FACULTE DES SCIENCES /97/
THESE
PRESENTEE A L'ECOLE DES GRADUES
DE L'UNIVERSITE LAVAL
POUR L'OBTENTION DU GRADE DE
DOCTEUR ES-SCIENCES par YVES FAQUIN B. Sc., UNIVERSITE LAVAL ETUDE DE LA DECOMPOSITION THERMIQUE DE L'AZOMETHANE Août 1971 /
de VüniveAAlte Lavat
REMERCIEMENTS
Je
déAiAe AemeActeA:
mon6Ze.uA. te. pAofie66e.uA Wendett
FOR ST,qui a dtAigé
cetAaooJX,
meA6teuAA
F.BouchaAd,
R.CAépautt et G.
MaAzeApouA t'atde techni
que qu'ilA ont appoAtêe à ta con6tAuction de t'appaAeittage
,
Ze6
étudtantA gAaduéA du dépaAtement de chimie atnAi que madame
M. 1/ezZZeax pouA la.
dactytognapkie de ta théAe,
te tabonatotAe du V
aV. Rou66eau du dépaAtement de chimie de
VUniveA6tté de MontAéat ainàt que te CentAe de AecheAche pouA ta
défienbe de VatcaAXZeA pouA te6 6pectAe6 de mcu>6e qu’ÜA ont efifiec-
tué6,
te Con6eit nationat de AecheAcheA ain6i que ta compagnie C.l.L.
pouA t'octAoi de bouA6e6 po6t-gnade.
Page
RESUME... vi
LISTE DES SYMBOLES... viii
INTRODUCTION... 1
CHAPITRE 1: METHODE EXPERIMENTALE... 8
1.1 Préparation de l'azométhane... 8
1.2 Appareillages... 11
CHAPITRE 2: PRODUITS DE LA DECOMPOSITION DE L’AZOMETHANE... 15
2.1 Produits "légers"... 16 2.1.1. Colonne I... 16 2.1.2. Colonne II... 18 2.2 Produits "lourds"... 20 2.2.1. Colonne III... 20 2.2.2. Colonne IV... 24 2.2.3. Colonne V... 25 2.2.4. Colonne VI... 27
2.2.5. Identification des produits "lourds" de poids .... moléculaires de 72... 27
2.2.6. Identification des produits "lourds” de poids moléculaires de 86... 32
2.2.7. Identification d'autres produits "lourds" ... 39
2.2.8. Produits non formés au cours de la pyrolyse de 1 ' azométhane... 41
2.2.9. Résumé... 43
CHAPITRE 3: RESULTATS EXPERIMENTAUX... 45
IV
Page3.2 Etude de la décomposition en fonction du temps... 47
3.3 Analyse des produits et bilan en carbone. ;... 49
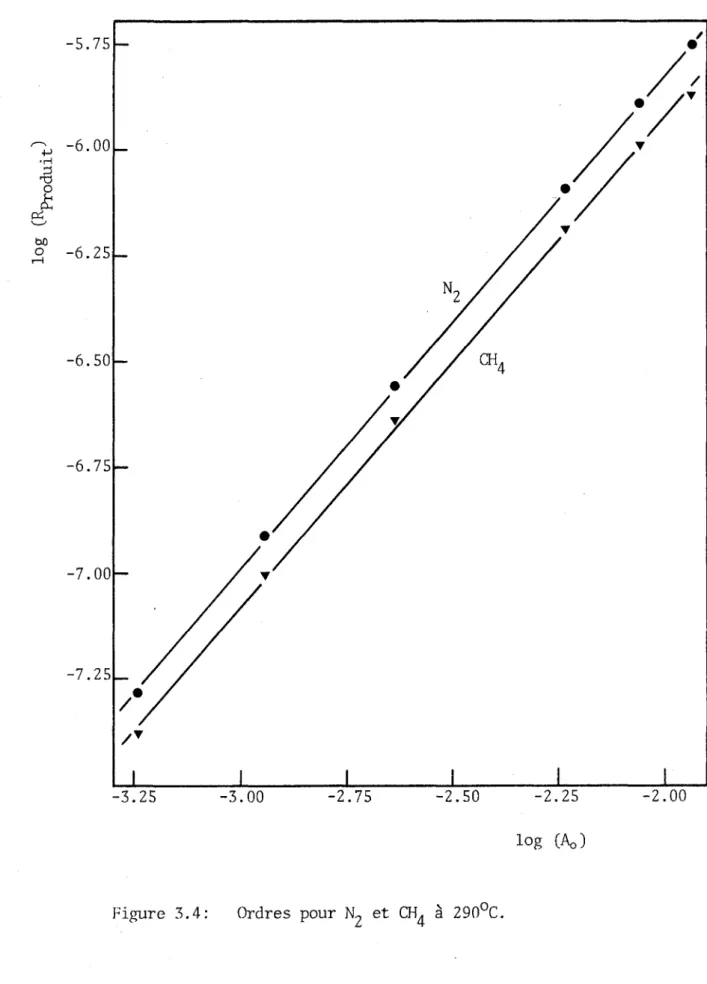
3.4 Variations de la vitesse de formation des produits en fonc tion de la pression de pyrolyse... 54
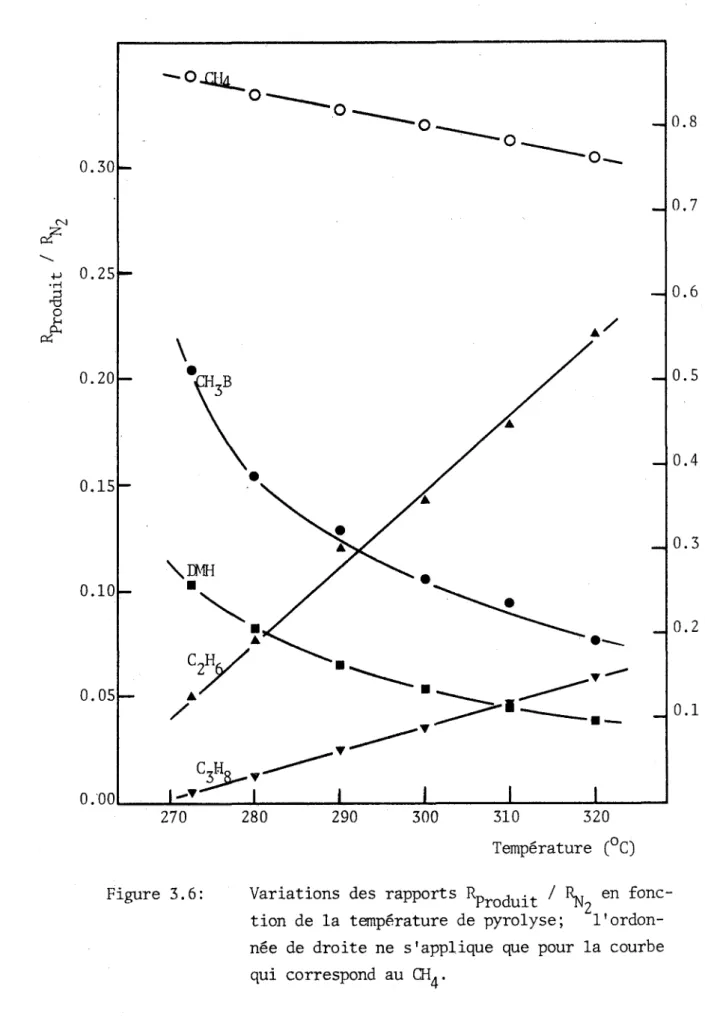
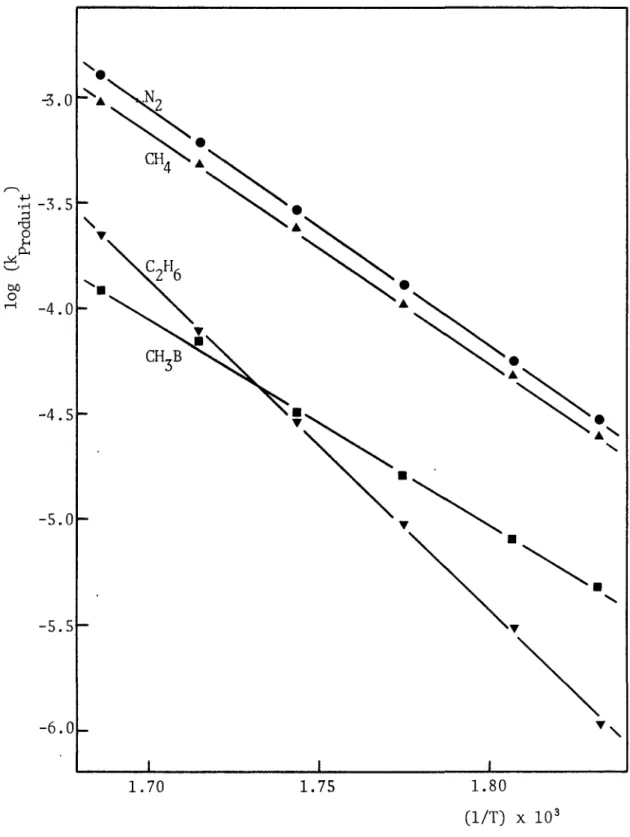
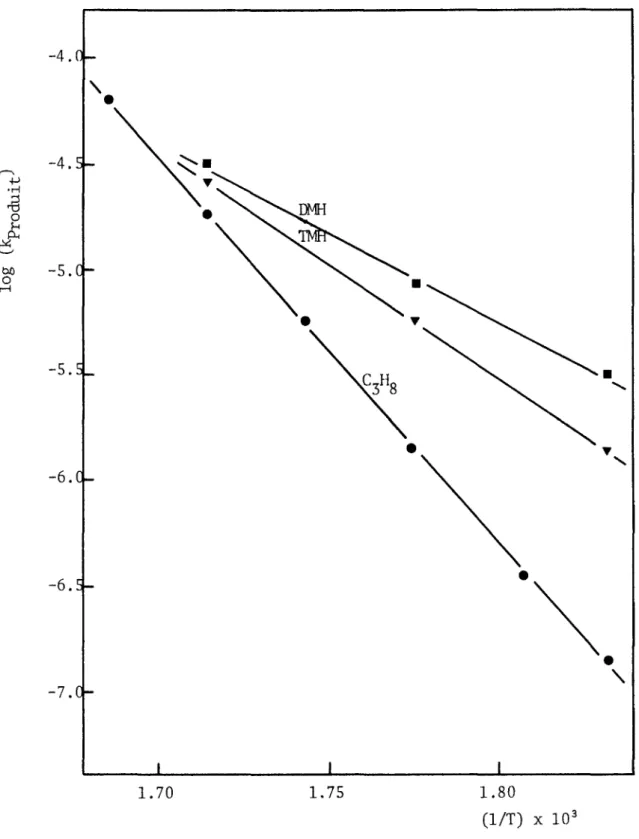
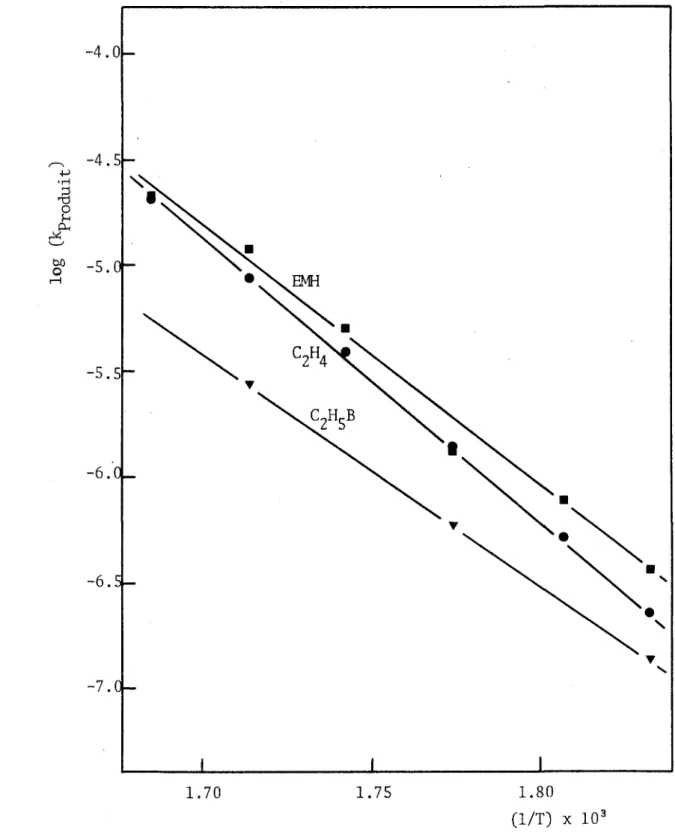
3.5 Variations de la vitesse de formation des produits en fonc tion de la température... 58
CHAPITRE 4: MECANISME DE LA DECOMPOSITION DE L'AZOMETHANE... 70
4.1 Les réactions principales de la décomposition de l'azométha ne... 71
4.1.1. Quelques aspects de la formation de l'azote... 71
4.1.2. Schéma I... 77
4.1.3. Formation de l'éthane... 81
4.2 Les réactions secondaires de la décomposition de 1'azométha-ne. ... 103
4.2.1. Schéma II... 105
4.2.2. Réactions possibles des radicaux CH^ ... 109
4.2.3. Réactions possibles des radicaux CH^A... 114
4.2.4. Formation du CH^B et du C?HgB... 124
4.2.5. Formation du C^H^... 132
4.2.6. Formation du DMH et du EMH...
135
4.2.7. Formation des radicaux C^H^...
141
4.3 Schéma global de la décomposition de 1 'azométhane... 153
4.3.1. Schéma III... 154
4.3.2. Energies d'activation et ordres relatifs à la forma tion des principaux produits ... 168
Page APPENDICE: DONNEES EXPERIMENTALES NECESSAIRES AU CALCUL DES ENER
GIES D'ACTIVATION... 185 BIBLIOGRAPHIE... 196
RESUME
La decomposition thermique de l'azométhane, CH^N^CH^, a été étudiée en système statique entre 250 et 320°C et de 5 à 402 torr. L'identifi
cation des produits formés au cours de la pyrolyse a été faite par chro matographie en phase gazeuse et par spectrométrie de masse. Les princi paux produits de décomposition sont, par ordre d'importance, l'azote, le méthane et, selon la température de pyrolyse utilisée, l'éthane ou le ^2^5^2®3" Nous avons aussi constaté la formation d'autres produits, en quantités moins importantes, soit des hydrocarbures en C2 et C, et des produits azotés tels des hydrazines, des hydrazones et un diimide.
La décomposition thermique de 1'azométhane comporte une courte chaîne dont la longueur varie en fonction de la température et de la près sion de pyrolyse. L'analyse des résultats expérimentaux confirme que la chaîne est propagée par cette réaction:
ch3 + ch3n2ch3 -*■ ch4 + ch2n2ch3
Notre travail apporte 1'évidence que le radical CH2N2CH3 se décompose en suite de cette façon :
CH
2
N
2
CH
3
■+
CH
2
+ N
2
+ CIIj
Les radicaux CH2, formés lors de la décomposition du radical CH2N2CH3, don nent lieu à la formation des produits secondaires et à la polymérisation
constatée à certaines températures et pressions. En effet, si la forma tion des produits principaux de la pyrolyse peut être expliquée par les diverses réactions possibles des radicaux CH^ entre eux ou avec 1'azomé thane, la formation des produits secondaires ne peut s'expliquer que par la présence de radicaux C^Hg au cours de la pyrolyse. Le mode de forma tion des radicaux C^Hg implique la formation d'un biradical qui provient de l'addition de radicaux Cl^ sur la double liaison de 1'azométhane. Ce biradical sert aussi d'intermédiaire à la polymérisation qui se produit
principalement à basse température ou à haute pression de pyrolyse.
Un mécanisme est proposé qui explique 1'ensemble des résultats à 1'intérieur de tout le domaine de température et pression que nous avons étudié.
LISTE DES SYMBOLES
Au cours de ce travail, il est fait mention de plusieurs produits et radicaux qui sont nommés par des symboles, afin de faciliter l'écritu re. La liste qui suit permet de voir â quel produit ou radical correspond
chaque symbole. PRODUITS A gh3b DMH TMH EMH C2H5B œ3p œ3 XN=Nx CH3 C2H5 VN=NV CH, Azométhane Ethylmêthyldiimide CH, CH ;n-n=œ2 CH, CH, CH3 XCH3 C2H5 CH. /xn-n=ch2 C3H7 N=N, CH, CH CH, XN-N.Z CH, CH' \N-N ^ CH,
\
CH, Diméthylhydrazone du formaldéhyde Tétraméthylhydrazine Ethylméthylhydrazone du formaldéhyde n-propy Iméthyldiimide PolymèreDIM^ : CH, CH, XN-NZ CH, CH,Z >-Nz J CH3 XCH3 DIM2 : CTL XN=N. ™2-™2x XN=Nx xch3 Œ3AB : CH, CH, CHZ CH N=N. ™3 B : RADICAUX •CH 2 XN=Nx -w ŒL=N-NX
X
^3
CH3A : CH, ŒL >n/ «3 ' P : ch3 Œ3 XN\ /Œ3 < ™>N-N !” CH^A : Q-L-N-N-CE, 3 i • 3 ÇH2INTRODUCTION
La décomposition thermique de l’azométhane a été longtemps considé rée comme un exemple parfait de réaction monomoléculaire (1), (2). Cette conclusion découlait de plusieurs études de la pyrolyse de 1 *azomêthane faites à l'aide de mesures manométriques. En effet, déjà en 1927,
Ramsperger (3), (4), étudiant la décomposition thermique de 1'azomêthane à des températures comprises entre 278 et 330°C et à des pressions de 26 à 710 torr avait conclu que celle-ci était homogène et monomoléculaire. D'après Ramsperger, la décomposition de 1'azomêthane devait se faire pres- qu'entièrement selon la réaction suivante:
+ N,
Des travaux subséquents de Rice et Ramsperger (5), (6), ayant pour objet l'étude des théories sur les échanges d'énergie associés aux réactions mono moléculaires, de même que des études analogues faites par Kassel (7), ont aussi porté à croire que la décomposition de 1'azomêthane était un bon exemple de réaction monomoléculaire.
Cependant, en 1933, Leermakers (8) puis, Evering et Rice (9) ont mis en évidence la formation de radicaux CH^ lors de la pyrolyse de l'azo- méthane. Les travaux de Sickman et Allen (10) devaient d'ailleurs confir mer ce fait par l'étude de la décomposition de 1'acétaldéhyde induite par les radicaux CH^ produits par la décomposition de 1'azomêthane au cours de
pyrolyses de mélanges azométhane-acétaldéhyde. De plus, Rice et Sickman (11) ont étudié la polymérisation de l'éthylène et du propylène induite par les radicaux CH^ formés lors de pyrolyses de mélanges azométhane-oléfine. Plus tard, en 1936, Rice et Sickman (12), (13), étudiant l'effet de diffé rents gaz inertes sur la décomposition thermique de 1'azométhane, se sont interrogés sur la possibilité de la présence d'une chaîne au cours de la pyrolyse de 1'azométhane, mais sans toutefois en établir ou en réfuter le
fait. Puis, Riblett et Rubin (14) ayant montré la complexité des produits de la décomposition de 1'azométhane, on a commencé à douter de la signifi cation des mesures manométriques et de la monomolécularité de la réaction. En effet, d'après les analyses faites par Riblett et Rubin, le principal produit de la réaction est le CH^ et non le C^H^; leurs analyses ont mon tré qu'il se formait aussi un produit de poids moléculaire élevé contenant de l'azote.
Les travaux de Jahn et Taylor (15) confirmèrent les résultats obte nus par Riblett et Rubin. Selon Jahn et Taylor, le produit azoté de poids moléculaire élevé serait la tétraméthylhydrazine. Celle-ci serait formée par l'addition de deux radicaux CH^ sur la double liaison de 1'azométhane puis, pourrait être décomposée fournissant ainsi une étape intermédiaire au cours de la pyrolyse de 1'azométhane. Cette idée de Jahn et Taylor leur permettait aussi d'expliquer les résultats de pyrolyses faites en présence de NO (16) où 1'absence d'hydrocarbures, en particulier de C«H^, contre disait définitivement l'hypothèse de Ramsperger de la décomposition direc te de 1'azométhane en une molécule d'azote et une molécule de C^H^ formée intramoléculairement. L'idée de Jahn et Taylor peut être résumée ainsi:
3
Œ3N2CH3 •> 2Œ3 + N2
2CH3 + Œ3N2CH3 -*■ tétramëthylhydrazine
CH3 + NO Œ3NO
Même si Jahn et Taylor ont noté une diminution de la vitesse de décomposi tion de 1'azométhane en présence de NO, ils n'ont cependant pas relié ce phénomène à la présence d'une chaîne.
En 1953, Page, Pritchard et Trotman-Dickenson (17) ont étudié la pyrolyse de l’azométhane en système dynamique en employant le toluène comme gaz porteur. Ils ont suivi la décomposition de 1'azométhane par la for mation de l'azote et les résultats qu'ils ont obtenus ne différaient pas sensiblement des résultats alors connus. Ils ont cependant soulevé la pos sibilité de la formation du par recombinaison de radicaux CHg. En 1956, McCoy (18) a étudié, à l'aide d'un spectromètre de masse, la décompo sition de 1'azométhane en système statique en suivant la disparition du pic parent de 1'azométhane. McCoy, par cette méthode, ne pouvait pas réelle ment savoir si tout 1'azométhane disparu s'était décomposé car une partie de 1'azométhane pouvait disparaître par réaction avec des radicaux présents au cours de la pyrolyse. Selon McCoy, la décomposition de 1'azométhane ne se ferait pas par un mécanisme en chaîne impliquant des radicaux, même si des radicaux sont formés au cours de cette pyrolyse. McCoy a aussi analysé les produits formés quand un échantillon d'azométhane est décomposé. Cette analyse a révélé la présence d'un grand nombre de produits : de l'azote, des hydrocarbures contenant de 1 à 4 atomes de carbone, du HCN, du ŒLCN et du (Œ3)3N. Il faut cependant noter que 1'identification de ces produits ayant été faite à partir d'un spectre de masse du mélange après pyrolyse,
il est difficile de savoir si certains de ces produits, comme par exemple le HCN, ont été réellement formés au cours de la pyrolyse ou s'ils sont dus à la fragmentation de 1'azométhane lui-même dans le spectromètre de masse. En 1968, Gay (19) a utilisé la même technique que McCoy (18) et il a aussi trouvé qu'à une pression de 0.31 torr et à 332°C il n'y a pas de chaîne lors de la décomposition de 1'azométhane. Gay a aussi constaté qu'un effet de surface affecte la décomposition de 1'azométhane aux pressions inférieures à 3 torr; cet effet de surface serait dû à 1'adsorption de 1'azométhane sur
les parois du réacteur.
Lee (20), en 1959, a étudié, à l'aide d'un tube de choc, la décompo sition de 1'azométhane en présence d'argon et d'hélium. Il a trouvé, à basse concentration d'azométhane dans le mélange argon-hélium, que la dé composition était monomoléculaire. Bauer et ses collaborateurs (21) sont arrivés à la même conclusion, eux aussi en étudiant la décomposition de 1'azométhane par la technique du tube à choc. D'après ces derniers, au cours de la décomposition de mélanges de 1 à 3% d'azométhane dans l'argon, s'il y a une chaîne, elle est négligeable par rapport au processus mono moléculaire. Dans ces conditions expérimentales de hautes températures, la réaction de décomposition produit des radicaux CIL qui se recombinent pres- qu'entièrement. Il y a très peu de réactions secondaires. En effet, plus récemment, en 1970, Dove et ses collaborateurs (22), au cours de l'étude de la pyrolyse de 1'azométhane dans un tube à choc, ont aussi montré l'im portance de la réaction de recombinaison des radicaux Gif,. Ces études ont été faites à des températures de 500 à 1200°C pour des mélanges d'azométha
ne dans le krypton ou l'argon. La discussion de ces travaux a été faite par Kistiakowsky et ses collaborateurs (23) en 1971. Ils ont conclu que dans ces conditions la formation des produits secondaires n'implique pas la
5
participation de 11azomêthane. La principale réaction secondaire est l’abstraction d'un hydrogène de 1'éthane par un radical CH^ pour donner un radical qui se décompose ensuite pour former du C^H^.
Les travaux faits dans des tubes à choc ont donc montré qu'aux tem pératures élevées, la décomposition de l'azométhane ne donne pas lieu à la présence d’une chaîne. Par contre, à des températures beaucoup plus basses, de l'ordre de 300°C, la situation est différente comme le montrent les étu des de la décomposition de l'azométhane faites en système statique par Steel et Trotman-Dickenson (24) qui suivaient 1'évolution de la réaction par 1'analyse de l'azote. Ces travaux ont montré que le propylène avait un effet inhibiteur sur la réaction, en particulier aux pressions d'azométhane les plus hautes. Ils ont conclu à un double effet du propylène: accrois sement de la vitesse de la réaction due à un accroissement de 1'activation par collision et inhibition d'une courte chaîne au cours de la pyrolyse. La présence de certains effets de surface semblait confirmer 1'existence d'une chaîne initiée et terminée à la surface. De plus, ils ont noté que
la longueur de la chaîne était plus grande aux pressions les plus hautes.
Plus réceiment, en 1963, Forst et Rice (25) ont étudié la pyrolyse de l'azométhane en présence de différents inhibiteurs tels 1'éthylène, le propylène et le NO. Ils ont montré qu'il y a effectivement une courte chaî ne au cours de la pyrolyse de l'azométhane et que 1'éthylène et le propy
lène ne sont pas, pour cette réaction, des inhibiteurs adéquats. Par con tre, ils ont trouvé que 1'addition de faibles quantités de NO réduisait la formation d'hydrocarbures de façon appréciable; le rendement du CH^, du
et du peut être réduit pratiquement à zéro quand la quantité de NO ajoutée est suffisante alors que la formation de l'azote se trouve ré duite à un minimum. Cependant, 1 'addition de quantités de plus en plus im
portantes de NO fait ensuite croître la vitesse de la réaction mesurée par la formation de l'azote. Forst et Rice ont expliqué ce fait par la forma tion d'une faible quantité d'azote due à la décomposition du NO lui-même et à la décomposition de 1'azométhane induite par le NO. Ils ont alors corri gé pour ces deux effets la vitesse de décomposition inhibée et ils ont con clu qu'elle correspondait au processus monomoléculaire initial suivant:
CH3N2CH3 + 2CH3 + N2
Forst et Rice ont ainsi mesuré une énergie d'activation de 55.5 kcal mole-1 pour cette réaction. La longueur de chaîne qu'ils ont mesurée varie d'en viron 1.5 à 3.3 à 1'intérieur du domaine de température et de pression
qu'ils ont étudié. Ils ont remarqué que la longueur de chaîne diminue quand la température de pyrolyse augmente et qu'elle augmente quand la pres sion de pyrolyse est plus grande.
Puis, en 1966, Forst (26) a discuté de la décomposition monomolécu laire de 1'azométhane en fonction de la version quantique de la théorie Marcus-Rice. Il a alors montré qu'en dépit de la façon indirecte dont la partie monomoléculaire de la décomposition de 1'azométhane a été étudiée, il se dégage une image cohérente de la monomolécularité de la réaction in hibée même si le domaine de pression qui a été étudié est petit. Par con tre, comme Forst (27) l'a mentionné, le mécanisme de la réaction non in hibée est encore à l'état d'ébauche. Ceci provient du fait, qu'en dépit du grand nombre d'études de la décomposition de 1'azométhane, aucune étude systématique de tous les produits de la pyrolyse non inhibée n'a été faite. Notre but sera donc de trouver quels sont les produits qui se forment au cours de la décomposition de 1'azométhane et ensuite d'étudier l'évolution de la formation de ces produits en fonction de la température et de la
7
pression de pyrolyse afin de proposer un mécanisme de la décomposition de 1 *azométhane. Nous pourrons alors comparer certains de nos résultats à ceux trouvés par Rice et Chang (28) pour 1'azométhane-d^ et à ceux de
METHODE EXPERIMENTALE
1.1 Préparation de 11azométhane
L1azométhane a été préparé par la méthode décrite par Renaud et Leitch (30) qui consiste à oxyder la diméthyl-1,2 hydrazine par l'oxyde mercurique.
La diméthyl-1,2 hydrazine est obtenue en mettant en solution 10 g du sel chloré de la diméthyl-1,2 hydrazine, CH^NHNHCH^•2HC1 (Aldrich Chemi cal Co., Inc., Milwaukee, Wis.), dans une solution de 10 g de NaOH dans 50 cc d'eau distillée. La solution d'hydrazine ainsi obtenue est placée dans 1'ampoule à décantation A, (voir figure 1.1), reliée par un joint rodé au ballon B qui contient une dispersion de 100 g d'oxyde mercurique jaune, HgO, qui est maintenu en état de dispersion à l'aide d'un agitateur magnétique situé sous le ballon B. Le système schématisé à la figure 1.1 étant totalement clos, on doit créer une légère dépression d'environ 100 torr dans le système afin de pouvoir ajouter facilement la solution d'hy drazine. Celle-ci est alors ajoutée goutte à goutte dans le ballon B, ce qui se traduit par une lente augmentation de la pression mesurée à l'aide du manomètre M. Au moment où la pression à 1 ' intérieur du ballon B est à peu près revenue à la pression atmosphérique, on arrête d'ajouter la
solu-9
A:B:
C:M:
R:ampoule à décantation qui con tient la solution de dime thyl -1,2 hydrazine
ballon qui contient l'oxyde mercurique en suspension dans l'eau
P: piège refroidi à la tempé rature du mélange acétone- glace carbonique
R,: pièges refroidis à la tem pérature de l'azote liqui de
ballon de stockage de l'azo- méthane
manomètre à mercure
robinet
T^: tube en "IJ" contenant des pastilles de KOH
Tg: tube en "U" contenant des billes de verre saupou drées de P^Or
Figure 1.1: Schéma de l'appareil utilisé pour la prépa ration de 1'azométhane.
tion d'hydrazine, et on crée de nouveau une légère dépression dans le bal
lon B en ouvrant le robinet R pour évacuer un peu du mélange azométhane-air vers le piège P préalablement évacué et maintenu à la température du mélan ge acétone-glace, carbonique. Alors, on recommence le processus jusqu'à ce que toute la solution d'hydrazine soit ajoutée, ce qui dure environ trois heures. Ensuite, le ballon B est lentement réchauffé jusqu'à une tempéra ture de 70°C afin de dégager lentement 1'azométhane pour le condenser dans le piège P. Finalement, le ballon B est lentement évacué à travers le piè ge P, les tubes en "U" et Tg, et les pièges P^ et P^ placés à la tempé rature de l'azote liquide. L'azométhane condensé dans le piège P est dis tillé à travers le tube en "U" T^, contenant du KOH, vers le piège P^, puis du piège P^ à travers le tube en "U" contenant du P^Or jusqu'au piège
P2 *
L'azométhane obtenu est débarrassé de toute trace d'air par pompage à la température de l'azote liquide et de traces de CJHU par pompage à la température du mélange azote liquide-alcool isopropylique (-129°C). L'ana lyse par chromatographie de 1'azométhane ainsi préparé n'a révélé la pré sence que d'une légère impureté. Le spectre de masse de cette impureté était le même que celui du CH^Cl donné dans la littérature (31); l'injec tion d'un peu de CH^Cl sur la colonne à chromatographie a confirmé 1'iden tification de cette impureté. Le CH^Cl a aussi été trouvé par Gay (19) en très petite quantité dans 1'azométhane préparé par la même méthode que nous avons utilisée.
L'azométhane étant sensible à une décomposition photochimique, il a toujours été conservé dans un ballon teinté en rouge C et gardé à la tem pérature de l'azote liquide.
11
1.2 Appareillages
Nous avons utilisé pour nos expériences un système statique classi que consistant d’un réacteur sphérique placé dans un four comme on peut le voir à la figure 1.2. L'évacuation du système est assurée par une pompe à palette et une pompe à diffusion à mercure qui permettent d'atteindre des pressions de 1CT5 torr telles que mesurées à l'aide de la jauge McLeod Mc. Le système de mesure de pression comprend aussi un manomètre m.
Le réacteur R utilisé est un ballon de Pyrex qui est relié au robinet B par un tube semi-capillaire afin de réduire le volume mort. Le volume de l'ensemble formant le réacteur, qui est de 308.3 cm3, a été calibré à l'eau distillée. Le réacteur est situé dans un cylindre d'aluminium de 12 pouces de longueur et de 2 pouces d'épaisseur dont les extrémités sont fermées par des plaques d'aluminium. Le cylindre d'aluminium est lui-même placé dans un cylindre d'amiante d'une épaisseur de .75 pouce autour du quel est enroulé un fil d'une résistance de 70 ohms. Le tout est placé dans une boîte d'amiante de 24 x 24 x 28 pouces remplie de laine minérale pour assurer une bonne isolation. La température du réacteur est contrô
lée par un autotransformateur permettant de varier le voltage appliqué aux bornes du filament chauffant. La lecture de la température du réacteur est effectuée à l'aide d'un thermocouple placé dans une cavité qui se pro longe jusqu'au centre du réacteur, et d'un potentiomètre de la maison Ru bicon Instruments (No 2745). Nous avons constaté que le système utilisé avait une grande inertie thermique, ce qui nous a permis d'obtenir des tem pératures qui demeuraient constantes pour des périodes de temps plus gran des que les temps de pyrolyse les plus longs qui sont d'environ deux heures. La lecture de la température de la pyrolyse a été faite à plus ou moins 0.1°C.
A: ballon de stockage de 1'azométhane B et B ' : robinets C: système de mesure de la quantité d'azométhane à pyrolyser P: . piège froid
m: manomètre à mercure R: réacteur
Me : jauge McLeod T: pompe Toepler
M: burette à gaz V: valves d'introduction dans
le chromatographe
Figure 1.2: Schema de l'appareil utilise pour pyro- lyser 1'azométhane.
13
L'azométhane qui doit être pyrolysé est amené du ballon de stockage A au système de mesure C. Ce système de mesure est constitué de trois pe
tits ballons reliés à un réservoir de mercure. Le mercure peut être amené à une des trois marques gravées sous chacun des ballons à partir desquelles les volumes compris entre chacune de ces marques et les robinets B et B' sont connus exactement par calibration au mercure. La lecture de la pression d'azométhane dans un de ces volumes de référence est faite en mesurant la hauteur de la colonne de mercure dans la branche de gauche du système de mesure C par rapport à la marque de référence utilisée. Une mesure de pres sion avant 1 'introduction de l'azométhane dans le four et une seconde mesure de pression après 1'introduction permet de connaître la quantité d'azométha ne dans le four. L'opération de mesure de l'azométhane et son injection dans le four sont faites dans les plus brefs délais afin de limiter le plus possible le temps d'exposition de l'azométhane à la lumière.
Après pyrolyse, l'azométhane est évacué du four directement dans le piège P préalablement refroidi à la température de l'azote liquide. Nous
avons aussi utilisé un piège de type LeRoy (32) à la place du piège P; ce piège à doubles parois est entouré d'un fil chauffant qui permet de varier la température du piège et ainsi d'effectuer une distillation de mélanges gazeux. La pompe de type Toepler T permet ensuite de pomper les fractions distillées à différentes températures vers le système de mesure M consti tué d'une burette à gaz calibrée au mercure. Du système de mesure M, cha que fraction peut être injectée dans un chromatographe à gaz à l'aide du système de valves V ou peut être recueillie pour analyse par spectrométrie de masse. Le système de valves V est constitué de valves en acier inoxyda ble afin d'éviter 1'emploi de graisse. Ces valves de marque Hoke, à poin teau, qui sont rapides à manipuler, permettent de contrôler l'évacuation
de la boucle où circule l'hélium et d'introduire les échantillons dans un chromatographe de la maison F & M (modèle 700) équipé d'un détecteur à filaments chauds (les filaments sont du type Gow-Mac, WX). L'analyse quan titative du mélange des produits a été faite sur trois colonnes à chromato graphie différentes placées en parallèle dans le chromatographe et reliées entre elles par une valve à trois voies permettant l'injection des produits directement dans la colonne désirée.
CHAPITRE 2
PRODUITS DE LA DECOMPOSITION DE L'AZOMETHANE
Lors d'une pyrolyse de 1'azométhane, on peut prévoir, à priori, que le mélange à analyser contiendra des hydrocarbures saturés et insatu- rés d'un à quatre ou cinq atomes de carbone, de l'azométhane qui n'a pas été décomposé, différents produits azotés et de l'azote élémentaire. La formation de quelques-uns de ces produits, tels l'azote, le méthane, l'étha- ne, l'éthylène et le propane, au cours de la décomposition de 1'azométhane, a déjà été prouvée antérieurement (14), (18), (20) et (25). Par contre, la formation d'autres produits comme la tétraméthylhydrazine et l'éthylméthyl- diimide a été suggérée, mais elle n'a pas été vérifiée directement par ana lyse. D'autres produits, tels différentes amines, des nitriles ou de l'aci de cyanhydrique pourraient aussi être éventuellement formés au cours d'une pyrolyse de 1'azométhane. Il est donc nécessaire, si on veut proposer un mécanisme cohérent pour la décomposition de 1'azométhane, de savoir exacte ment lesquels de ces produits sont formés et de connaître 1' importance de chacun des produits formés.
La première partie de notre travail a donc consisté en une recherche systématique des différents produits de la pyrolyse de 1'azométhane, ce qui inclut aussi, la vérification de l'absence d'autres produits dont la forma
tion pourrait être possible. C'est ainsi que nous avons été amenés à étu dier plusieurs colonnes à chromatographie et à identifier par spectrométrie de masse les produits élués sur ces colonnes. Ce chapitre comprend la lis te de ces colonnes à chromatographie, la description de leurs caractéristi ques et des produits pouvant être analysés sur chacune d'elles ainsi que l'identification de plusieurs des produits par spectrométrie de masse. Nous avons divisé ce chapitre en deux sections. La première traite des produits "légers", c'est-à-dire, de l'azote et des hydrocarbures gazeux. La seconde section traite des produits "lourds", c'est-à-dire, de 1'azométhane et des autres produits azotés qui sont de poids moléculaires semblables ou supé rieurs à celui de 1'azométhane. Au cours de la recherche des produits de pyrolyse, les analyses sur les différentes colonnes ont été faites sans condensation préalable des produits de pyrolyse afin d'éviter que certains des produits restent absorbés sur les parois d'un piège froid.
2.1 Produits "légers"
2.1.1 Colonne I: colonne d'alumine (25)
Cette colonne qui peut être employée pour séparer l'azote, le métha ne et les hydrocarbures contenant jusqu'à trois atomes de carbone, ne con tient que de l'alumine de type Alcoa F-l (The Coast Engineering Laboratory, Hermosa Beach, Californie). C'est une colonne en cuivre d'une longueur de
26 pieds et d'un diamètre extérieur de \ de pouce. La température d'uti lisation de cette colonne est de 42°C à un débit d'hélium de 40 cc par mi
nute .
Par injection des produits purs sur cette colonne, nous avons obtenu les temps de rétention de quelques produits ; ces temps de rétention sont
17
donnés au tableau 2.1. On y remarque que la séparation des produits injec tés sur la colonne I est très bonne ; la séparation de 1'azote et du métha ne permet d'effectuer facilement des calibrations pour ces deux gaz car leurs pics d*élution sont symétriques et très étroits. Nous avons tracé pour chacun de ces produits une courbe de calibration obtenue par des injec tions successives de quantités différentes du produit pur sur la colonne et nous avons vérifié que ces courbes de calibrations sont des droites qui pas sent par 1'origine. Nous avons aussi vérifié que les courbes de calibra tion de l’azote pur et du méthane pur étaient les mêmes que celles obtenues par injection de différents mélanges d'azote et de méthane de compositions
connues.
L'analyse sur la colonne I d'une partie d'un mélange obtenu après la pyrolyse de l'azométhane révèle la présence d'azote, de méthane, d'étha- ne, de propane et d'éthylène; 1'importance relative de chacun de ces pro duits est dans l'ordre dont les produits ont été nommés. Les analyses fai tes après plusieurs pyrolyses effectuées sous différentes conditions expéri mentales montrent que ces cinq produits sont les seuls qui sont élués sur
la colonne I.
TABLEAU 2.1
Analyse sur la colonne I
Produit Temps de rétention (minutes)
N. '2 5.4 16.0
21.6
6.7 50.02.1.2 Colonne II: colonne de carbonate de propylène sur alumine (33)
Cette colonne qui sert principalement pour la séparation des hydro carbures en C? et C^ contient 20.8% en poids de carbonate de propylène
(Eastman Organic Chemicals, Rochester, N.Y.) sur de 1 'alumine du type Alcoa F-l. 1'alumine est la même que celle utilisée pour la colonne I.
L’éther éthylique a été employé comme solvant pour déposer le carbonate de propylène sur 1’alumine. La tempérautre d'utilisation de la colonne II est de 42°C à un débit d'hélium de 40 cc par minute. Les temps de rétention de plusieurs produits donnés au tableau 2.2 ont été obtenus par l'injection des produits purs sur la colonne II.
La méthode utilisée pour déposer la phase liquide sur le support so lide a été la même pour la fabrication de toutes nos colonnes. Cette métho de consiste à dissoudre la phase liquide dans un solvant qui est plus vola til et à ensuite mélanger la solution obtenue et le support solide en em ployant suffisamment de solvant pour que le support solide baigne complète ment dans la solution. Le solvant est alors évaporé à l'aide d'un évapora-
teur rotatif de façon à former un dépôt homogène de la phase liquide sur le support solide. La colonne est ensuite remplie en la tenant verticalement et en ayant soin d'utiliser un vibrateur pour s'assurer de l'homogénéité du matériel de remplissage. Finalement, le conditionnement de la colonne est effectué par la circulation à 1'intérieur de celle-ci d'un léger flux d'hélium à une température légèrement supérieure au point d'ébullition du solvant employé. Le temps nécessaire au conditionnement d'une colonne dé pend de la nature des solvants et des phases liquides utilisés.
L'analyse, à l'aide de la colonne II, des produits de pyrolyses faites à différentes conditions expérimentales, n'a pas permis de découvrir
19
TABLEAU 2.2
Temps de rétention de différents produits sur la colonne II*
Produit Temps de rétention (minutes)
CH, + N, 5.0 C2H6 + C2H4 6.5 C3H8 8.9 ^«6 10.6 ^°‘C4H10 12.8 n“C4H10 15.6 néo-C5H12 18.0 l-C,Hg 19.2 19.9 tyumô-2-C,Hg 21.9 cc4-2-C,Hg 25.0 ch3ci 29.7 CH3N2CH3 45.8
Il faut noter que ce tableau ne correspond pas à une analyse des / produits de pyrolyses mais qu'il est une liste des produits qu'rVi peut séparer a l'aide de la colonne II.
y
archives
ET LIVRES RARES
des quantités importantes d'hydrocarbures autres que ceux trouvés à l’aide de la colonne I. En effet, les chromatogrammes alors obtenus comportent quatre pics situés à 5.0 minutes (Ng + CH^), 6.5 minutes (C^H^ + C^H^j, 8.9 mi nutes (CjHg) et à 45.8 minutes (CHÿ^CHg). Comme on peut constater d'après le tableau 2.2 que la colonne II permet l’analyse d'hydrocarbures en et Cg, on doit conclure que les seuls hydrocarbures formés au cours de la pyro lyse sont ceux analysés sur la colonne I. Cependant, nous avons noté la formation de propylène et de n-butane à l’état de traces au cours de certai nes pyrolyses.
2.2 Produits "lourds"
2.2.1 Colonne III: colonne de diméthoxytétraéthylène glycol sur Fluoropak.
Cette colonne contient 20.0% en poids de diméthoxytétraéthylène gly col déposé sur du Fluoropak 80 (ces deux produits sont de la maison Applied Science Laboratories Inc., State College, P.A.). Le chloroforme a été uti lisé comme solvant. Cette colonne est en cuivre et elle a une longueur de 6 pieds et un diamètre extérieur de l de pouce alors que sa température d’utilisation est de 25°C à un débit d’hélium de 40 cc par minute.
Nous avons utilisé cette colonne pour faire les analyses de diffé rentes pyrolyses effectuées sous diverses conditions expérimentales. Les chromatogrammes ainsi obtenus comportent cinq pics dont les temps de ré tention sont donnés au tableau 2.3.
Le pic situé à 1.2 minutes a été identifié par injection des pro duits purs comme étant celui du couple N^-CH. qui sont élués en même temps sur cette colonne. De même façon, le pic à 1.8 minutes a été identifié
21
Analyse sur la colonne III
TABLEAU 2.5
Produit Temps de rétention (minutes)
N
2
+ CH4
1.2C2H4 + C2H6 1.8
C3H8 3.4
8.5
III-A 19.5
comme étant dû au CLH. et au alors que les pics à 3.4 et 8.5 minutes proviennent du CL1L et de 1 *azométhane respectivement. Le produit élué à 19.5 minutes n’a pu être identifié à aucun des produits purs que nous possédions dans notre laboratoire et qui ont été injectés sur la colonne ; nous l'avons appelé produit III-A.
Dans les cas où il nous était impossible d'identifier les produits élués sur diverses colonnes, nous avons eu recours à 1'analyse de ces pro duits par spectrométrie de masse après en avoir recueilli à la température de l'azote liquide les pics chromatographiques correspondants. La techni que qui a été utilisée pour recueillir ces produits est la suivante: au moment désiré, à l'aide d'un système de valves placées à la sortir du chro- matographe, le flux d'hélium pouvait être dirigé dans un tube en "U" con
tenant de la laine de verre. Plusieurs de ces tubes en "U", préalablement purgés à l'hélium et refroidis à la température de l'azote liquide,
pou-valent être disposés de façon à ce que le flux d’hélium puisse à tout mo ment être dirigé indifféremment dans n’importe lequel des tubes. Nous pouvions de cette façon obtenir séparément les produits élués à des temps différents sur une colonne donnée et ensuite les analyser par spectrométrie de masse. Tous les spectres de masse que nous avons obtenus l'ont été à une énergie de 70 électrons volts sur un appareil de type cycloidal (Con solidated Electrodynamics Corporation, modèle 149400).
Afin de vérifier notre technique, nous avons piégé le pic d'azométha- ne élué à 8.5 minutes sur la colonne III et nous avons pris son spectre de masse. On peut voir, d'après le tableau 2.4, que le spectre de masse de l'azométhane ainsi piégé se compare très bien à celui d'un échantillon d'azo- méthane pur injecté directement dans notre spectromètre de masse. La seule différence notable entre ces deux spectres de masse se trouve à m/e = 44 où le spectre de masse de l'azomêthane piégé possède un pic qui provient sans doute du CO^ contenu dans l'hélium et donc piégé en même temps que 1'azométhane. Le spectre de masse du CO^ possède en effet un pic très in tense à m/e = 44 (34). Nous avons aussi inclus dans le tableau 2.4 les spectres de masse de 1'azométhane cités dans la littérature (35), (36). Les spectres de masse a et b du tableau 2.4 qui ont été obtenus pour 1'azométha ne sur notre appareil montrent une fragmentation analogue aux spectres de masse ç et d; les différences d'intensité qui existent entre les spectres de masse donnés au tableau 2.4 sont dues en partie au fait que les spec- tromètres qui ont été utilisés pour les obtenir sont différents.
Après avoir vérifié que le spectre de masse de 1'azométhane piégé à la sortie de la colonne III était semblable au spectre de masse de 1'azomé thane pur, nous avons décidé d'employer la même méthode pour identifier par
23
TABLEAU 2.4
Spectres de masse de 1 ’ azomé thane ; a: azomé thane piégé à 8.5 mi nutes sur la colonne III; b: azométhane pur; c: Stief et Ausloos
(35); d: Prdsil et Forst (36). m/e Abondance relative en % ion a 70 eV b 70 eV c 70 eV d 75 eV 14 7.6 8.0 10.0 7.5 ®2+ 15 100.0 100.0 100.0 100.0 CH3+ 26 1.0 1.0 1.6 1.4 %' 27 7.6 7.8 12.8 12.0 HCN+ 28 22.7 17.8 28.5 31.6 CH/ 29 1.9 1.3 1.8 2.0 CH3N+, 30 . 2.6 2.4 2.7 3.6 %* 42 4.5 3.7 5.6 6.8 43 26.2 26.4 34.1 42.6 44 11.0 57 2.5 1.0 1.5 2.4 58 10.3 12.6 12.9 23.0
%
spectrométrie de masse la nature des produits inconnus élués sur diverses colonnes. Nous avons ainsi pris le spectre de masse du produit III-A élue à 19.5 minutes sur la colonne III. On remarque d'après ce spectre donné au tableau 2.8 que le pic situé à la masse la plus haute est celui à m/e = 72. Donc, ce produit aurait un poids moléculaire de 72 si ce pic correspond à l'ion parent. Comme un hydrocarbure en Cc. de poids moléculaire de 72 de vrait être élué avant 1'azométhane sur la colonne III, on doit donc conclu re que le produit III-A, élué 11 minutes après 1'azométhane, n'est pas un hydrocarbure, mais probablement un produit semblable à 1'azométhane conte nant de l'hydrogène, du carbone et de l'azote, seul autre élément disponi ble à 1'intérieur de notre système. La nature exacte de ce produit sera discutée plus loin, à la section 2.2.5.
2.2.2 Colonne IV: colonne de carbonate de propylène sur Firebrick (33)
Cette colonne contient 20.0% en poids de carbonate de propylène (Eastman Organic Chemicals, Rochester, N.Y.) sur du Firebrick de type Gas- Chrom R, 30-60 mesh (Applied Science Laboratories, Inc., State College, P.A.). Cette colonne est en cuivre et elle a une longueur de 9 pieds et un diamètre extérieur de l de pouce. La température d'utilisation de la colonne est de 25°C à un débit d'hélium de 40 cc par minute. L'éther éthy lique a été employé pour déposer la phase liquide sur le Firebrick.
Nous avons utilisé cette colonne pour faire des analyses de diffé rentes pyrolyses effectuées à diverses conditions expérimentales. Les chromatogrammes ainsi obtenus comportent cinq pics dont les temps de ré tention sont donnés au tableau 2.5.
25
TABLEAU 2.5
Analyse sur la colonne IV
Produit Temps de rétention (minutes)
N% + C^ + C% 2.1 à 2.2
C3H8 3.0
0
%%
10.4IV-A 19.0
IV-B 42.4
Les produits élues à 2.1, 3.0 et 10.4 minutes ont été identifiés par injection de produits purs sur la colonne. Les produits IV-A et IV-B élués à 19.0 et 42.4 minutes respectivement ont été piégés de la façon habituelle afin d'obtenir leurs spectres de masse qui sont donnés aux tableaux 2.8 et 2.11 respectivement. La nature du produit IV-A sera discutée à la section 2.2.5, tandis que celle du produit IV-B sera discutée à la section 2.2.6.
2.2.3 Colonne V: colonne de Carbowax 400 sur Fluoropak (37)
Cette colonne contient 20.0% en poids de Carbowax 400 déposé à l'aide du méthanol sur du Fluoropak 80 (les deux produits sont de la maison Applied Science Laboratories, Inc., State College, P.A.). Cette colonne est en cui vre et elle a une longueur de dix pieds et un diamètre extérieur de l de pouce, alors que sa température d'utilisation est de 65°C à un débit d'hé lium de 40 cc par minute.
Cette colonne a été employée pour analyser des pyrolyses faites à diverses conditions de température et de pression. Les chromatogrammes ainsi obtenus comportaient cinq pics dont les temps de rétention sont don nés au tableau 2.6. De plus, nous avons remarqué sur les chromatogrammes, la présence de deux autres produits élués sous forme de traces à 51.0 et 58.0 minutes. Dans le but d'obtenir suffisamment de ces deux produits pour les analyser, nous avons effectué une pyrolyse pendant un temps très long. La nature des produits alors formés était la même que lors des pyrolyses plus courtes, mais la quantité des produits formés était beaucoup plus gran de. L'injection sur la colonne de plusieurs produits purs ne nous a pas permis de trouver l'identité des produits élués à 21, 27, 40, 51 et 58 mi nutes sur la colonne V.
TABLEAU 2.6
Analyse sur la colonne V
Produit Temps de rétention (minutes)
2.0 CH3% 4.0 V-A 21.0 V-B 27.0 V-C 40.0 V-D 51.0 V-E 58.0
27
analysés par spectrométrie de masse. Les spectres de masse des produits élués à 21, 51 et 58 minutes sont donnés au tableau 2.12 et la nature de ces produits sera discutée à la section 2.2.7. Les spectres de masse des produits élués à 27 et 40 minutes sont donnés aux tableaux 2.8 et 2.11 res pectivement alors que la nature de ces deux produits sera discutée aux sec tions 2.2.5 et 2.2.6 respectivement.
2.2.4 Colonne VI: colonne de Carbowax sur Firebrick (38)
Cette colonne contient 40.0% en poids de Carbowax 400 déposé, à l'aide du méthanol, sur du Firebrick de type Cas Chrom R, 30-60 mesh (les deux produits proviennent de la maison Applied Science Laboratories, Inc., State College, P.A.). Cette colonne est en cuivre et elle a une longueur de six pieds et un diamètre extérieur de l de pouce. La température d'uti lisation de la colonne est de 100°C à un débit d'hélium de 33 cc par minu te. Plusieurs analyses de pyrolyses faites à diverses conditions expéri mentales ont été effectuées à l'aide de cette colonne. Les chromatogrammes alors obtenus comportent six pics dont les temps de rétention sont donnés au tableau 2.7. L'identification des produits VI-A et VI-D sera faite à la section 2.2.6, tandis que la nature des produits VI-C et VI-B sera dis cutée aux sections 2.2.5 et 2.2.7 respectivement.
2.2.5 Identification des produits "lourds" de poids moléculaires de 72
L'analyse par spectrométrie de masse des échantillons piégés à la sortie de diverses colonnes à chromatographie a montré que quatre de ces échantillons, III-A, IV-A, V-B et VI-C, ont un spectre de masse où le pic situé à la plus haute valeur de m/e est celui à m/e = 72. Ainsi, si ces
Analyse sur la colonne VI
TABLEAU 2.7
Produit Temps de rétention (minutes)
N2 + C1 + C2 + C3 1.6 3.7 VI-A 5.2 VI-B 11.4 VI-C 13.6 VI-D 19.0
pics à m/e = 72 correspondent aux pics parents, ces quatre spectres de masse, qui sont donnés au tableau 2.8, sont probablement ceux de produits de poids moléculaires de 72. Les temps de rétention des produits III-A, IV-A, V-B et VI-C étant plus grands que le temps de rétention de l'azomé- thane sur leurs colonnes à chromatographie respectives, ces produits ne peuvent être des hydrocarbures car, sur ces colonnes à chromatographie, des hydrocarbures de poids moléculaires de 72 seraient élués avant l'azomé thane lui-même. Comme l'azote est le seul élément autre que le carbone et l'hydro gène qui soit disponible dans notre système, les produits III-A, IV-A, V-B et VT-C doivent donc être composés d'azote en plus du carbone et de l'hydro gène .
L'étude des quatre spectres de masse donnés au tableau 2.8 nous per met de constater que les spectres V-B et VI-C sont presqu'exactement les
29
mêmes et que les spectres III-A et IV-A montrent aussi une grande simili tude entre eux. La différence entre les spectres III-A et IV-A à m/e = 28 peut être due à la présence d'un peu d'air dans l'échantillon III-A. Le
fait que les pics les plus importants sont plus intenses pour le spectre IV-A que pour le spectre III-A est dû à l'effet de normalisation à une va leur de 50 de 1'intensité des pics à m/e = 72. Les quantités de produits analysés étant très faibles, nous avons dû utiliser notre spectromètre de masse à la limite de sa sensibilité. Comme à cette sensibilité le spectre de fond de l'appareil est important et qu'il varie dans le temps, la correc tion des spectres de masse des échantillons par soustraction du spectre de fond entraîne des erreurs qui sont relativement plus grandes aux valeurs de m/e dont les intensités sont les plus faibles.
Donc, les spectres de masse III-A et IV-A seraient tous les deux celui d'un même produit azoté de poids moléculaire de 72 alors que les spec tres de masse V-B et VI-C seraient tous les deux les spectres d'un second produit azoté ayant aussi un poids moléculaire de 72. La recherche de pro duits contenant de l'azote, du carbone et de l'hydrogène et ayant un poids moléculaire de 72 nous amène à considérer finalement deux structures diffé rentes possibles: Œ: 3 N=N \ nN-N=CH.
2
CH:
3ê thylmé thy ldi imide diméthylhydrazone du formaldéhyde
Le spectre de masse de 1'éthylméthyldi imide n'étant pas donné dans la lit térature, il nous faut essayer d'en déterminer les principales caractéris tiques à la lumière des spectres de masse de produits dont les structures
sont semblables.
Au tableau 2.9, nous avons réuni les spectres de masse de deux diimi- des symétriques, 11azométhane et l'azoéthane, et d'un diimide non symétri que, le Aec-butyléthyldiimide. L'examen de ces spectres révèle que les pics les plus intenses dans les spectres de masse de diimides symétriques, RN
2
R, ou non symétriques, R'N^R, sont ceux qui correspondent aux ions R+ ou R,+. De plus, ces spectres de masse ont des pics importants qui corres pondent aux ions RN^^ et R1^*. Finalement, on doit noter dans ces spectres la présence du pic parent et l'absence de pic intense correspondant à l'ion parent amputé d'un atome d'hydrogène. Ainsi, par analogie avec les spectres de masse des diimides donnés au tableau 2.9, on peut prévoir quels sont les pics les plus caractéristiques que devrait contenir le spec tres de masse de 1'éthylméthyldiimide. Ceux-ci seraient les suivants: en premier lieu, deux pics intenses à m/e = 15 et 29 correspondant aux ions
CH^"1" et CzHg^, puis, des pics à m/e = 43 et 57 correspondant aux ions 0-1^2+ et et finalement, le pic parent à m/e = 72. On devrait no
ter de plus 1'absence de pic intense à m/e = 71. Cette description des pics les plus caractéristiques qui devraient être présents dans les spec tres de masse de 1'éthylméthyldiimide correspond très bien aux spectres III-A et IV-A où les pics importants à m/e = 15, 27, 28, 29, 43, 57 et 72 peuvent être attribués aux divers ions dus à la fragmentation du CH-N^CJHr. Ainsi, les spectres de masse III-A et IV-A sont très probablement tous les
deux celui de 1'éthylméthyldiimide qui a été piégé à la sortie de deux co lonnes à chromatographie différentes.
A cause de l'absence d'un pic important à m/e = 29 et de la présence de pics intenses à m/e = 30, 42 et 71, les spectres de masse V-B et VI-C ne
31
TABLEAU 2.8
Spectres de masse des produits "lourds" de poids moléculaires de 72. Tous ces spectres de masse ont été normalisés de façon à avoir une intensité relative de 50 à la masse 72.
m/e
Abondance relative en %
III-A
IV-A
V-B
VI-C
14 35.8 52.3 16.4 13.3 15 208.0 319.1 124.7 99.0 26 26.7 24.9 2.4 1.5 27 150.8 209.4 18.5 16.2 28 351.0 275.0 26.9 20.4 29 199.2 236.2 6.8 4.9 30 19.9 23.4 25.4 29.6 42 26.9 28.9 68.7 68.9 43 286.1 327.6 40.3 35.1 57 39.2 46.4 16.9 17.6 58 18.9 28.5 11.5 13.4 71 1.2 3.2 27.6 28.2 72 50.0 50.0 50.0 50.0
peuvent être dus au ®
3
^2
^2^5" seconde structure considérée étant celle de la diméthylhydrazone du formaldéhyde, le spectre de masse de ce produit est connu (voir tableau 2.10). On remarque que les spectres de masse V-B et VI-C ont les mêmes caractéristiques que le spectre de masse de la dimé thylhydrazone du formaldéhyde: la fragmentation est la même et l'intensité des pics à m/e = 72, 71, 57, 42, 30, 28 et 27 est presque identique dans les deux cas. Les différences d'intensité à m/e = 15 et 43 entre le spec tre de la diméthylhydrazone du formaldéhyde et les spectres V-B et VI-G s'expliquent par la présence d'un peu d'azométhane dans les échantillons V-B et VI-C comme 1'indique le pic à m/e = 58 des spectres V-B et VI-C. Donc, il n'y a pas de doute que les spectres de masse V-B et VI-C sont tousles deux celui de la diméthylhydrazone du formaldéhyde qui a été piégé à la sortie de deux colonnes à chromatographie différentes.
L'analyse par spectrométrie de masse des échantillons piégés à la sortie de diverses colonnes à chromatographie a donc révélé la présence de 1'êthylméthyldiimide et de la diméthylhydrazone du formaldéhyde dans les mélanges analysés après différentes pyrolyses de 1'azométhane. La formation de 1'êthylméthyldiimide au cours de la pyrolyse de 1'azométhane qui avait été suggérée par quelques auteurs, (18), (28) et (63), se trouve ainsi con firmée. Par contre, la formation de la diméthylhydrazone du formaldéhyde au cours de la pyrolyse de 1'azométhane n'a jamais été prise en considéra tion.
2.2.6 Identification des produits "lourds" de poids moléculaires de 86
Nous avons réuni au tableau 2.11 les spectres de masse IV-B, VI-A, V-C et VI-D dont le pic situé à la plus haute valeur de m/e est dans chaque
33
Spectres de masse de différents diimides; spectre de masse de l'azo- méthane sur notre appareil, de 11azoéthane d'après Clark (39) et du Aec-butyléthyldiimide d'après Strausz, Berkley et Gunning (40).
TABLEAU 2.9
azométhane azoéthane Acc-butyléthyldiimide m/e intensité ion intensité ion intensité ion
15 100 œ3+ 6 ch3+ 27 8 HCN+ 32 C2< HCN+ 13 C2< HCN+ 28 18 CH/ 24
C2V
29 100 100 41 11 CHN/ 50 %' CHN/ 42 4 9 %+ 7 C3H6+ ch2n2+ 43 26 CH^N/ 5 %' 56 10 %' 57 13 45 C4< C2H5N2+ 58 13 CH^CH^ 71 6 85 5 86 14 :2
W2
^ 114 8TABLEAU
2.10-Spectres de masse de 11éthylhydrazone de l'acétaldéhyde (40), de la diéthylhydrazone de l'acétaldéhyde (40) et de la diméthylhydrazone du formaldéhyde (41). intensité m/e ch3œ=n-n-hc2h5 ch3œ=n-n(c2h5)2 ch2=n-n(œ3)2 15 21 30 27 23 32 13 28 59 30 29 16 67 30 85 51 22 41 16 42 49 43 68 43 14 44 16 34 56 38 57 16 71 100 32 26 72 50 86 74 99 100 114 37
35
cas le pic à m/e = 86. Les quantités des produits IV-B, VT-A, V-C et VI-D qui sont formés au cours des pyrolyses de l'azomé thane étant très faibles, il en résulte que les spectres de masse qui correspondent à ces produits sont difficiles à mesurer avec précision pour deux raisons principales. La première raison est due à 1'importance du spectre de fond du spectromètre de masse dont plusieurs des pics sont du même ordre de grandeur que les pics correspondants des spectres de masse des produits. La seconde difficulté provient de la contamination des échantillons par des produits formés en quantités plus grandes et qui sont élués plus rapidement sur les colonnes à chromatographie. A cause de la forme allongée de plusieurs des pics d'élu- tion, on piège en même temps que l'échantillon un peu du produit qui le pré cède sur la colonne. Il serait donc illusoire de chercher à comparer de façon rigoureuse les intensités des pics des spectres de masse IV-B, VI-A, V-C et VI-D. Il convient plutôt de comparer les tendances caractéristiques de ces spectres de masse afin de voir s'ils correspondent aux mêmes pro duits .
Même s'il existe des différences quantitatives entre les spectres de masse IV-B et VI-A, il apparaît que les pics importants sont les mêmes dans
les deux cas. En effet, les spectres de masse IV-B et VI-A sont tous les deux caractérisés par des pics très intenses à m/e = 15, 27, 28, 29, 41, 42 et 43 et par d'autres pics à m/e = 39, 57, 72 et 86. La similitude entre les spectres de masse V-C et VI-D est aussi à noter. Ces deux spectres sont caractérisés par des pics à m/e = 15, 27, 30, 42, 43, 57, 71, 85 et 86.
Même si l'intensité des pics â m/e = 85 est très faible, il faut remarquer leur présence dans les spectres V-C et VT-D. Le pic à m/e = 85 devient une caractéristique très importante des spectres V-C et VI-D car il fut impossi ble de trouver une contribution à m/e = 85 pour les spectres IV-B et VI-A.
Spectres de masse des produits "lourds" de poids moléculaires de 86. Tous ces spectres ont été normalisés de façon à avoir une intensité relative de 13.8 à la masse 86.
TABLEAU 2.11
m/e
abondance relative en %
IV-B VI-A V-C VI-D
14 464.1 132.8 24.0 35.7 15 511.1 1725.0 194.3 250.9 26 33.2 43.1 10.5 5.5 27 339.8 370.9 43.8 34.4 28 370.0 487.0 52.5 12.0 29 208.5 224.3 62.3 17.0 30 4.1 51.3 36.7 30.1 39 27.6 51.7 6.9 1.2 41 100.2 89.7 23.7 1.8 42 45.0 110.4 58.4 65.7 43 454.8 733.1 75.4 89.1 57 45.0 44.8 33.3 32.1 58 17.9 13.8 21.6 56.7 71 7.7 2.3 33.6 30.3 72 35.3 32.8 9.9 17.0 85 1.9 1.7 86 13.8 13.8 13.8 13.8
37
Il faut donc conclure que les spectres IV-B et VI-A correspondent à un pro duit azoté de poids moléculaire de 86 tandis que les spectres V-C et VI-D correspondent à un second produit azoté ayant aussi un poids moléculaire de
86
.
Nous avons trouvé trois structures possibles pour des produits azo tés de poids moléculaires de 86. Celles-ci sont les suivantes:
C2H5 CH,
X
N=N x.X
N=N> C2H5 C3H7 CH-C2H5^
n
-
n
=
œ
2
azoéthane éthylméthylhydrazone n-propylméthyldiimide du formaldéhydeUn examen des spectres de masse donnés au tableau 2.11 nous montre qu'aucun de ces spectres de masse ne correspond à celui de 1'azoéthane donné au ta bleau 2.9. Dans le cas des spectres IV-B et VI-A, 1'importance des pics à m/e = 15, 27, 39, 41, 42, 43 et 72 est incompatible avec le spectre de masse de l'azoéthane tandis que dans le cas des spectres V-C et VI-D, l'intensité des pics à m/e = 15, 27, 42, 43, 71 et 72 et aussi la présence d'un pic à m/e = 85 sont incompatibles avec le spectre de masse de 1'azoéthane.
Les pics importants du spectre de masse du n-propylméthyldiimide peu vent être déduits par analogie avec les spectres des diimides donnés au ta bleau 2.9. Ainsi, on peut prévoir que le spectre de masse du CH^NgC^Hy de vait posséder des pics importants à m/e = 15 (CH^) et 43 (C^H^*) et qu'il devrait aussi contenir les pics dus à la fragmentation de l'ion C^Hy+. La fragmentation de cet ion donne des pics caractéristiques à m/e = 42 (C^H^+), 41 (C3H5+), 39 (C3H3+), 29 (C^H^), 28 (CgH^) et 27 (C^) comme on peut
(43) qui contiennent tous les deux un pic très intense à m/e = 43 (C^Hy+).
De plus, le spectre de masse du CE^C^Hy devrait aussi posséder des pics à m/e = 71 (CgHyN^^) et
86
(ion parent) et il ne devrait pas par contre avoirde pic à m/e = 85. Cette description du spectre de masse du CH^N^C^Hy s'ap plique très bien aux spectres IV-B et VI-A où les pics les plus importants sont situés à m/e = 15 (CH^) et à m/e = 27, 28, 29, 39, 41, 42 et 43 (pics de la fragmentation des ions C^Hy*) tandis que le pic à m/e = 71 correspond à l'ion CjHyN
2
+ et que celui à m/e =86
est dû à l'ion parent. La présenced'un pic à m/e = 72 dans les spectres de masse IV-B et VI-A ne peut être expliquée que par la contamination des échantillons IV-B et VI-A par un peu du (m/e = 72) élué lui aussi sur les mêmes colonnes à chromato
graphie .
La dernière structure à considérer est celle de 1'éthylméthylhydra- zone du formaldéhyde. Il nous faut voir si les spectres de masse V-C et VI-D peuvent être dus à cette hydrazone dont les principales caractéristi ques de son spectre de masse peuvent être déduites de celui de la diméthyl- hydrazone du formaldéhyde. D'après le tableau 2.10, le spectre de masse de la diméthylhydrazone du formaldéhyde, qui est de formule générale H
2
C=N-N(R)2
, contient les pics suivants: m/e = 72 (pic parent), 71(HC=N-N(R)2+), 57 (H2C=N-NR+), 42 (H2C=N-N+), 30 O^H^) et 15 (R+). Ainsi,
le spectre de masse de 1'éthylméthylhydrazone du formaldéhyde, qui est une hydrazone de formule H?C=N-N(R)r', devrait posséder des pics à m/e =
86
(pic parent), 85 (HC=N-N(R)R,+), 71 (I^CrN-NR^), 57 (H2C=N-NR+), 42
(HgCzN-N^), 30 (CgH^), 29 (R,+) et 15 (R+). Cette fragmentation s'applique
bien aux spectres V-C et VI-D sauf pour 1' importance des pics à m/e = 58, 43 et 15 qui peuvent être dus à une contamination par 1'azométhane. Le pic à m/e = 72 correspondrait à l'ion (N-N(R)R,+), ce qui est possible si on
39
compare avec les spectres de masse des autres hydrazon.es du tableau
2
.10
.Il se forme donc au cours de la pyrolyse de l'azométhane deux pro duits azotés de poids moléculaires de
86
qui sont le n-propylméthyldiimide et1
'éthylméthylhydrazone du formaldéhyde.2.2.7 Identification d'autres produits "lourds"
L'analyse par spectrométrie de masse des divers échantillons piégés a permis 1'identification de trois autres produits azotés. Les spectres de
masse de ces produits sont donnés au tableau 2.12. On y remarque que les spectres de masse VT-B et V-A sont très semblables sauf pour les pics à m/e = 28, 32 et 44 du spectre V-A qui sont dus à la présence d'air et de CO
2
dans 1'échantillon V-A. L'intensité des pics à m/e = 15, 27, 43 et 58 plus importante pour le spectre VI-B que le spectre V-A est probablement due à la contamination de l'échantillon VI-B par un peu d'azométhane. Les spectres VI-B et V-A possèdent des pics importants à m/e = 15, 30, 42, 43, 46, 73 et88
qui sont conformes à la fragmentation de la tétraméthylhydra- zine où en particulier, l'intensité des pics à m/e = 42, 46, 73 et88
est caractéristique du spectre de masse de la tétraméthylhydrazine tel que cité dans la littérature (44). Les échantillons VI-B et V-A sont donc de la tétraméthylhydrazine qui a été piégée à la sortie de deux colonnes à chro matographie différentes.Corne on l'a vu à la section 2.2.3, les produits V-D et V-E sont tous les deux formés à l'état de traces au cours de la pyrolyse de l'azo méthane. Nous avons tenu à les identifier et pour y arriver nous avons dû pyrolyser l'azométhane pendant un temps beaucoup plus long que le temps cor respondant à un taux de décomposition inférieur à 10%. A l'exception du
Spectres de masse des produits V-A, VI-B, V-D et V-E
TABLEAU 2.12
m/e abondance relative en % VT-B V-A V-D V-E 14 30.6 25.9 15.3 15.7 15 289.8 75.810.0
26 7.1 4.3 18.7 27 38.8 5.3 18.0 28 78.0 240.0 16.7 149.1 29 18.4 22.4 7.0 30 40.8 53.4 4.8 31 9.212.0
32120.0
38 10.4 39 18.5 4012.0
49.4 41 28.6100.0
42100.0
100.0
6.7 43 145.0 43.1 6.7 9.0 44200.0
41.7 45 14.3 13.8 46 45.9 44.8 5211.2
53 6.7 54 62.2 55 9.7 57 14.3 58 10.2 3.5 71 8.1 72 61. 5.2 73 59.2 58.6 88 63.3 75.841
pic à m/e = 44 dû au CO?, le spectre de masse V-D est exactement le même que celui du cyanure de méthyle tel que donné dans la littérature (45) tan dis qu'à l'exception de pics à m/e = 28, 32 et 44 dus à l'air et au CO?, le spectre de masse V-E correspond au spectre du cyanure d'éthyle tel que cité dans la littérature (46).
Nous avons aussi effectué par spectrométrie de masse 1'analyse du mé lange des produits après pyrolyse. Ces analyses ont été faites pour les di verses fractions du mélange des produits distillé à des températures diffé rentes à l'aide d'un piège de type LeRoy. Le but de ces analyses était la recherche de produits "lourds" autres que ceux déjà trouvés par chromato graphie. Nous avons pu ainsi identifier la présence des produits "lourds" déjà trouvés par chromatographie ; nous avons même vu ainsi le pic parent des produits "lourds" de poids moléculaires de
86
qui sont présents en très faibles quantités dans les mélanges à analyser. Ces analyses par spectro métrie de masse n'ont pas permis de découvrir de produits importants autres que ceux déjà trouvés. Cependant, la présence dans ces spectres de masse d'un pic très faible à m/e =102
révèle la formation à l'état de traces d'un nouveau produit que nous avons identifié à1
'hydrazine suivante :C2H5 CH3 )N-N(
ch
3
ch3
2.2.8 Produits non formés au cours de la pyrolyse de 1'azométhane
L'analyse des produits de décomposition de 1'azométhane a aussi por té sur la recherche de certains produits spécifiques. Nous allons simple ment mentionner quels produits ont fait l'objet d'une recherche et quelles
colonnes à chromatographie ont été utilisées pour essayer de mettre leur présence en évidence.
Wacks (47), au cours d'une étude de la décomposition thermique de l'azométhane en système dynamique, a identifié par spectrométrie de masse la méthylamine et l'acide cyanhydrique comme deux des produits de décompo sition de l'azométhane. Même si ces études ont été faites à des pressions beaucoup plus petites que celles que nous avons étudiées et à des tempé ratures beaucoup plus hautes que celles auxquelles nous avons travaillé, nous avons tenu à vérifier si la décomposition de l'azométhane, sous les conditions expérimentales auxquelles nous l'avons étudié, pouvait aussi don ner lieu à la formation de HCN et d'amines.
La recherche du HCN a été effectuée à l'aide d'une colonne de gel de silice, telle que celle décrite par Lichtin et ses collaborateurs (48), et employée par eux pour la séparation de mélanges contenant des hydrocarbures et du HCN. Nous avons noté le temps de rétention du HCN par injection du HCN pur sur cette colonne. Les analyses de pyrolyses faites sous diverses conditions expérimentales n'ont jamais montré aucune trace de HCN, même lorsque certaines pyrolyses ont été faites à des taux de décomposition beau coup plus grands que ceux normalement utilisés.
Pour la recherche des amines, nous avons utilisé une colonne de 5% de THEED et de 15% de TEP sur du chromosorb W, telle que décrite par Borke et ses collaborateurs (49) comme étant spécifique pour les amines. Sur cette colonne nous avons injecté diverses amines telles que (CH^)^N,
(CH^jgNH, CH^NHg et C^H^NH^, afin de trouver leurs temps de rétention. L'ana lyse des pyrolyses sur cette colonne ne nous a pas permis de trouver de pro duits correspondant à ces amines. Ceci confirme nos analyses faites sur
43
d'autres colonnes où aucun des échantillons piégés n'a pu être identifié à une amine quelconque.
Kerr et Calvert (50), ayant identifié la triméthylhydrazine comme étant un des produits formés au cours de la photolyse de l'azométhane en présence d'acétaldéhyde ou de monoxyde de carbone, nous avons vérifié si cette hydrazine se formait au cours de la pyrolyse de l'azométhane. L'ana lyse des produits de la décomposition de l'azométhane à l'aide de la colonne VI spécifique pour les hydrazines, n'a jamais montré la formation de trimé thylhydrazine au cours de la pyrolyse de l'azométhane. Les analyses par spectrométrie de masse n'ont jamais, non plus, permis de mettre en évidence la formation de triméthylhydrazine dont le spectre de masse (44) possède un pic très caractéristique à m/e =74.
Nous avons aussi vérifié, à l'aide d'une colonne de tamis molécu laire de type 5A telle que décrite par Lard et Hom (51), que la décompo sition de l'azométhane, sous les conditions expérimentales que nous avons employées, ne donnait pas lieu à la formation d'hydrogène comme l'a suggé ré Lee (20).
2.2.9 Résumé
La recherche des produits de décomposition de l'azométhane nous a permis de mettre en évidence la formation de N^, CH^, CgH^, C^H., CH
3
N2
C2
H5, GH3
N2
C3
Hy, (CH3
)2
N2
CH2, CH3
(C2
H5
)N2
CH2
et de (CHjgN (OL)^. Nous avons aussi noté la formation de traces de C^H^, , CHgCN,CgHgCN et d'une hydrazine de poids moléculaire de 102. Nous avons aussi vérifié que la décomposition de l'azométhane ne donne pas lieu à la forma tion de Hg, HCN, d'amines ou de triméthylhydrazine.
Les analyses quantitatives des produits de décomposition, au cours de ce travail, ont alors été effectuées sur la colonne I pour l'azote et les hydrocarbures, sur la colonne IV pour les diimides et sur la colonne VI pour les hydrazones et la tétraméthylhydrazine.
CHAPITRE 3
RESULTATS EXPERIMENTAUX
3.1 Reproductibilité des résultats
Les pyrolyses de l'azométhane étant faites dans un réacteur en py rex, le conditionnement du réacteur ne pose pas de problème particulier
(28). En effet, après trois ou quatre pyrolyses d’essai, les résultats deviennent reproductibles. Le conditionnement du réacteur a été fait cha que fois que de l'air était admis dans le réacteur. Les produits de dé composition obtenus lors du conditionnement du réacteur servaient ensuite à conditionner les colonnes à chromatographie, en particulier la colonne VI utilisée pour l'analyse des hydrazones.
Des contrôles de reproductibilité des résultats furent effectués à diverses périodes de temps et ont montré que la vitesse de décomposition de l'azométhane mesurée par la vitesse de formation de l'azote était cons tante tout au long de ce travail pour des pyrolyses faites dans des condi tions identiques. Le tableau 3.1 donne deux exemples qui illustrent la constance des données recueillies dans des conditions identiques. On y remarque que les résultats sont satisfaisants sauf pour le EMH et le C.HgB. Ces deux produits étant formés en quantités très faibles et ayant les temps de rétention les plus longs sur leurs colonnes à chromatographie respectives,
TABLEAU 3.1
Exemples de reproductibilité d’analyses de produits de pyrolyses faites aux mêmes conditions expérimentales; la pression initiale d'azométhane est de 80.1 torr dans un cas et de 201.5 torr dans l'autre cas, tandis que la température est de 290°C dans les deux
cas.
Produit
Concentration en mole litre
"1
x106
80.1 torr 80.1 torr 201.5 torr 201.5 torr




![Figure 4.2: Droites d'Arrhenius pour le rapport [RCH4/(A°^R(10)C2H6)2]**](https://thumb-eu.123doks.com/thumbv2/123doknet/3724003.111280/109.876.119.793.87.1061/figure-droites-arrhenius-rapport-rch-c-h.webp)
![Etude expérimentale et modélisation de la décomposition thermique de l'exo-tricyclo[5.2.1.0(2.6)]décane.](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)