ABINIT: Overview and focus on selected capabilities
Texte intégral
Figure




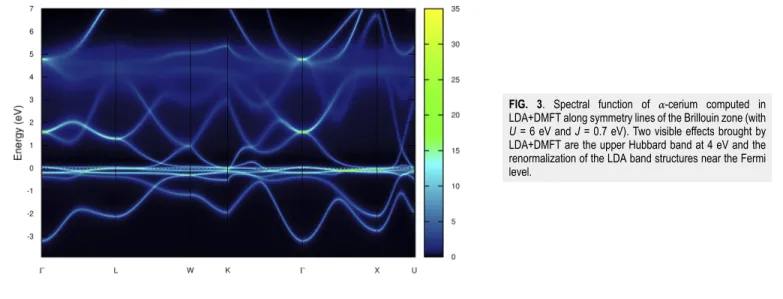
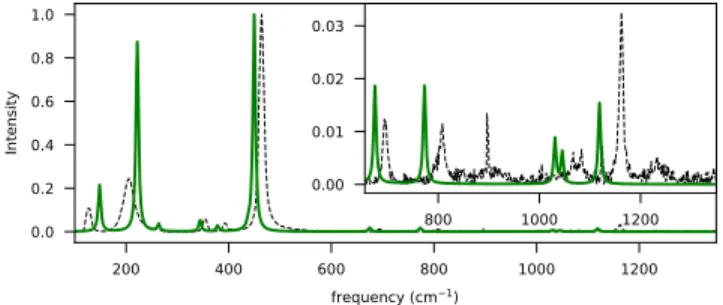
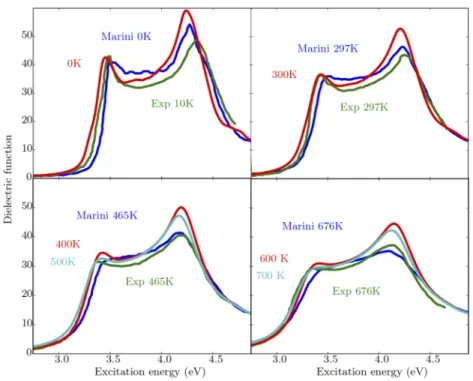
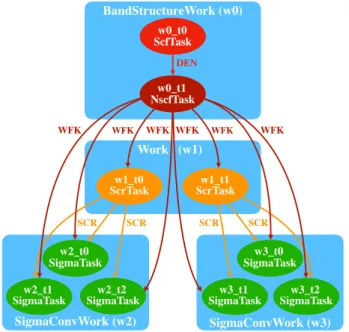
Documents relatifs
I describe below how such an exact functional can be formally constructed for a correlated electron model (irrespective, e.g of dimensionality), hence leading to a local
Because the original Wannier construction 17 is based on a decompo- sition of the extended Bloch states into a superposition of rather localized orbitals, it appears that
The Embassy of France in Japan strongly supports French–Japanese cooperation on materials for energy through numerous scientific seminars and events such as the
2014 Argonne’s Intense Pulsed Neutron Source, IPNS, was dedicated as a major user-oriented neutron.. scattering facility two
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
The current state- of-the-art of Enterprise Modeling (EM) methods and frameworks that address organi- zational structures may be efficient, yet, not optimal for this task compared to
The conclusions on optimization capacity are based on the results of studies published over the past few years, and on the multi-core data processing capabilities in the
In this paper, we investigated the exploratory capabilities of uncertainty sampling (US) in combination with different support vector machines (SVMs).. We described an
