patch clamp permet d’enregistrer le courant ionique transitant au travers de la membrane des cellules. La technique utilisée de nos jours a été mise au point à la fin 1970 par Erwin Neher et Bert Sakmann et ils ont obtenu le prix Nobel de médecine en 1991.
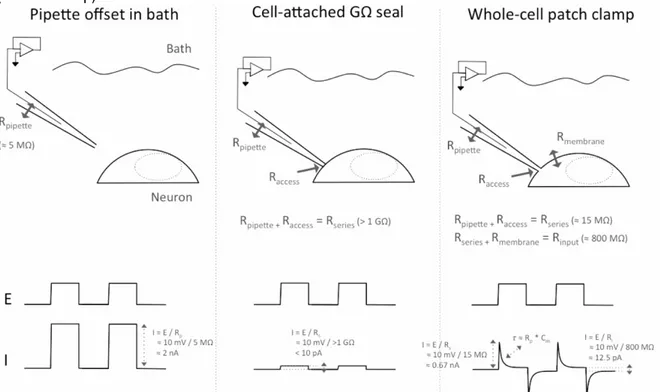
Les courants peuvent être enregistrés grâce à une pipette de verre remplie d’une solution ionique, dont la concentration varie suivant les types cellulaires ou les canaux d’intérêt. Cette pipette de verre doit être accolée à la membrane de la cellule, permettant ainsi d’enregistrer le courant d’un canal unique (configuration cellule attachée). Il existe d’autres configurations comme la configuration en cellule entière (Figure 9), obtenue par perforation de la membrane cellulaire au niveau de la jonction avec la pipette. Cette perforation donne accès au milieu intracellulaire, qui est dilué due au volume important de la solution intrapipette. Cette configuration permet d’’enregistrer le courant des canaux sur l’ensemble de la cellule. Il existe deux modes, le voltage imposé (voltage-clamp) et le courant imposé (current-clamp).
Figure 9 : Schéma représentant les 3 étapes de la configuration cellule entière. Les variations de l’oscilloscope sont montrées pour chaque étape. Tirée du protocole “Whole-cell Patch-clamp recordings for characterizing Neuronal Electrical Properties” de la compagnie Axol Bioscience.
En mode voltage imposé, l’expérimentateur impose un voltage à la cellule et permet d’enregistrer le courant membranaire. À l’inverse, en mode courant imposé, l’expérimentateur impose des charges qui vont par la suite faire varier le voltage, qui sera alors enregistré.
Différents protocoles sont utilisés afin de déterminer les propriétés biophysiques des canaux ioniques. Ces protocoles vont jouer sur le voltage, sa durée et le nombre d’impulsion. Grâce à ces différents protocoles, il est possible de déterminé entre autres l’activation, l’inactivation et la réactivation des canaux.
Canaux sodiques
Les canaux sodiques sont des protéines membranaires qui permettent le passage des ions sodium. Ils ont un rôle essentiel dans la physiologie en permettant la régulation de la concentration intracellulaire en sodium mais également en déclenchant des cascades de signalisation. Toutes les cellules du corps humain possèdent des canaux sodiques. Cependant, même si leur action commune est de modifier la concentration intracellulaire de sodium, leurs effets sont en revanche très différents selon le type cellulaire. Ces canaux se différencient en plusieurs types : le canal de fuite NALCN, les canaux sodiques épithéliaux ENac, les canaux sensibles au pH ASIC et les canaux voltages- dépendants (Nav).
Les canaux sodiques voltages-dépendants
La famille des Nav comprend 9 membres (isoformes) connus (Nav1.1 à Nav1.9) qui sont exprimés dans
différents tissus excitables3 (Tableau 1) et sont codés par les gènes SCN1A à SCN11A (SCN6A et
SCN7A étant associés à une sous-famille). C’est une protéine membranaire glycosylée ayant un poids moléculaire d’environ 220 kDa et d’une longueur d’environ 2000 acides aminés. Les structures sont très conservées entre les différentes isoformes. Elles sont constituées d’une sous-unité 𝛼 de 4 domaines transmembranaires (D), chacun constitué de 6 segments transmembranaires. Les 4 premiers segments (S1-S4) de chaque domaine, constituent le senseur de voltage (VS) alors que les deux derniers (S5 et S6) forment le pore du canal. C’est le S4 qui est chargé positivement et qui va réagir à la différence de potentiel. La boucle intracellulaire entre le DIII et le DIV sert de porte d’inactivation rapide grâce au motif isoleucine-phénylalanine-méthionine (IFM). Ce motif vient s’insérer dans une poche hydrophobe formée par plusieurs segments de plusieurs domaines. Les parties C-Terminales et N-C-Terminales viennent stabiliser cette liaison hydrophobe. Des mutations peuvent venir
perturber cette liaison et donc affecter, voire abolir, l’inactivation rapide. Le canal sodique Nav1.5, codé
par le gène SCN5A, est essentiel au bon fonctionnement cardiaque. Il génère le courant sodique qui initie le PA après la dépolarisation membranaire et constitue le canal sodique prédominant au niveau cardiaque4,5. Le courant sodique (INa) est donc majoritairement dû au canal Nav1.5. Sa sélectivité pour
le Na+ est due à 4 domaines Asp373-Glu901-Lys1421-Ala1713 (DEKA)6–8. Le stimulus électrique doit
atteindre un seuil pour que l’ouverture des canaux sodiques se produise. Le canal peut aussi interagir avec une ou plusieurs sous-unité(s) 𝛽 (1-4)9. Ces sous-unités 𝛽 ont un poids moléculaire d’environ 30
à 35 kDa et sont constituées d’un seul segment transmembranaire. Elles permettent de moduler la dépendance au voltage, les cinétiques d’activation, d’inactivation, la localisation à la surface de la cellule ainsi que la densité des canaux Nav. Ces derniers s’activent rapidement après la dépolarisation
Table 1 : Liste des différents membres de la famille des Nav ainsi que de leur localisation10.
Ils s’inactivent très rapidement, bloquant l’entrée de Na+ et ont une période réfractaire, empêchant leur
réactivation même après une nouvelle dépolarisation. Lors du potentiel de repos, les Nav sont fermés
(Figure 9). La porte d’activation intracellulaire est formée par les terminaisons intracellulaires des S6 qui contrôlent l’ouverture et la fermeture du canal et sont couplées aux changements de conformation voltage-dépendants du VS (via la boucle intracellulaire entre S4 et S5).
Expression des canaux sodiques voltage-dépendants dans les différents tissus
Canal Expression dans les tissus
Nav1.1 SNC et SNP Nav1.2 SNC et SNP Nav1.3 SNC et SNP Nav1.4 Muscle squelettique Nav1.5 Muscle cardiaque Nav1.6 SNC et SNP Nav1.7 SNP Nav1.8 SNP Nav1.9 SNP
SNC : Système Nerveux Central ; SNP : Système Nerveux Périphérique
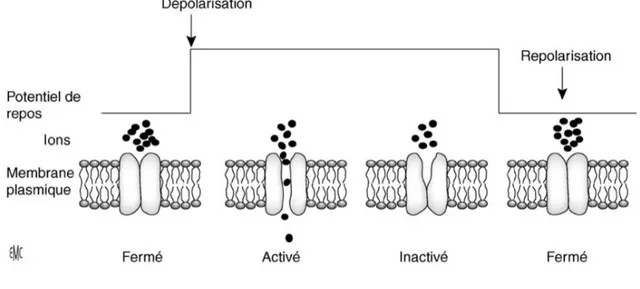
Figure 10 : Schéma des différents états de canaux voltage-dépendants. Lors du potentiel de repos, le canal est fermé. Il s’active lors de la dépolarisation membranaire, puis va s’inactiver, même si la dépolarisation persiste. La fin de la dépolarisation ramène le canal en configuration fermée, mais accuse une période réfractaire pendant laquelle il ne peut être réactivé11.
Une ouverture de 5,6 Å permet à l’eau et aux ions de passer à travers le pore du canal (Figure 10A et B) mais pas de façon optimale. Des mouvements des boucles intracellulaires entre le S4 et le S5 vers l’axe du pore permettent de former un anneau de résidus hydrophobes (Val413, Leu941, Ile1471 et Ile1773) empêchant le passage de l’eau et des ions (Figure 10C). Le canal Nav1.5 s’inactive en
l’espace de 1 à 2 ms. Cette inactivation rapide fait diminuer l’influx de Na+ et permet aux canaux
potassiques de s’activer et de repolariser le potentiel de membrane. C’est le motif IFM, présent sur la boucle intracellulaire entre DIII et DIV, qui est responsable de cette inactivation rapide et vient interagir avec les résidus d’acides aminés au niveau de la boucle intracellulaire entre le S4 et le S5 des domaines DIII et DIV. L’IFM interagit également avec le S5 et le S6 du DIV. L’IFM vient s’insérer dans une poche formée par le S6 du DIII et les S5 et S6 du DIV (Figure 10A). Le motif IFM a été bien résolu par cristallographie et présente une densité électronique non ambigüe (Figure 10D). Le motif IFM pénètre dans une poche hydrophobe d’environ 8 Å formée par la boucle intracellulaire entre le S4 et le S5 du DIV, le S5 du DIII et les S5 et S6 du DIV (Figure 10E). De plus, d’autres interactions viennent stabiliser le motif IFM, impliquant Asn1661 avec Phe1488 et Gln1320 avec Thr1490, formant deux liaisons hydrogène (Interaction du S6 du DIII avec la boucle intracellulaire entre S4 et S5 du DIII) (Figure 10F et 10G). Afin d’avoir une parfaite stabilité, Asn1474 et Gln1478 du S6 du DIII interagissent
avec Gly1338 et Alaa1328 (Figure 10G). Ces interactions permettent de comprendre que des mutations affectant ces résidus ou ceux situés à proximité peuvent engendrer des problèmes d’inactivation du canal Nav1.5.
Figure 11 : Portes d’activation et d’inactivation rapides. (A) Vue du dessous de la porte d’activation de rNav1.5C. Le motif IFM (Ile1485-Phe1486-Met1487) et les chaînes latérales des résidus d’acides
aminés qui ferment partiellement la porte d’inactivation sont montrés en bâtonnets avec la surface de van der Waals en transparent. (B) Vue rapprochée de la porte d’activation de rNav1.5C alignée avec
la structure en état ouvert de NavAb-D40 (PDB : 5VB8) coloriée en magenta. (C) Vue rapprochée de la porte d’activation de rNav1.5C alignée avec la porte d’activation à l’état de repos du canal rNav1.5C
calculé par MODELER. Le modèle de repos est colorié en orange avec les résidus clés fermant la porte d’activation montrés en bâtonnet et la surface de van der Waals en transparent. (D) Vue de côté du motif IFM montré en bâtonnet. (E) Vue rapprochée des liaisons du motif IFM avec la poche hydrophobe montrées en jaune. Les points rouges correspondent aux liaisons hydrogènes. (F) Interaction de la boucle intracellulaire entre le DIII et le DIV avec le DIII et la boucle intracellulaire entre le S4 et le S5 du DIV. « (O) » indique une interaction importante avec les groupes carboxyles. Les points rouges correspondent aux liaisons hydrogène. (G) Interaction du S6 du DIII avec la boucle intracellulaire entre le S4 et le S5 du DIII. « (O) » indique une interaction importante avec les groupes carboxyles. Les points rouges correspondent aux liaisons hydrogène12.
Le S4, situé dans le VS contient de 4 à 8 acides aminés chargés positivement, lui conférant la capacité de pouvoir se déplacer vers l’extérieur lors des changements de polarisation. Ce mouvement est limité et ne permet en aucun cas de créer un mouvement transmembranaire d’eau ou d’ions, mais permet toutefois l’activation du canal12. Ceci est dû aux arginines et aux lysines (chargées positivement) qui
se déplacent vers le site de restriction hydrophobe (HCS), sous l’influence d’un champ électrique et viennent se lier à des acides aminés chargés négativement. Des mutations peuvent survenir au niveau du VS et engendrer des dysfonctionnements, en altérant les changements de conformation naturelle. Ces résidus mutés vont alors interagir avec des acides aminés proches d’eux et donc perturber le mouvement du VS. De plus, ces mutations peuvent mener à un courant de porte lorsqu’une arginine est substituée par un résidu non chargé. Ceci provoque alors un pore dans le VS où l’eau peut s’introduire. Cette « porte » à travers le VS n’est présente qu’à l’état activé du canal, pas au repos12.
La régulation de l’activité du canal Nav1.5 est gérée par de nombreuses protéines. Les sous-unités 𝛽1
et 𝛽2 ne s’associent pas de façon covalente avec le canal Nav1.5, contrairement aux autres canaux
sodiques exprimés dans les nerfs ou les muscles squelettiques. La co-expression du canal Nav1.5
avec les sous-unités 𝛽 permet d’en augmenter l’expression, mais les sous-unités 𝛽 ne modulent pas la dépendance au voltage ni les cinétiques du canal13,14. La sous-unité 𝛽1 s’associe aux Nav en
formant une liaison hydrogène. Cependant, le canal Nav1.5 ne contient pas de tyrosine en position 304
mais il possède une leucine en position 316. Ce changement d’acide aminé permet de diminuer l’interaction entre la sous-unité 𝛽1 et le canal Nav1.5. Contrairement à la sous-unité 𝛽1, il y a une
Cependant, les canaux Nav1.5 et Nav1.8 forment des ponts disulfure grâce à une leucine en position
869 et une alanine en position 822, respectivement. Ces éléments indiquent que la structure du canal Nav1.5 permet d’éviter la liaison avec les sous-unités 𝛽 et donc, sa modulation12.
Figure 12 : Structure cristalline Cryo-EM de rNav1.5C. (A) Vue de côté de l’ensemble de la
reconstruction Cryo-ME de rNav1.5C. (B) Représentation de l’ensemble de la structure de rNav1.5C.
vert, beige et orange. (C) Vue de côté de rNav1.5C avec la surface coloriée en gris. (D) Vue de côté
du complexe Nav1.7- 𝛽1- 𝛽2 humain. La sous-unité 𝛼 est coloriée en gris et les sous-unités 𝛽 sont
coloriées en magenta (𝛽1) et en or (𝛽2). (E) Vue rapprochée des sites d’interaction de 𝛽1(panneau du haut) et de 𝛽2 (panneau du bas). (F) Alignement du SF entre rNav1.5C et Nav1.7 (Gris). Les chaînes
du motif DEKA (DEKA : Asp373-Glu901-Lys1421-Ala1713) sont montrées en bâtonnets. (G) Comparaison du SF du canal rNav1.5C résistant à la TTX (coloré) et du canal Nav1.7 sensible à la TTX
(gris). La TTX est montrée en jaune12.
La tétrodotoxine (TTX) est une toxine provenant du poisson Fugu, qui occasionne entre 20 à 100 morts par an au Japon, à cause de la consommation de sa chair. C’est un bloqueur spécifique des canaux sodiques qui est très utilisé comme outil dans l’étude des canaux sodiques15,16.
Cependant, concernant le canal Nav1.5, la TTX ne présente qu’une faible affinité (2 𝜇M) comparé aux
Nav dans les nerfs et muscles squelettiques (4-10 nM). Curieusement, son site de liaison, le filtre de
sélectivité (SF), est identique à celui du canal Nav1.7 qui lui, a une grande affinité pour la TTX (Figure
11F). Cependant, rNav1.5C présente une différence d’acide aminé sur le côté extracellulaire, alors que
le canal Nav1.7 montre la présence d’une Tyr362, aidant à conserver une interaction forte avec la TTX
par interaction d’empilement p-p (Figure 11G). Cette interaction étant manquante chez le canal Nav1.5,
il montre donc une interaction 500 fois moins importante pour la TTX que le canal Nav1.717,18. De fait,
ce résidu est le déterminant structurel qui différencie les Nav TTX-résistants (Nav1.5, Nav1.8 et Nav1.9)
présentant une cystéine ou une sérine, des Nav TTX-sensibles (Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6
et Nav1.7) présentant plutôt une tyrosine ou une phénylalanine.
D’autres molécules ont fait l’objet d’études quant à leur affinité avec le canal Nav1.5 et leur capacité à
diminuer ou bloquer le courant sodique. Ces molécules sont utilisées en médecine afin de supprimer diverses arythmies auriculaires ou ventriculaires. Ces médicaments sont appelés antiarythmiques et se distinguent en 3 classes. La classe I se subdivise en 3 sous-classes (IA, IB, IC) et cible plus ou moins spécifiquement les Nav19. La classe IA élargit le QRS et prolonge l’intervalle QT (Ex : quinidine,
hydroquinidine). La classe IB n’affecte que très peu le QRS et raccourcit légèrement l’intervalle QT (Ex : lidocaïne, mexilétine). La classe IC tend à élargir significativement le QRS et à prolonger légèrement l’intervalle QT (Ex : flécaïnide, propafénone). La classe II concerne les 𝛽-bloquants qui ralentissent la fréquence cardiaque ainsi que la conduction auriculoventriculaire (Ex : propanolol,
métoprolol). La classe III vise principalement les canaux potassiques et prolonge l’intervalle QT significativement en retardant la repolarisation (Ex : dofétilide).
La flécaïnide (Figure 12B) par exemple, est un antiarythmique de classe 1C utilisé pour bloquer les canaux sodiques cardiaques dans le cadre d’arythmies auriculaires ou ventriculaires.
Figure 13 : La structure cristalline Cryo-EM du canal rNav1.5C et le complexe de la flécaïnide. (A) Vue
de côté de l’ensemble de la reconstruction Cryo-ME de rNav1.5C du complexe de la flécaïnide. (B)
Structure chimique de la flécaïnide. (C) Densité EM pour la poche de fixation de la flécaïnide avec des résidus clés pour la fixation de la flécaïnide montrées en bâtonnets jaunes. Les densités pour les résidus et la flécaïnide ont été contournés à 3 𝜎 et coloriés respectivement en bleu et en vert. (D) Vue du dessus et de côté de la structure rNav1.5C avec la structure de la flécaïnide montrée en bâtonnets
jaunes. (E) Vue de côté de la fenêtre entre DII-DIII12.
La flécaïnide se lie dans la cavité centrale de Nav1.5, du côté intracellulaire du SF, empêchant ainsi
les ions sodium de sortir du SF. La cavité centrale présente 4 fenêtres conduisant à l’intérieur de la bicouche lipidique entre les deux modules du pore (MP). La flécaïnide se lie au pore grâce à ses deux queues trifluoroéthyoxy hydrophobes qui s’insèrent au fond de la fenêtre, entre DII et DIII (Figure 12 C, D et E). Une autre fenêtre existe entre le DIII et le DIV, mais elle est significativement plus petite,
favorisant alors l’entrée de la flécaïnide par celle située entre DII et DIII12,20. Cette fenêtre a été
découverte grâce à la cristallisation du canal rNav1.5C (canal Nav1.5 exprimé chez le rat) obtenue grâce
à la technique Cryo-Microscopie Électronique (Cryo-ME) et à la construction d’une carte 3D d’une résolution de 3,5 Å, basée sur le critère de corrélation de la coquille de Fourier (Figure 12A).
Ce canal est connu également pour être impliqué dans de nombreuses pathologies comme le syndrome du QT long de type 3 ou le syndrome de Brugada; ce qui en fait un élément d’étude majeur21,22. Ces pathologies se traduisent par des dysfonctionnements biophysiques du canal Nav1.5.
Par exemple, un gain de fonction peut être responsable d’un retard de repolarisation (Ex : présence d’un courant sodique persistant), soit par une réouverture précoce et brève (Ex : période réfractaire perturbée).
Syndrome du QT long
Le syndrome du QT long (LQTS) est une maladie congénitale ou acquise, caractérisée par des anomalies électriques sur l’électrocardiogramme (ECG). Il est caractérisé par la prolongation de la repolarisation ventriculaire, visible grâce à un ECG (intervalle QT prolongé) ou lors de l’enregistrement d’un PA (phase 3 prolongée). Ce syndrome s’accompagne également d’un risque d’arythmies ventriculaires particulièrement malignes; les torsades de pointes, dégénérant souvent en fibrillations ventriculaires, syncopes, et éventuellement vers la mort subite.23
Historique
C’est en 1957 que Jervell et Lange-Nielsen ont pour la première fois décrit le LQTS24. Ils ont en effet
observé que chez une famille de six enfants, quatre d’entre eux présentaient une prolongation de l’intervalle du QT. Trois d’entre eux sont décédés subitement à l’âge de 4, 5 et 9 ans. La forme homozygote de la maladie, connue sous le nom de syndrome de Jervell et Lange-Nielsen, a été la première à être reconnue, dû à la plus forte sévérité du phénotype cardiaque, ainsi qu’à la présence concomitante de surdité congénitale; le même canal potassique mutant étant aussi exprimé dans l’oreille interne. Toutefois, la plupart des patients présentent une forme hétérozygote, sans aucun déficit auditif, appelée syndrome de Romano-Ward et décrite pour la première fois dans les années 196025,26.
Ce syndrome a, dès sa découverte, fait l’objet de nombreuses recherches, bien que celles-ci étaient initialement restreintes à l’étude des facteurs cliniques, tels que les symptômes et les paramètres des ECG. Par la suite, l’avènement des technologies de biologie moléculaire a alors permis d’améliorer les connaissances et la compréhension de cette maladie.
Prévalence
Originellement, ce syndrome était considéré comme très rare. Toutefois, selon différentes études plus récentes qui se sont intéressées à mieux caractériser la prévalence de ce syndrome, cette dernière se situerait entre 1 :2500 et 1 :7000, selon la population considérée27,28.
Notamment, dans une étude de 2009, les auteurs ont évalué la prévalence du LQTS congénital dans la population caucasienne29. Ils ont utilisé pour leur étude les résultats des ECG de 44 596 enfants
caucasiens, âgés de 15 à 25 jours, et nés dans 18 maternités différentes. Leur étude a permis de mettre en évidence que le SQTL a une prévalence de 1 :2000 au sein de la population caucasienne.
Étiologie
Le LQTS congénital est une maladie d’origine génétique. Comme précédemment décrit, la forme homozygote est la plus sévère, mais c’est la forme hétérozygote qui est la plus prévalente. Il existe à l’heure actuelle 16 gènes ayant été identifiés pour provoquer diiférentes formes de LQTS congénital, désignées sous les noms LQT1 à LQT16. Chacune de ces formes est associée à une anomalie d’un type spécifique de canal ionique régulant le PA cardiaque ou à une protéine interagissant avec un canal (ex : calmoduline, ankyrine). Les formes les plus communes de LQTS (environ 90% des cas de LQTS congénital) sont les formes LQT1 (KCNQ1), LQT2 (KCNH2) et LQT3 (SCN5A) avec des prévalences respectives de 45 à 50%, 40 à 45% et 5 à 10%30–32. Les patients atteints de LQT1 sont
plus susceptibles de souffrir d’arythmies cardiaques au cours d’un exercice physique, notamment la natation. Les arythmies associées au LQT2 sont plutôt déclenchées par des stimuli auditifs brusques tels que les sonneries ou alarmes. Au contraire, les arythmies surviennent typiquement au repos chez les patients atteints de LQT3. Dans l’immense majorité des cas, les LQT1, LQT2 et LQT3, sont causés par des mutations dans les gènes codants pour leur sous-unité a, affectant respectivement la composante lente du courant potassique rectifiant retardé (IKs) du canal Kv7.1, la composante rapide
du courant potassique rectifiant retardé (IKr) du canal Kv11.1 et le courant sodique cardiaque rapide
ventriculaire et de l’intervalle QT sur un ECG33. Les mutations de KCNQ1 entraînent une perte de
fonction, qui se manifeste par une réduction de l’expression du canal, une expression d’un canal non fonctionnel, une activation à des potentiels plus positifs et/ou des cinétiques de désactivation plus rapides, par exemple. Concernant le LQT2, les mutations de KCNH2 sont caractérisées par une perte de fonction qui se manifeste par une diminution de la densité de courant, l’expression de canaux non fonctionnels, une inactivation et désactivation plus rapides. Enfin concernant le LQT3, les mutations de SCN5A entraînent généralement un gain de fonction qui se manifeste par une augmentation du courant persistant qui prolonge le plateau du PA et de surcroît l’intervalle QT. De plus, on observe également des augmentations ou diminutions du courant de fenêtre dues aux décalages de l’activation ou de l’inactivation. Le courant de fenêtre correspond à la zone délimitée par l’intersection des courbes d’activation et d’inactivation, ce qui correspond à l’état d’équilibre entre les différents états des canaux sodique (ouvert, inactivé, fermé). Le diagnostic du LQTS se base principalement sur la mesure du QT corrigé (QTc) à partir de l’ECG. Lorsque qu’un LQTS est suspecté, le diagnostic débute donc avec un ECG de surface. L’intervalle « normal » du QT se situe entre 350 ms et 440 ms. Schwartz et al, ont mis en place une table indicatrice afin de diagnostiquer le LQTS en absence de dépistage génétique34.
Physiopathologie et manifestations cliniques
Table 2 : Critère de diagnostic du LQTS. Le QTc est calculé suivant la formule de Bazett, QTc=QT/√𝑅𝑅. Un LQTS acquis est défini par un score LQTS ³4. Score : £1 point, faible probabilité d’un LQTS; 2 à 3 points, probabilité intermédiaire de LQTS; ³4 points, forte probabilité d’un LQTS34.
Critères de diagnostic du LQTS Points ECG A QTc ³480 ms1/2 3 460-470 ms1/2 2
450 ms1/2 (chez les hommes) 1
B Torsades de pointes 2
C Onde T alternante 1
D Onde T dentelée 1
E Faible rythme cardiaque (selon l’âge) 0,5
Historique clinique A Syncope Avec stress 2 Sans stress 1 B Surdité congénitale 0,5 Historique familial
A Membre(s) de la famille atteint(s) de LQTS 1
Cette table tient compte de la valeur du QT corrigé obtenue à partir de l’ECG et ajustée à la fréquence cardiaque, de l’historique clinique et de l’historique familial. Un QTc dont la valeur se situe au-dessus de 450 ms sera alors considéré comme un QTc long (Table 2).
Prise en charge
En raison de l’hétérogénéité de ce syndrome, le génotypage est souvent très utile afin de déterminer les décisions thérapeutiques à prendre. Par exemple, dans le cas de patients atteints de LQT1, il sera recommandé de limiter l’activité physique intense, afin de ne pas provoquer une élévation marquée de la fréquence cardiaque. Pour les patients atteints de LQT2 et de LQT3, la fréquence cardiaque trop basse peut entraîner des événements cardiaques néfastes35. L’utilisation d’un test d’effort physique
afin d’augmenter la fréquence cardiaque peut être un outil afin d’aider au diagnostic d’un LQTS36. Les
patients sous traitement 𝛽-bloquant, à la suite d’épisodes de syncope ou d’arrêts cardiaques, sont considérés à risque élevé et il est alors recommandé d’implanter un défibrillateur automatique. Dans de rares cas, il sera même recommandé d’effectuer une dénervation sympathique cervico-thoracique gauche. Concernant le LQT3, l’usage de la mexilétine est recommandé et permet de réduire les risques de survenue d’événements arythmiques graves. La mexilétine agit sur le cœur en bloquant sélectivement le courant sodique, facilitant ultimement la repolarisation cardiaque et raccourcissant ainsi l’intervalle QT.
Sujet d’étude
Deux nourrissons de familles différentes sont décédés des suites de fibrillations ventriculaires à 5 (Patient 1) et 12 semaines (Patient 2). Leurs QTc étaient de 600 ms et 700 ms respectivement et ils exprimaient un bloc auriculo-ventriculaire 2 :1 à la naissance. Le traitement par 𝛽-bloquant n’a eu aucun effet sur ces deux nourrissons. Suite à leur décès, les gènes impliqués dans le QT long ont été criblés et ont permis de révéler une substitution hétérozygote de la base G – vers – T à la position 4473 dans l’exon 23, se traduisant par une substitution du glutamate (E) – vers – histidine (H) au résidu 1491 (Patient 2) et une substitution hétérozygote de la base G – vers – T à la position 4442 de l’exon 23, se traduisant par une substitution de la glycine (G) – vers – valine (V) au résidu 1481 (Patient 1). Les deux substitutions sont localisées sur la boucle intracellulaire entre de le DIII et le DIV, à 4 acides aminés avant (Patient 1) et après (Patient 2) le motif isoleucine-phénylalanine-méthionine (IFM), jouant un rôle important dans l’inactivation des canaux sodiques voltage-dépendants.
Hypothèses et objectifs
De nombreux mutants découverts sur la boucle intracellulaire entre le DIII et le DIV ont engendré des perturbations de l’état inactif du canal Nav1.5. L’hypothèse avancée dans cette étude soutient l’idée
que ces mutants impactent l’état inactif du canal Nav1.5, puisqu’elles se situent de part et d’autre de
l’IFM, et qu’elles sont vraisemblablement responsables du phénotype de LQT3 ayant mené au décès de ces deux nourrissons.
La présente étude a été menée en suivant des objectifs bien précis. Dans un premier temps, la construction des mutants par mutagénèse dirigée a constitué le point essentiel pour le lancement de l’étude. Le second objectif de l’étude a été d’ exprimer ces mutants (ou le canal sauvage) ainsi que la sous-unité β1 (couplée au Enhance Green Fluorescent Protein (EGFP)), via la technique du phosphate
de calcium dans la lignée cellulaire HEK (Human Embryonic Kidney) 293, en raison de sa très faible densité en canaux sodiques endogènes37. Le troisième objectif de cette étude a été d’enregistrer et
d’analyser les caractéristiques biophysiques de ces mutants, grâce à la technique de patch-clamp en configuration cellule entière, et de les comparer à celles du canal sauvage (WT), afin d’arriver à déterminer si ces mutants ont pu jouer un rôle dans le décès des deux nourrissons.
Chapitre 1 : Novel G1481V and Q1491H SCN5A
mutations linked to long QT syndrome destabilize
the Na
v1.5 inactivation state
1.1 Résumé
Le canal Nav1.5, codé par le gène SCN5A, est le canal Na+ voltage-dépendant prédominant au niveau
cardiaque. Plusieurs mutants alléliques de ce gène ont été identifiés comme étant impliqués dans différents troubles du rythme cardiaque comme le syndrome du QT long de type 3 (LQT3), pouvant provoquer la mort subite. De nouveaux mutants de ce gène (G1481V et Q1491H), proches du motif Isoleucine-Phénylalanine-Méthionine (IFM) impliqué dans l’inactivation rapide du canal, ont été découverts chez des nourrissons décédés subitement à 5 et 12 semaines et ayant démontré des intervalles QT prolongés.
Le but de cette étude était de caractériser les propriétés biophysiques de ces nouveaux mutants via la technique de patch-clamp. Nous avons émis l’hypothèse selon laquelle les cinétiques d’inactivation du canal sont affectées, considérant l’emplacement de ces mutants
Le canal Nav1.5 de type sauvage (WT) et les mutants G1481V et Q1491H ont été reproduits in vitro.
Le canal WT et les mutants ont été respectivement co-transfectés dans des cellules Human Embryonic Kidney (HEK) 293 avec la sous-unité régulatrice b1. Les courants Na+ ont été enregistrés via la
technique de patch-clamp en configuration cellule entière.
Le canal mutant Q1491H produit une densité de courant plus faible, un courant persistant Na+, une
augmentation du courant fenêtre due au décalage de +20 mV de l’inactivation rapide, de +10 mV de l’activation, une inactivation lente plus rapide et une réactivation présentant 2 constantes de temps, une rapide et une lente. Le canal mutant G1481V produit une augmentation de la densité de courant et un décalage de +7 mV de l’inactivation rapide.
Les nourrissons décédés à 5 et 12 semaines présentaient un prolongement de l’intervalle QT. Nos analyses des effets électrophysiologiques cellulaires des mutants G1481V et Q1491H corrèlent avec le diagnostic clinique. Les dysfonctions biophysiques engendrées par ces deux mutants sur le canal sodique cardiaque sont probablement responsables de la mort subite des deux nourrissons.
1.2 Abstract
Nav1.5, which is encoded by the SCN5A gene, is the predominant voltage-gated Na+ channel in the
heart. Several mutants of this gene have been identified and have been reported to be involved in several cardiac rhythm disorders, including type 3 long QT syndrome (LQT3),that can cause sudden cardiac death. We analyzed the biophysical properties of two novel mutants of the Nav1.5 channel
(Q1491H and G1481V) detected in 5- and 12-week-old infants diagnosed with a prolonged QT interval. The Nav1.5 wild-type (WT) and the Q1491H and G1481V mutant channels were reproduced in vitro.
WT or the mutant channels were co-transfected in HEK 293 cells with the beta 1 regulatory subunit. Na+ currents were recorded using the whole-cell configuration of the patch-clamp technique.
The Q1491H mutant channel exhibited a lower current density, a persistent Na+ current, an enhanced
window current due to a +20-mV shift of steady-state inactivation, a +10-mV shift of steady-state activation, a faster onset of slow inactivation, and a recovery from fast inactivation with fast and a slow time constants of recovery. The G1481V mutant channel exhibited an increase in current density and a +7-mV shift of steady-state inactivation. The observed defects are typical of gain-of-function-associated LQT3 mutants.
The 5- and 12-week-old infants displayed prolonged QT intervals. Our analyses of the Q1491H and G1481V mutants correlated with the clinical diagnosis. The observed biophysical dysfunctions associated with these two mutants are most likely responsible for the sudden deaths of the two infants.
Novel G1481V and Q1491H SCN5A mutations linked to long QT
syndrome destabilize the Nav1.5 inactivation state
Plumereau Quentin MSc.
1, Theriault Olivier. Ph.D.
1, Pouliot Valérie. BSc.
1, Moreau
Adrien. Ph.D.
2, Morel Elodie Ph.D.
3, Fressart Véronique M.D. Ph.D.
4, Denjoy
Isabelle M.D.
5, Delinière Antoine M.D.
3, Bessière Francis M.D.
3, Chevalier Philippe
M.D.
3,6,7, Gamal El-Din M. Tamer Ph.D.
8and Chahine Mohamed Ph.D.
1,91CERVO Brain Research Center, Quebec City, QC, Canada
2Inserm U1046, CNRS UMR 9214, Université de Montpellier, Montpellier, France
3Lyon Reference Center for Inherited Arrhythmias, Louis Pradel Cardiovascular Hospital, Bron, France
4Centre de génétique moléculaire et chromosomique, Hôpital Pitié-Salpêtrière, Paris, France
5Hôpital Bichat Claude Bernard, Paris, France
6Department of Rhythmology, Louis Pradel Cardiovascular Hospital, Lyon, France
7Université de Lyon, Lyon, France
8Department of Pharmacology, University of Washington, Seattle, WA 98195, USA.
9Department of Medicine, Faculty of Medicine, Université Laval, Quebec City, QC, Canada
1.3 Abbreviations
AP Action potential
BS Brugada syndrome
cDNA Complementary DNA DI-DIV Sodium channel domain 1-4 ECG Electrocardiogram
EADs early after depolarizations
HEK293 Human embryonic kidney cell line LQTS Long QT interval syndrome LQT3 Long QT interval syndrome type 3
PCCD progressive cardiac conduction disorder QTc QT interval corrected for heart rate
SIDS sudden infant death syndrome.
TTX Tetrodotoxin
VT / VF Ventricular tachycardia / ventricular fibrillation
1.4 BACKGROUND: Nav1.5, which is encoded by the SCN5A gene, is the predominant voltage-gated
Na+ channel in the heart. Several mutations of this gene have been identified and have been reported
to be involved in several cardiac rhythm disorders, including type 3 long QT syndrome (LQT3), that can cause sudden cardiac death. We analyzed the biophysical properties of two novel variants of the Nav1.5
channel (Q1491H and G1481V) detected in 5- and 12-week-old infants diagnosed with a prolonged QT interval.
METHODS: The Nav1.5 wild-type (WT) and the Q1491H and G1481V mutant channels were
reproduced in vivo. WT or the mutant channels were co-transfected in HEK 293 cells with the beta 1 regulatory subunit. Na+ currents were recorded using the whole-cell configuration of the patch-clamp
technique.
RESULTS: The Q1491H mutant channel exhibited a lower current density, a persistent Na+ current, an
enhanced window current due to a +20-mV shift of state inactivation, a +10-mV shift of steady-state activation, a faster onset of slow inactivation, and a recovery from fast inactivation with fast and a slow time constant of recovery. The G1481V mutant channel exhibited an increase in current density and a +7-mV shift of steady-state inactivation. The observed defects are characteristic of gain-of-function mutations typical of LQT3.
CONCLUSION: The 5- and 12-week-old infants displayed prolonged QT intervals. Our analyses of the Q1491H and G1481V mutations correlated with the clinical diagnosis. The observed biophysical dysfunctions associated with both mutations were most likely responsible for the sudden deaths of the two infants.
Brief Summary:
Nav1.5, encoded by the SCN5A gene is the predominant sodium channel in the heart. Mutations in this
gene cause type 3 long QT syndrome (LQT3). We report here on two Nav1.5 mutations, Q1491H and G1481V found in 5- and 12-week-old infants diagnosed with a prolonged QT interval and died suddenly. The biophysical characterization revealed major defects that are characteristic of gain-of-function mutations, typical of LQT3 and therefore explain the sudden death on these two infants.
KEY WORDS: SCN5A, Nav1.5, Voltage-gated sodium channels, Long QT syndrome, Variant, LQT3,
1.5 Introduction:
Voltage-gated Na+ channels are crucial for the amplitude and upstroke of cardiac action potentials
(AP), which are important determinants for driving AP propagation and conduction velocity throughout the working myocardium1. Mutations in SCN5A, the gene that encodes Nav1.5, the predominant cardiac
Na+ channel, have been implicated in rare familial forms of cardiac arrhythmias such as type 3 long QT
syndrome (LQT3), Brugada syndrome (BS), progressive cardiac conduction disorder (PCCD), atrial fibrillation (AFib), and sudden infant death syndrome (SIDS). Another SCN5A mutation has recently been reported to be involved in dilated cardiomyopathy, a structural heart disease2,3. In addition to their
role in changing gating characteristics, there is growing recognition that such mutations may also be associated with alterations in channel protein trafficking and expression levels. SCN5A gene is also the only gene to have a definitive evidence, for clinical validity of BS4.
Long QT syndrome (LQTS) is an inherited cardiac channelopathy that may lead to syncope and even sudden cardiac death as a result of polymorphic ventricular tachycardia known as torsade de pointes. LQTS manifests as a prolonged corrected QT (QTc) interval exceeding 450 ms on 12-lead electrocardiograms (ECG). To date, seventeen genes have been linked to inherited LQTS but only three have shown a definitive evidence as a genetic cause for typical LQTS. Those genes are KCNQ1, KCNH2 and SCN5A5.
LQT3 is caused by mutations in SCN5A genes mapped to chromosome 3q21-24, which encodes the α-subunits of the cardiac Na+ channel6,7. SCN5A mutations have been identified in about 10% of
genotyped LQTS patients8. When a patient presents a LQTS, a genetic testing is used to identify the
gene that cause the disease. If the gene is identified, then the clinician knows the type of LQTS (LQT1, LQT2 or LQT3)9. The development of arrhythmias is often associated with bradycardia during sleep or
relaxation, when the QTc interval is prolonged. The most common mechanism for QT interval prolongation in LQT3 is due to a persistent or a late Na+ current leading to an increase in AP plateau
duration6. This can be a substrate for early after depolarizations (EADs), which can potentially trigger
ventricular tachyarrhythmias. Most mutations linked to LQT3 are located in exons 23, 26, and 28, which encode the III-IV linker or inactivation gate, the voltage sensor domain, and the C-terminus of the Na+ channel, all of
which are involved in fast inactivation. These mutations impede the proper closure of the channel during this critical process, resulting in an increase in the number of channels that are unable to reach a stable inactivated state. This leads to a rise in persistent Na+ currents unlike the normal inactivation of WT Na+ channels.
Although SIDS is the leading cause of death in the first year of life, its cause is still unknown. The only recommended preventive measure is to avoid placing infants on their stomachs or sides for sleep10.
Several studies have linked SIDS to LQTS11 and have shown that 50% of infants who die of SIDS have
a prolonged QTc interval and that a prolonged QTc interval over 440 ms in the first week of life increases the risk of SIDS by a factor of 4112,13.
We report here on two infants who presented with a prolonged QTc interval and who died suddenly, most likely after experiencing ventricular fibrillation. Although the 5- and 12-week-old infants were within the age range during which the incidence of SIDS peaks, their deaths were not attributed to SIDS. A sequencing analysis revealed a heterozygous G‐to‐T base substitution at position 4473 in exon 23 that resulted in a glutamate (E)‐to‐histidine (H) substitution at residue 1491 and a heterozygous G‐to‐T base substitution at position 4442 in exon 23 that resulted in a glycine (G)‐to‐valine (V) substitution at residue 1481. We recorded macroscopic Na+ currents in transfected mammalian cells
using the whole-cell configuration of the patch-clamp technique. The data revealed a marked shift of inactivation to more positive potentials, the presence of a persistent Na+ current, and a large increase
in the window current. All these characteristics point toward a gain-of-function due to the mutations.
1.6 Methods
Institutional Committee on Human Research
The study was conducted in accordance with the principles of the Declaration of Helsinki and the protocol was approved by the local ethics committee. The parents provided written informed consent.
Cell cultures
Human embryonic kidney 293 (HEK 293) cells were used. The cells were grown in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% streptomycin at 37°C in a 5% CO2 atmosphere. The cells were transfected with the pcDNA3.1 vector containing
either WT Nav1.5 cDNA (1 𝛍g) or Nav1.5/Q1491H and G1481V mutants with the pIRES2/EGFP vector
containing β1 subunit cDNA (1 𝛍g) in 10-cm cell culture dishes using the calcium phosphate method as previously reported14.
Whole-cell patch-clamp recordings
Na+ currents were recorded using low-resistance, fire-polished electrodes (≈ 1MΩ) made from 8161
Corning borosilicate glass coated with HIPEC (Dow-Corning, Midland, MI, USA) to minimize electrode capacitance. An Axopatch 200 amplifier and pClamp software (Molecular Devices, Sunnyvale, CA, USA) were used to record Na+ currents. The series resistance was compensated to 80% to minimize
voltage-clamp errors. The cells were allowed to stabilize for 5 min after the whole-cell configuration was established. The membrane potential was held at –140mV before the currents were recorded. Sodium Currents were filtered at 5 kHz and digitized at 83.33 kHz. The liquid junction potential was not corrected. All the experiments were performed at room temperature (22°C).
Solutions
The intracellular solution was composed of 35 mM NaCl, 105 mM CsF, 10 mM EGTA, and 10 mM HEPES. The pH was adjusted to 7.3 with 2 M CsOH. The first external solution (Low Na+) was
composed of 35 mM NaCl, 115 NMDG, 2 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and
10 mM HEPES. The pH was adjusted to 7.4 with 2 M HCl. The second external solution (Full Na+) used
to record persistent Na+ currents was composed of 150 mM NaCl, 2 mM KCl, 1.5 mM CaCl2, 1 mM
MgCl2, 10 mM glucose, and 10 mM HEPES. The pH was adjusted to 7.4 with 2 M HCl. TTX (LATOXAN,
Portes-lès-Valence, France) was diluted to 75 𝛍M in 2% acetic acid. Data analysis
The slope factor (K) and the midpoint (V1/2) for activation and inactivation were calculated using
standard Boltzmann functions: 1/(1 + exp ((V1/2activation – V)/kactivation)) for activation and (1 - C)/(1 + exp
((V – V1/2inactivation)/kinactivation)) + C) for inactivation. V is the voltage. The window current was obtained
using equation 1: (1/(1 + exp ((V1/2activation – V)/kactivation)) x ((1 - C)/(1 + exp ((V – V1/2inactivation)/kinactivation))
+ C) x 100, which is the probability of having the channel in the open state. Statistical analysis
Results are expressed as means ± S.E.M. Statistical comparisons were performed using a one-way ANOVA in GraphPad Prism for statistical comparisons. Differences were considered significant at P<0.05.