Studying cross-talk between different transcriptional pathways controlling azole resistance in Candida albicans
85
0
0
Texte intégral
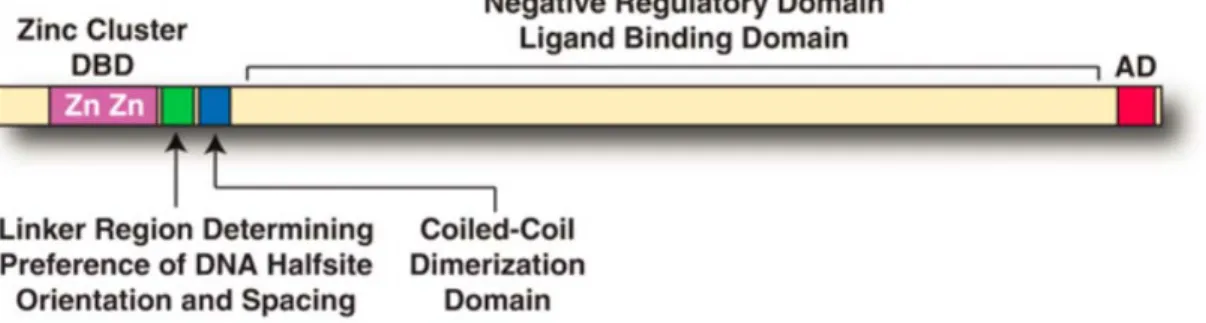
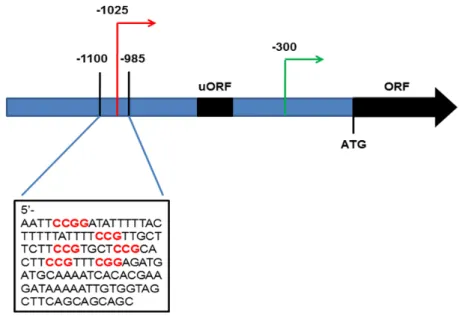
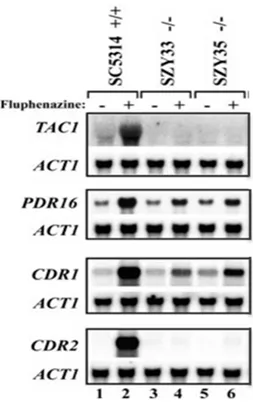
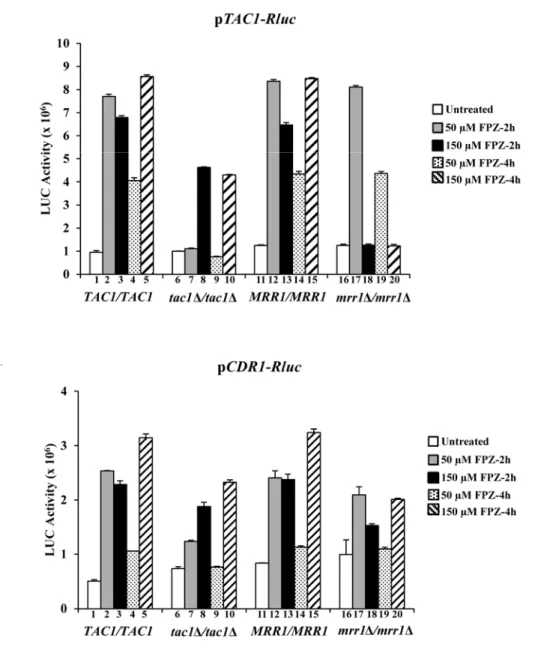
Figure

+7

Documents relatifs