Analyse du sécrétome des leucocytes de patients
suspectés d'être atteints de syndromes
auto-inflammatoires chroniques
Mémoire
Marie-Pier Longchamps
Maîtrise en microbiologie-immunologie - avec mémoire
Maître ès sciences (M. Sc.)
Résumé
Les syndromes auto-inflammatoires (AIS) sont des affections héréditaires caractérisées par une inflammation récurrente et une hypersécrétion de cytokines par les cellules immunitaires innées. Les faibles taux de détection de mutations, la ressemblance des symptômes aux maladies rhumatismales auto-immunes systémiques (SARD) et l’absence d’information sur les cytokines anormalement sécrétées par chaque individu compliquent le diagnostic, menant à un traitement inapproprié et à des complications graves. Nous avons émis l’hypothèse que la sécrétion de cytokines par les leucocytes du sang périphérique de patients AIS suspectés pourrait orienter le diagnostic et le traitement par biothérapie. Le plasma, les cellules mononucléées du sang périphérique (PBMC) et les neutrophiles polymorphonucléaires (PMN) ont été isolés chez des donneurs sains, des patients SARD et des patients AIS suspectés. Les PBMC et les PMN ont été stimulés avec différents activateurs de l’inflammation et les concentrations de cytokines ont été analysées par dosage multiplex. Les concentrations de cytokines détectées dans le plasma étaient similaires entre les trois groupes. En réponse à l'activation des inflammasomes NLRP1/3, NOD1/2, AIM2 et pyrin, les PBMC et les PMN des patients AIS ont sécrété plus d’IL-1α, d’IL-18, d’IFNγ et d’IL-12 que les cellules des patients SARD et des contrôles sains. Une analyse détaillée des cytokines sécrétées par les leucocytes sanguins de chaque patient AIS a montré que certains patients sécrétaient une quantité excessive d’IL-1α ou d’IL-1β en absence de l’antagoniste IL-1RA, suggérant que des antagonistes de l’IL-1 pourraient être utilisés, alors que d'autres patients ont sécrété des concentrations élevées d'IFNγ, d'IL-12 et d'IL-18, soulignant l'utilité des inhibiteurs des janus kinases, d’anti-IL-12/IL-23 ou d’anti-IL-18, présentement en cours de développement. Cette étude démontre que les analyses de la sécrétion de cytokines par les leucocytes sanguins fournissent un aperçu de l’environnement pro-inflammatoire anormal, en plus d’être un indicateur potentiel d’options de biothérapie pour les patients AIS suspectés.
Abstract
Autoinflammatory syndromes (AIS) are inherited conditions characterized by recurrent inflammation and hypersecretion of cytokines by innate immune cells. Diagnosis and treatments are challenging as detection of mutations rates is low, symptoms are reminiscent of other autoimmune diseases, especially Systemic Autoimmune Rheumatic Diseases (SARDs), and the cytokines abnormally secreted by each individual are unknown. Thus, patients can be misdiagnosed, leading to inappropriate treatment and severe complications. We hypothesized that the secretion of cytokines by peripheral blood leukocytes of suspected AIS patients could guide the diagnosis and treatment to the most suitable biotherapy. Plasma, peripheral blood mononuclear cells (PBMCs) and polymorphonuclear neutrophils (PMNs) were isolated from healthy controls, SARD patients and suspected AIS patients. PBMCs and PMNs were stimulated with well-known immune activators and levels of cytokines were analyzed by multiplex assays. On the one hand, cytokine levels detected in the plasma were similar between healthy donors, SARD patients and suspected AIS patients. On the other hand, PBMCs and PMNs from AIS patients secreted more IL-1α, IL-18, IFNγ and IL-12 than cells from SARD patients or healthy controls in response to NLRP1/3, NOD1/2, AIM2 and pyrin inflammasome activation. Detailed analysis of the cytokines secreted by PBMCs and PMNs from each suspected AIS patient showed that some suspected AIS patients secreted excessive IL-1α or IL-1β in the absence of the antagonist IL-1RA, suggesting blockers of IL-1 could be used as biotherapeutic approach. Cells from other suspected AIS patients secreted high levels of IFNγ, IL-12 and IL-18, pointing to the usefulness of treatments such as janus kinase inhibitors, the anti-IL-12/IL-23 antagonist and/or the novel anti-IL-18 in clinical development. This study demonstrates that analyses of cytokine secretion by blood leukocytes can provide an overview of the abnormal pro-inflammatory environment, in addition to being a potential indicator for biotherapy treatment options for patients with suspected AIS.
Table des matières
Résumé ... ii Abstract ... iii Liste des figures ... vi Liste des tableaux ... vii Liste des abréviations ... viii Remerciements ... xii Introduction ... 1 1. Le système immunitaire ... 11.1 L’immunité innée, mécanismes de défense non spécifique ... 2
1.1.1 Les phagocytes et leurs rôles dans l’inflammation ... 3
1.1.2 Les autres cellules de l’immunité innée et leurs fonctions ... 3
1.1.3 Les récepteurs spécialisés dans la reconnaissance de pathogènes ... 5
1.1.4 Les cytokines, des médiateurs protéiques solubles essentiels ... 5
1.1.4.1 La superfamille de l’interleukine-1 ... 6
1.1.4.2 Le facteur de nécrose tumorale et l’interleukine-6 ... 9
1.1.4.3 La famille des interférons ... 11
1.1.4.4 Les interleukines-12 et -23 ... 11
1.1.5 Le pont entre l’immunité innée et adaptative ... 12
1.2 L’immunité adaptative, mécanismes de défense spécifique ... 12
1.2.1 Les cellules de l’immunité adaptative et leurs fonctions ... 13
2. Le système immunitaire en conditions pathologiques ... 14
2.1 Maladies auto-immunes vs auto-inflammatoires ... 14
2.2 Les syndromes auto-inflammatoires, aussi appelés cytokinopathies ... 16
2.2.1 Historique et classification des syndromes auto-inflammatoires ... 17
2.2.2 Quatre grands mécanismes physiopathologiques impliqués dans les maladies auto-inflammatoires ... 19
2.2.2.1 Hyperréactivité intrinsèque des PRR ... 19
2.2.2.1.1 La fièvre méditerranéenne familiale – FMF ... 19
2.2.2.1.2 Les syndromes périodiques associés à la cryopyrine – CAPS ... 20
2.2.2.1.3 Le syndrome de Blau ... 21
2.2.2.2 Stress intracellulaire menant à l’inflammation ... 21
2.2.2.2.1 Le syndrome périodique associé au récepteur du TNF – TRAPS ... 22
2.2.2.2.2 Le syndrome d’hyper IgD avec fièvres périodiques – HIDS ... 22
2.2.2.2.3 Le syndrome d’arthrite pyogène associé au pyoderma gangrenosum et à l’acné – PAPA ... 23
2.2.2.2.4 Le syndrome de dermatose neutrophilique atypique chronique avec lipodystrophie et température élevée – CANDLE ... 23
2.2.2.3 Déficience d’antagonistes de la réaction inflammatoire ... 24
2.2.2.3.1 Le syndrome associé à la déficience de l’IL-1RA – DIRA ... 24
2.2.2.3.2 Le syndrome associé à la déficience de l’IL-36RA – DITRA ... 25
2.2.2.4 Altération des voies de signalisation associées aux récepteurs ... 25
2.2.3 Les traitements anti-inflammatoires ... 25
2.2.3.1 Les anti-inflammatoires non stéroïdiens ... 26
2.2.3.2 Les anti-inflammatoires stéroïdiens ... 26
2.2.3.3 Les DMARD synthétiques et les biothérapies ... 26
2.2.3.3.1 Les biothérapies ... 27
3. Problématique ... 29
3.1 Complications au niveau du diagnostic clinique ... 30
3.2 Complications au niveau du diagnostic génétique ... 30
4. Hypothèse et objectifs ... 33 4.1 Hypothèse ... 33 4.2 Objectifs ... 33 4.2.1 Objectifs généraux ... 33 4.2.2 Objectifs spécifiques ... 33 Chapitre 1 : Matériel et méthodes ... 34
1. Recrutement des patients et donneurs sains ... 34
2. Prélèvements sanguins et isolement des cellules ... 36
3. Stimulation des cellules ... 37
4. Quantification des cytokines ... 39
5. Analyses statistiques ... 42
Chapitre 2 : Résultats ... 43
Chapitre 3 : Discussion et perspectives ... 55
Conclusion ... 66
Liste des figures
Figure 1 : Les cellules du système immunitaire et leur répartition au sein des deux grandes classes de mécanismes de défense de l’hôte, soit le système immunitaire inné et le système immunitaire adaptatif. ... 2 Figure 2 : Spectre des maladies auto-immunes et auto-inflammatoires. ... 15 Figure 3 : Schéma représentant la diversité des agents biologiques pouvant être utilisés selon la ou les cytokine(s) surexprimée(s) par les cellules des patients atteints de maladies auto-inflammatoires. ... 32 Figure 4 : Schéma illustrant les stimuli utilisés et les voies de signalisation activées lors de la réponse inflammatoire et la réponse interféron de type II. ... 37 Figure 5 : Principe du dosage d’analytes selon la technologie LuminexTM. ... 41 Figure 6 : Les patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques se distinguent des donneurs sains et des patients auto-immuns par la quantification des concentrations d’IL-1α, d’IL-18, d’IFNγ et d’IL-12 dans les surnageants des PBMC suivant l’activation des inflammasomes/signalosomes. .. 46 Figure 7 : Les patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques se distinguent des donneurs sains et des patients auto-immuns par la quantification des concentrations d’IL-1α, d’IL-18, d’IFNγ et d’IL-12 dans les surnageants des PMN suivant l’activation des inflammasomes/signalosomes. ... 48 Figure 8 : La stimulation des PBMC avec diverses cytokines pro-inflammatoires ne permet pas de distinguer les patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques des donneurs sains et des patients auto-immuns. ... 49 Figure 9 : La stimulation des PMN avec diverses cytokines pro-inflammatoires ne permet pas de distinguer les patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques des donneurs sains et des patients auto-immuns. ... 50 Figure 10 : L’analyse du sécrétome des PBMC et PMN des patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques révèle une hypersécrétion de cytokines de la superfamille de l’IL-1. ... 53 Figure 11 : L’analyse du sécrétome des PBMC et PMN des patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques révèle une hypersécrétion d’IFNγ et d’IL-12. ... 54
Liste des tableaux
Tableau I : Liste partielle des syndromes auto-inflammatoires et leurs principales caractéristiques. ... 18 Tableau II : Données démographiques, manifestations cliniques et traitements des donneurs sains, des patients auto-immuns et des patients suspectés d’être atteints d’un syndrome auto-inflammatoire chronique au moment du prélèvement sanguin. ... 35 Tableau III : Détails expérimentaux des stimuli utilisés afin de quantifier le sécrétome des PBMC et des PMN. ... 38 Tableau IV : Les concentrations de cytokines mesurées dans le plasma sont similaires entre les donneurs sains, les patients auto-immuns et les patients suspectés d’être atteints de syndromes auto-inflammatoires chroniques. ... 44
Liste des abréviations
ADN Acide désoxyribonucléique
AIM2 Absent in Melanoma 2
AIS Syndrome auto-inflammatoire (Autoinflammatory Syndrome)
ALR AIM2-Like Receptor
ANOVA Analyse de variance (Analysis of Variance)
APLAID Autoinflammation and PLAID
ARN Acide ribonucléique
ASC Apoptosis-Associated Speck-Like Protein CARD Domain
ATP Adénosine triphosphate
BCR Récepteur spécifique d’antigènes des lymphocytes B (B Cell
Antigen Receptor)
BP Binding Protein
CaCl2 Chlorure de calcium
CANDLE Syndrome de dermatose neutrophilique atypique chronique
avec lipodystrophie et température élevée (Chronic Atypical
Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature)
CAPS Syndrome périodique associé à la cryopyrine
(Cryopyrin-Associated Periodic Syndrome)
CD2BP1 CD2-Binding Protein 1
cGAMP Cyclic [G(3’,5’)pA(3’,5’)p]
CLR Récepteur lectine de type C (C-Type Lectin Receptor)
CPA Cellule présentatrice d’antigènes
CRCHU Centre de recherche du centre hospitalier universitaire
CRP Protéine C-réactive (C-Reactive Protein)
DAMP Motif moléculaire associé aux dangers (Damage-Associated
Molecular Pattern)
DC Cellule dendritique (Dendritic Cell)
DIRA Syndrome associé à la déficience de l’antagoniste du
récepteur de l’IL-1 (Deficiency of the Interleukin-1 Receptor
DITRA Syndrome associé à la déficience de l’antagoniste du récepteur de l’IL-36 (Deficiency of Interleukin Thirty-Six
Receptor Antagonist)
DMARD Disease-Modifying Antirheumatic Drug
ESR Vitesse de sédimentation des érythrocytes (Erythrocyte
Sedimentation Rate)
FCAS Syndrome auto-inflammatoire familial associé au froid
(Familial Cold Autoinflammatory Syndrome)
FMF Fièvre méditerranéenne familiale (Familial Mediterranean
Fever)
G-CSF Facteur de croissance des granulocytes (Granulocyte-Colony
Stimulating Factor)
HEPES 4-(2-Hydroxyethyl)-1-Piperazineethanesulfonic Acid
HIDS Hyper immunoglobulinémie D avec fièvres périodiques
(Hyper Immunoglobulinemia D Syndrome with Periodic
Fever)
IFN Interféron
IFNAR IFN-α/β Receptor
IFNLR IFN-λ Receptor
Ig Immunoglobuline
IL Interleukine
IL-1R Récepteur de l’IL-1
IL-1RA IL-1 Receptor Antagonist
IL-36RA IL-36 Receptor Antagonist
IP-10 Interferon Gamma-Induced Protein 10
IPAF Ice Protease-Activating Factor
IQR Écart interquartile (Interquartile Range)
JAK Janus kinase
LAID LYN-Associated Autoinflammatory Disease
LPS Lipopolysaccharide
LUBAC Linear Ubiquitination Assembly Complex Deficiency
MAS Macrophage Activation Syndrome
M-CSF Facteur de croissance des monocytes et des macrophages
(Macrophage-Colony Stimulating Factor)
MDP Muramyl dipeptide
MgCl2 Chlorure de magnésium
MWS Syndrome Muckle-Wells (Muckle-Wells Syndrome)
NFκB Nuclear Factor κB
NIH National Institutes of Health
NK Natural Killer
NLRC4 NLR Family CARD Domain-Containing Protein 4
NLR NOD-Like Receptor
NLRP NLR Protein
NOD Nucleotide Oligomerization Domain
NOMID Syndrome inflammatoire multi-systémique débutant à
l’enfance (Neonatal Onset Multisystem Inflammatory
Disease)
NSAID Non-Steroidal Anti-Inflammatory Drug
PAMP Motif moléculaire associé aux pathogènes
(Pathogen-Associated Molecular Pattern)
PAPA Syndrome d’arthrite pyogène associé au pyoderma
gangrenosum et à l’acné (Pyogenic Arthritis, Pyoderma
Gangrenosum and Acne)
PLAID PLCγ2-Associated Antibody Deficiency and Immune
Dysregulation
PBMC Cellule mononucléée du sang périphérique (Peripheral Blood
Mononuclear Cell)
PMN Neutrophile polymorphonucléaire (Polymorphonuclear
Neutrophil)
Poly(dA:dT) Acide poly(deoxyadenylic-deoxythymidylic) Poly(I:C) Polyinosinic-Polycytidylic Acid
PRR Récepteur de reconnaissance de motifs moléculaires (Pattern
Recognition Receptor)
RA Polyarthrite rhumatoïde (Rheumatoid Arthritis)
RIP-2K Receptor-Interacting Protein-2 Kinase
RLR Récepteur de type RIG-I (RIG-I-Like Receptor)
ROS Forme réactive de l'oxygène (Reactive Oxygene Specie)
SAA Sérum amyloïde A (Serum Amyloid A)
SAID Steroidal Anti-Inflammatory Drug
SA-PE Streptavidine conjuguée à la phycoérythrine
SARD Maladie rhumatismale auto-immune systémique (Systemic
Autoimmune Rheumatic Disease)
SAVI STING-Associated Vasculopathy with Onset in Infancy
SJIA Arthrite juvénile idiopathique systémique (Systemic Juvenile
Idiopathic Arthritis)
SLE Lupus érythémateux disséminé (Systemic Lupus
Erythematosus)
SNC Système nerveux central
STAT Signal Transducer and Activator of Transcription
STING Stimulator of Interferon Genes
S.-u. Sous-unité
TA Température ambiante
TACE TNF-α-Converting Enzyme
TCR Récepteur d’antigènes des lymphocytes T (T Cell Antigen
Receptor)
TLR Récepteur de type Toll (Toll-Like Receptor)
TNF Facteur de nécrose tumorale (Tumor Necrosis Factor)
TNFR TNF Receptor
TRAPS TNF Receptor Associated Periodic Syndrome
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche, le Dr Martin Pelletier, et mon codirecteur de recherche, le Dr Philippe Tessier, pour leur confiance, leur encadrement, leur dynamisme et la chance qu’ils m’ont donnée de réaliser ma maîtrise dans leurs laboratoires. Leur dévouement, leurs conseils et leur passion pour la science ont été d’une aide précieuse durant ces deux années.
Un énorme merci à Nathalie Pagé puisque son dévouement, sa présence à mes côtés et son aide furent déterminants dans la réussite de ma maîtrise. Je tiens également à remercier mes collègues de laboratoire qui ont partagé mes journées et qui m’ont aidée de près ou de loin avec mes expériences et mes analyses. Merci à Anthony Kerever, Asmaa Lachhab, Maude Leclerc, Joan Defrene, Julie-Christine Lévesque et Guillaume St-Pierre.
Merci à mes parents, Charles et Chantal, à ma sœur et à mon frère, Mélanie et Charles-Antoine, à mes amies préférées, Maude, Blandine, Alexandra, Amélie, Mathilde, Audrey-Jade, Ann-Marie, Ariane, Daphnée et Véronique, d’avoir illuminé mes journées, d’avoir toujours cru en moi et de m’avoir constamment encouragée dans mes études, surtout dans mes moments les plus difficiles. C’est grâce à vous que je suis une meilleure version de moi-même à chaque jour.
Je tiens également à remercier très spécialement mon amoureux, Maxime Lavoie, sans qui je ne me serais jamais rendue aussi loin. Son amour, son amitié et ses encouragements constants ont été une source d’inspiration inépuisable.
Je remercie aussi tous nos collaborateurs sans qui cette étude n’aurait pas pu être réalisée : Dr Paul R. Fortin, Dr Alexandra Albert, Dr Anne-Laure Chetaille, Dr Laëtitia Michou, Dr Louis Bessette, Dr Ivona Aksentijevich et Anne-Sophie Julien. Et finalement, merci aux organismes subventionnaires : Rare Disease Foundation, La Fondation du grand défi Pierre Lavoie et la Fondation du CHU de Québec.
Introduction
1. Le système immunitaire
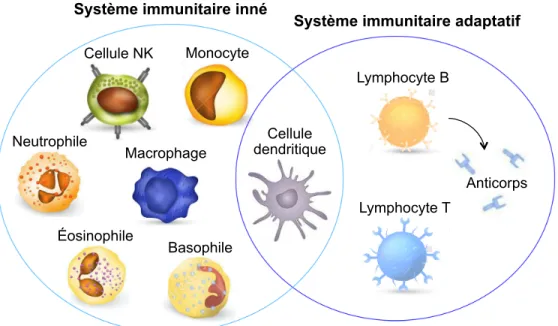
Les leucocytes, communément appelés globules blancs, sont les effecteurs principaux du système immunitaire, un système biologique qui englobe l’ensemble des mécanismes de protection de l’organisme en réponse à une agression. Il existe différents types de leucocytes regroupés au sein de deux grandes classes de mécanismes de défense : l’immunité innée et l’immunité adaptative. Les granulocytes polymorphonucléaires, incluant les neutrophiles, les éosinophiles et les basophiles; les phagocytes mononucléés, incluant les monocytes et les macrophages; les mastocytes; les thrombocytes, également appelés plaquettes; et les cellules NK (Natural Killer – NK) appartiennent au système immunitaire inné. L’immunité adaptative, quant à elle, repose sur les fonctions des lymphocytes T et B, ainsi que sur les anticorps que produisent les lymphocytes B. Un anticorps est une protéine complexe qui a la capacité de reconnaitre et neutraliser spécifiquement un antigène donné, i.e. un composé susceptible de déclencher une réponse immunitaire. Finalement, les cellules dendritiques (Dendritic Cells – DC) sont des cellules sentinelles qui font le pont entre l’immunité innée et l’immunité adaptative.1 À noter que les autres cellules des tissus, comme les fibroblastes, les cellules épithéliales et les cellules endothéliales, sont aussi des composantes importantes du système immunitaire puisqu’elles peuvent participer à la réaction inflammatoire en interagissant avec les cellules immunitaires (Figure 1).2
Inspiré de Dranoff (2004)3
Figure 1 : Les cellules du système immunitaire et leur répartition au sein des deux grandes classes de mécanismes de défense de l’hôte, soit le système immunitaire inné et le système immunitaire adaptatif.
L’immunité innée (gauche) repose, entre autres, sur les fonctions des monocytes, des macrophages, des neutrophiles, des éosinophiles, des basophiles, des mastocytes (non illustrés), des thrombocytes (non illustrés) et des cellules NK. L’immunité adaptative (droite) repose sur les fonctions des lymphocytes B, qui sont responsables de la réponse humorale via la production d’anticorps, et des lymphocytes T qui assurent la réponse cellulaire. Les cellules dendritiques font le pont entre l’immunité innée et adaptative (centre). Les cellules des tissus comme les fibroblastes, les cellules épithéliales et les cellules endothéliales (non illustrées) participent aussi à la réaction immunitaire de par leur communication avec les leucocytes.
1.1 L’immunité innée, mécanismes de défense non spécifique
Le fonctionnement du système immunitaire inné dépend de mécanismes de défense non spécifique qui s’activent immédiatement à la suite d’une agression. Ce système de défense ancien et remarquablement sophistiqué repose principalement sur la présence de barrières épithéliales externes, comme la peau et les muqueuses, ainsi que sur les fonctions qu’exercent les cellules de l’immunité innée lorsque leurs récepteurs spécialisés reconnaissent les grands groupes de pathogènes.4,5
Lymphocyte B
Cellule dendritique
Système immunitaire inné
Éosinophile Basophile Monocyte Cellule NK Macrophage Anticorps Lymphocyte T Neutrophile
1.1.1 Les phagocytes et leurs rôles dans l’inflammation
Grâce à des récepteurs spécialisés dans la reconnaissance de motifs moléculaires spécifiques, les pathogènes sont reconnus dès leur passage à travers l’épithélium par les cellules résidentes des tissus, soit par les macrophages, les mastocytes et les DC immatures. Ces cellules s’activent alors et amorcent le recrutement massif des phagocytes, des acteurs cellulaires essentiels dans la réaction inflammatoire. Elles y parviennent, entre autres, en sécrétant des cytokines, des médiateurs protéiques solubles essentiels qui seront abordés dans la section 1.1.4.6,7
Les neutrophiles polymorphonucléaires (Polymorphonuclear neutrophils – PMN) sont les leucocytes les plus abondants dans la circulation sanguine (constituant 50-70% des leucocytes totaux)8 et sont les premiers phagocytes à être recrutés massivement au site inflammatoire.9 Ils sont caractérisés par la présence d’un noyau multilobé, des vésicules sécrétoires et des granules cytoplasmiques. Les fonctions assurées par les PMN sont la phagocytose, la dégranulation (libération d’enzymes de dégradation et de protéines antimicrobiennes), la sécrétion de cytokines, la modulation de la réponse immunitaire acquise, la flambée oxydative (libération de formes réactives de l'oxygène [Reactive Oxygene Species – ROS]) et la NETose (libération de fibres extracellulaires principalement composées d'acides désoxyribonucléiques (ADN), d’histones et de protéines granulaires qui lient et détruisent les agents pathogènes).10,11 Les monocytes sanguins sont par la suite recrutés en grand nombre au site inflammatoire où ils pourront se différencier en macrophages et ainsi éliminer les cellules mortes de l’organisme, en plus de capturer, internaliser et détruire les pathogènes, principalement les bactéries et les champignons. Les macrophages résidents des tissus ont également la capacité d’amorcer la réponse immunitaire adaptative en présentant des antigènes des pathogènes aux lymphocytes T.12,13
1.1.2 Les autres cellules de l’immunité innée et leurs fonctions
Dans la famille des granulocytes polymorphonucléaires, on retrouve aussi les éosinophiles et les basophiles qui participent à la lutte contre les parasites
extracellulaires qui envahissent les tissus. Ces cellules contribuent, entre autres, à la défense contre les vers (éosinophiles) et les parasites tels que les helminthes (basophiles). Tout comme les PMN, elles ont la capacité de phagocyter et de libérer des granules remplies d’enzymes, de protéines toxiques et de médiateurs inflammatoires solubles afin de détruire les pathogènes.14
Les mastocytes, quant à eux, sont considérés comme des cellules auxiliaires dans l’inflammation. Ils sont retrouvés à la proximité des vaisseaux sanguins de tous les tissus et favorisent le développement de la réaction inflammatoire en libérant davantage de médiateurs pro-inflammatoires. Ils possèdent également des granules remplies de médiateurs pro-inflammatoires solubles qui peuvent être libérés rapidement à la suite de la rencontre d’un pathogène. Ces cellules seraient principalement impliquées au niveau de l’immunité contre les parasites.15
Les thrombocytes, qui ne sont pas des cellules mais plutôt des fragments dérivés de mégacaryocytes de la moelle osseuse, favorisent aussi le développement de la réaction inflammatoire puisqu’ils participent à la coagulation et qu’ils ont la capacité d’activer le système du complément, d’augmenter la perméabilité des vaisseaux capillaires et de recruter davantage de leucocytes au site inflammatoire.16
Finalement, les cellules NK se distinguent des autres cellules de l’immunité innée par leur activité cytotoxique et leur capacité à reconnaitre et tuer les cellules infectées par un virus ainsi que celles qui sont en prolifération tumorale.17 Elles
jouent un rôle primordial dans la lutte contre les pathogènes intracellulaires et dans le contrôle des maladies infectieuses et du cancer.18
En somme, selon la nature des microorganismes envahisseurs reconnus par les récepteurs spécialisés, différents leucocytes seront recrutés à différents moments afin d’assurer les fonctions protectrices qui leur sont propres.19
1.1.3 Les récepteurs spécialisés dans la reconnaissance de pathogènes
Les récepteurs mentionnés précédemment sont des récepteurs de reconnaissance de motifs moléculaires (Pattern Recognition Receptors – PRR) qui reconnaissent des motifs moléculaires associés aux pathogènes (Pathogen-Associated Molecular
Patterns – PAMP) ou des motifs moléculaires associés aux dangers (Damage-Associated Molecular Patterns – DAMP). Les acides nucléiques viraux et
bactériens, les composants des flagelles, comme la flagelline, et des parois des bactéries, comme le lipopolysaccharide (LPS) et le peptidoglycane, sont tous des exemples de PAMP.12 Les DAMP, quant à eux, correspondent à tout ce qui est libéré dans le milieu extracellulaire par les cellules endommagées; les alarmines, les mitochondries, l’adénosine triphosphate (ATP) et l’acide ribonucléique (ARN) ou l’ADN en sont des exemples.20 Il existe différents types de PRR selon l’endroit où ils sont exprimés – surface cellulaire, endosome ou cytoplasme – et la nature des composés qu’ils reconnaissent. Les récepteurs de type Toll (Toll-Like
Receptors – TLR), les récepteurs de type NOD (Nucleotide Oligomerization Domain-Like Receptors – NLR), les récepteurs lectines de type C (C-type Lectin Receptors – CLR) et les récepteurs de type RIG-I (RIG-I-Like Receptors – RLR)
correspondent aux quatre grandes familles de PRR identifiées à ce jour.21 La
plupart des TLR et des CLR se retrouvent à la surface des cellules, alors que les récepteurs intracellulaires incluent les NLR, les RIG-I, les récepteurs de type AIM2 (Absent In Melanoma 2-Like Receptors – ALR), ainsi que les TLR3, 7, 8 et 9. Finalement, les récepteurs NLRP3/NALP3 (NLR Proteins – NLRP), NLRP1, NLRC4/IPAF (NLR family CARD domain-containing protein 4/Ice
Protease-Activating Factor), AIM2, et pyrine sont les cinq récepteurs actuellement connus
qui activent les inflammasomes (abordés dans la section 1.1.4.1).22
1.1.4 Les cytokines, des médiateurs protéiques solubles essentiels
Les cytokines sont des molécules de communication intercellulaire qui agissent à la suite de la liaison à leur récepteur spécifique, ce qui mène à l’activation de voies de signalisation intracellulaire et permet à la cellule de réaliser ses fonctions. Elles sont regroupées au sein de différentes classes sur la base de leur homologie de
structure. On retrouve notamment les interférons (IFN), les interleukines (IL), la superfamille du facteur de nécrose tumorale (Tumor Necrosis Factor – TNF) et les chimiokines.23 Les interférons, nommés ainsi par leur capacité à interférer avec la
réplication virale, ont une activité antivirale, antibactérienne et antiproliférative, en plus de permettre l’activation de cellules comme les macrophages, les cellules NK et les lymphocytes.24 Les interleukines sont une grande famille de cytokines aux fonctions diversifiées qui participent entre autres à l’inflammation, à la maturation, à la différenciation et à la prolifération cellulaire. L’IL-1β et l’IL-6, par exemple, participent à l’inflammation grâce à leurs activités pro-inflammatoires, alors que l’IL-2 est un facteur de croissance qui entraine la maturation et la prolifération des lymphocytes T.25 Il existe aussi d’autres facteurs de croissance comme celui des monocytes et des macrophages (Macrophage-Colony Stimulating Factor – M-CSF) et celui des granulocytes (Granulocyte-Colony Stimulating Factor – G-CSF). Le TNF, en plus d’agir comme médiateur inflammatoire au même titre que l’IL-1β et l’IL-6, participe également aux réactions cytotoxiques.23 Finalement, les chimiokines sont des cytokines chimiotactiques qui dirigent le mouvement des leucocytes à travers tout le corps, permettant à ces derniers de passer de la circulation sanguine aux différents tissus et vice versa.26 Les molécules
CXCL8/IL-8 et CCL2/MCP-1 sont des exemples de chimiokines importantes puisqu’elles attirent respectivement les neutrophiles et les monocytes au site inflammatoire.27,28
1.1.4.1 La superfamille de l’interleukine-1
La superfamille de l’IL-1 regroupe 11 membres sur la base de leur homologie de structure : l’IL-1α, l’IL-1β, l’IL-18, l’IL-33, l’IL-36α, l’IL-36β et l’IL-36γ qui sont des ligands agonistes des récepteurs, l’IL-1RA (IL-1 Receptor Antagonist) et l’IL-36RA (IL-36 Receptor Antagonist) qui sont des antagonistes des récepteurs, ainsi que l’IL-37 et l’IL-38 qui ont des effets inflammatoires. Ces cytokines pro- et anti-inflammatoires modulent les voies de signalisation intracellulaire en se liant à différents membres de la famille des récepteurs de l’IL-1 (IL-1R), incluant l’IL-1RI, IL-1RII, IL-1Rrp2 (IL-36R), ST2, IL-18Rα et IL-18BP (Binding Protein – BP), et
leurs corécepteurs, incluant l’IL-1RAcP et IL-18Rβ. Toutes les cellules de l’immunité innée comme les monocytes, les macrophages et les neutrophiles, ainsi que les cellules des tissus et des organes vont sécréter et/ou être affectées par cette superfamille de cytokines. Dans le cadre de ce mémoire, seuls quelques membres de la superfamille de l’IL-1 seront détaillés. Pour une description complète de la superfamille de l’IL-1 et leurs récepteurs, voir l’article de revue de Garlanda et ses collaborateurs.29
L'IL-1 est un médiateur central du système immunitaire inné qui a un rôle clé dans l'inflammation et l’induction de la fièvre, en plus de participer à la régulation des réponses immunes.30 Il en existe trois types : l’IL-1α et l’IL-1β qui sont des cytokines pro-inflammatoires, et l’IL-1RA qui est une cytokine anti-inflammatoire. L’IL-1α et l'IL-1β sont synthétisées dans le cytoplasme des cellules sous la forme de précurseurs, appelés respectivement pro-IL-1α et pro-IL-1β. Les principaux producteurs sont les phagocytes mononucléés, les lymphocytes, les fibroblastes et les kératinocytes.31 La pro-IL-1α peut être clivée sous sa forme mature par la
calpaïne, une protéase à cystéine liée à la membrane cytoplasmique qui est dépendante du calcium, et sécrétée dans les milieux extracellulaires par la cellule, mais la majorité de celle-ci demeure attachée à la membrane cytoplasmique ou est entreposée dans le noyau. La pro-IL-1α est biologiquement active à la suite de sa production, i.e. qu’elle a la capacité de se lier à son récepteur et d’activer les cellules. La pro-IL-1β quant à elle, doit d’être clivée sous sa forme mature par une enzyme de conversion, la caspase-1, avant d’être sécrétée et de pouvoir exercer ses fonctions pro-inflammatoires.32 Bien que la caspase-1 soit abondante dans les
cellules hématopoïétiques, elle existe sous la forme d’un précurseur inactif, la procaspase-1, qui doit être préalablement clivé par un inflammasome. Un inflammasome est un complexe multi-protéique oligomérique qui s’assemble dans le cytoplasme des cellules à la suite de la reconnaissance de PAMP ou de DAMP.33 Il en existe différents types selon la nature des stimuli rencontrés mais, de manière générale, ces derniers sont essentiellement composés d’un récepteur de type NOD, d’une protéine adaptatrice ASC (Apoptosis-associated Speck-like
protein CARD domain), de la caspase-1 et parfois de la caspase-5.34 Deux signaux sont nécessaires pour que l’IL-1β bioactive soit sécrétée dans le milieu extracellulaire par la cellule. Le premier signal consiste en la reconnaissance de pathogènes ou de signaux de danger qui vont induire la transcription de la pro-IL-1β. Le LPS, constituant essentiel de la membrane externe des bactéries gram-négatives, est un stimulus commun pour l’induction de ce signal. Le deuxième signal mène à la formation de l’inflammasome qui va libérer la caspase-1 active et permettre la production subséquente d’IL-1β. L'ATP, un nucléotide riche en énergie essentiel à la réalisation des fonctions cellulaires, peut induire ce deuxième signal.35 En se liant à son récepteur, l’IL-1RI, l’IL-1 permet le recrutement du corécepteur IL-1RAcP et l’activation subséquente des voies de signalisation intracellulaire du facteur nucléaire κB (Nuclear Factor κB – NFκB) et des MAP Kinases (Mitogen-Associated Protein Kinase – MAPK). Ces dernières, en modulant l’expression de différents gènes, vont permettre aux cellules de survivre, croître et d’effectuer leurs fonctions. La voie du NFκB est impliquée dans les réponses inflammatoires et immunes en induisant la production de plusieurs cytokines pro-inflammatoires comme le TNF, l’IL-1β et l’IL-6, ainsi que dans le développement et l'apoptose, i.e. la mort cellulaire programmée.36 La voie des MAPK régule les processus de différenciation, de prolifération, de survie et d’apoptose cellulaire.37 Finalement, l’IL-1RA agit comme un inhibiteur spécifique de
l’IL-1 puisqu’elle lie l’IL-1RI avec plus d’affinité, sans avoir la capacité d’activer les cellules. Elle est principalement synthétisée par les macrophages et les neutrophiles activés.38 L'équilibre entre les concentrations d'IL-1 et d'IL-1RA dans les tissus assure les conditions physiologiques alors qu’un débalancement au niveau de ces dernières peut contribuer à diverses pathologies comme la polyarthrite rhumatoïde (Rheumatoid Arthritis – RA) et le cancer.39
La famille de l’IL-18 regroupe l’IL-18, une cytokine pro-inflammatoire, et son antagoniste, l’IL-18BP. L’IL-18 joue un rôle majeur dans la défense contre les pathogènes intracellulaires en induisant la production d’IFNγ et d’autres cytokines pro-inflammatoires comme le TNF, l’IL-1β et le CXCL8/IL-8, en plus d’activer les
fonctions cytotoxiques des cellules NK et de certains lymphocytes T.40 Tout comme l'IL-1β, l’IL-18 est synthétisée sous la forme d'un précurseur biologiquement inactif, la pro-IL-18, qui est clivé par la caspase-1 et sécrété sous sa forme mature dans le milieu extracellulaire. Cette cytokine est sécrétée par plusieurs cellules immunitaires incluant les monocytes, les macrophages et les DC, ainsi que par d’autres cellules des tissus comme les fibroblastes et les kératinocytes.41 En se liant à son récepteur, l’IL-18Rα, l’IL-18 permet le recrutement du corécepteur IL-18Rβ et l’activation subséquente des mêmes voies de signalisation intracellulaire que l’IL-1, i.e. les voies NFκB et MAPK.42 L'IL-18BP, quant à elle, est un inhibiteur soluble naturel de l’IL-18 qui se lie à la forme mature de cette dernière avec une affinité élevée et qui empêche son interaction avec les récepteurs de surface cellulaire. Les cellules endothéliales, les monocytes et les macrophages représentent des sources importantes d’IL-18BP. Il en existe quatre isoformes chez l’humain : l’IL-18BPa, présentant la plus grande affinité pour l'IL-18, ainsi que l’IL-18BPb, c et d. Un débalancement entre les concentrations d’IL-18 et d’IL-18BP est aussi à l’origine de diverses conditions pathologiques.43
La famille de l’IL-36 regroupe l’IL-36α, l’IL-36β, l’IL-36γ et l’IL-36RA. L’IL-36α, β et γ amplifient les réponses du système immunitaire inné en induisant la production de cytokines pro-inflammatoires, de chimiokines et de molécules de co-stimulation qui permettent la différenciation des lymphocytes T en sous-types Th1 et Th17. Ces cytokines sont principalement sécrétées par les macrophages, les DC et les lymphocytes. En se liant à l’IL-1Rrp2 (IL-36R), l’IL-36 permet le recrutement du corécepteur IL-1RAcP et l’activation subséquente des voies de signalisation intracellulaire NFκB et MAPK. L’IL-36RA, quant à elle, empêche l’activation des cellules par l’IL-36 de la même façon que le fait l’IL-1RA avec l’IL-1.44,45
1.1.4.2 Le facteur de nécrose tumorale et l’interleukine-6
Le TNF et l’IL-6 sont d’autres médiateurs pro-inflammatoires importants dans la phase aiguë de l’inflammation. Ils sont synthétisés par plusieurs types cellulaires
incluant les phagocytes mononucléés, les fibroblastes et certains lymphocytes T. Accompagnées de l’IL-1β, ces cytokines permettent le recrutement des leucocytes au site inflammatoire ainsi que l’activation de leurs fonctions pro-inflammatoires. De plus, elles stimulent les hépatocytes du foie à produire des protéines de la phase aiguë de l’inflammation comme la protéine C-réactive (C-Reactive Protein – CRP) et le sérum amyloïde A (Serum Amyloid A – SAA) dont le rôle est d’amplifier la réaction inflammatoire. La quantification de ces molécules plasmatiques est une méthode couramment utilisée pour suivre l’inflammation. Finalement, le TNF, l’IL-6 et l’IL-1β, toutes trois reconnues comme des pyrogènes endogènes (inducteurs de la fièvre), peuvent également induire la réponse immunitaire adaptative lorsque la réponse immunitaire innée ne parvient pas à résoudre l’inflammation.46,47
Le TNF est une protéine trimérique qui existe sous deux formes biologiquement actives; la première étant attachée à la membrane cellulaire sous forme d’un précurseur du TNF, le pro-TNF, et la seconde étant sécrétée dans le milieu extracellulaire sous une forme soluble à la suite de son clivage protéolytique par l’enzyme TACE (TNF-α-Converting Enzyme).48 La liaison du TNF à ses
récepteurs, le TNFRI (TNF Receptor – TNFR) et le TNFRII, induit la trimérisation de ces derniers et l’activation subséquente des voies NFκB et MAPK. Les TNFR existent aussi sous une forme soluble qui agissent comme récepteurs leurres en séquestrant le TNF libre, permettant ainsi à la cellule de réguler finement l’activité biologique du TNF.49
Dans le cas de l’IL-6, la liaison de son récepteur, l’IL-6R, et le recrutement de la sous-unité de transduction de signal gp130 vont induire l’activation de la voie de signalisation des JAK/STAT (JAnus Kinases/Signal Transducers and Activators of
Transcription) qui, en modulant l’expression de plusieurs gènes, va permettre la
prolifération, la différenciation, l’apoptose et l’hématopoïèse des cellules.50,51 La formation du complexe IL-6/IL-6R/gp130 induit également la voie des MAPK.52
1.1.4.3 La famille des interférons
Il existe trois types d’IFN : les IFN de type I (IFNα et IFNβ) et de type III (IFNλ1, IFNλ2 et IFNλ3) qui sont des constituants majeurs de la réponse antivirale, ainsi que l’IFN de type II (IFNγ) qui intervient davantage comme immunomodulateur dans les réponses inflammatoires et immunes en permettant l’activation et la maturation de plusieurs cellules incluant les phagocytes et les lymphocytes.53,54 Il existe différents récepteurs des IFN selon le type : l’hétérodimère IFNAR1/IFNAR2 (IFN-α/β Receptor – IFNAR) pour les IFN de type I, l’hétérodimère IFNLR1/IL10R2 (IFN-λ Receptor – IFNLR) pour les IFN de type III, et l’hétérodimère IFNγR1/IFNγR2 pour l’IFN de type II. Dans tous les cas, la voie de signalisation intracellulaire induite est celle des JAK/STAT. Elle module entre autres l’expression de gènes qui codent pour des protéines de résistance à la réplication virale, en plus des gènes impliqués dans les processus de prolifération, de différenciation et d’apoptose cellulaire, ainsi que dans l’hématopoïèse.55
Le CXCL10/IP-10 (Interferon gamma-induced Protein 10) est produit par plusieurs cellules en réponse à l’IFNγ, incluant les monocytes et les neutrophiles, et joue un rôle dans diverses fonctions biologiques telles que la chimiotaxie, l’angiogenèse, l’apoptose, la croissance et la prolifération cellulaire. Il module principalement l’activité des DC, des macrophages, des cellules NK et des lymphocytes activés en se liant au CXCR3, un récepteur de la famille des récepteurs couplés aux protéines G, et en induisant la voie de signalisation associée à ces derniers.56
1.1.4.4 Les interleukines-12 et -23
L'IL-12 est un médiateur principal dans la défense contre les virus et les processus néoplasiques puisqu’elle mène à l’activation et à la prolifération des cellules NK et des lymphocytes T, ainsi qu’à la sécrétion d’IFNγ. Cette cytokine hétérodimérique, composée des sous-unités IL-12p35 et IL-12p40, est sécrétée par les DC, les lymphocytes B activés, les neutrophiles, les monocytes et les macrophages. En se liant à son récepteur, un hétérodimère formé des sous-unités IL-12Rβ1 et
IL-12Rβ2, l’IL-12 active la voie de signalisation des JAK/STAT.57 L’IL-23, quant à
elle, est impliquée dans la régulation de la réponse immunitaire. Cette cytokine hétérodimérique est composée des sous-unités IL-23p19 et IL-12p40. En se liant à son récepteur, un hétérodimère formé des sous-unités IL-12Rβ1 et IL-23R, l’IL-23 induit la voie de signalisation des JAK/STAT et permet la survie et l’expansion des cellules Th17. Les cellules Th17 représentent une sous-population particulière de lymphocytes T qui se caractérise par la production de l’IL-17A et de l’IL-17F, deux cytokines qui peuvent également s’associer pour former l’hétérodimère IL-17A/F. En se liant à leur récepteur, un hétérodimère composé des sous-unités IL-17RA et IL-17RC exprimé principalement par des cellules non hématopoïétiques comme les cellules épithéliales et mésenchymateuses, l’IL-17A, F et A/F activent les voies de signalisation intracellulaire NFκB et MAPK. Elles contribuent donc à la défense de l’hôte en induisant le recrutement et l’activation des leucocytes ainsi que la production de peptides antimicrobiens.58,59
1.1.5 Le pont entre l’immunité innée et adaptative
Tout comme les phagocytes, les DC immatures qui résident dans les tissus de l’organisme sont capables de reconnaitre, capturer et phagocyter les pathogènes. Cependant, une fois matures, elles se distinguent par leur capacité à présenter des antigènes provenant des pathogènes aux lymphocytes. Ainsi, elles permettent d’amorcer la réponse immune lorsque les mécanismes de défense du système immunitaire inné n’ont pas réussi à éradiquer les pathogènes.60
1.2 L’immunité adaptative, mécanismes de défense spécifique
L’immunité adaptative est une réponse immunitaire robuste et spécifique contre un antigène donné qui se développe plus lentement, i.e. environ 4-5 jours après la rencontre de l’antigène, et qui intervient lorsque les mécanismes de défense du système immunitaire inné ne réussissent pas à contrôler l’infection. Les effecteurs principaux sont les lymphocytes T et B qui agissent de façon complémentaire avec les cellules de l’immunité innée afin d’éradiquer les pathogènes.1
1.2.1 Les cellules de l’immunité adaptative et leurs fonctions
Les lymphocytes T sont les principaux acteurs cellulaires de la réponse immune grâce à leur capacité de reconnaitre et détruire les cellules infectées et cancéreuses. Ils reconnaissent ces dernières grâce à un récepteur spécifique exprimé à leur surface, le récepteur d’antigènes des lymphocytes T (T Cell antigen
Receptor – TCR), et les détruisent grâce à diverses fonctions cytotoxiques.61 Les lymphocytes T vont aussi agir comme intermédiaires dans la réponse immune en régulant ou en aidant les autres leucocytes à exercer leurs fonctions.62 À la différence des cellules de l’immunité innée qui sont activées immédiatement à la suite de la reconnaissance de motifs moléculaires étrangers, les lymphocytes T ne seront activés que par des cellules présentatrices d’antigènes (CPA), soit par les DC, les macrophages ou les lymphocytes B, et vont nécessiter plus de temps avant d’atteindre leur maturité et de pouvoir exercer leurs fonctions.1 Les signaux nécessaires à leur activation sont : 1) la présentation de l’antigène qui induit une cascade de signalisation intracellulaire dans le lymphocyte T via l’activation du CD3, la molécule associée au TCR63, 2) le signal de survie et de prolifération induit par l’interaction entre le CD28 (exprimé par le lymphocyte T) et les molécules CD80 et CD86 (exprimées par la CPA)64 et 3) le signal de différenciation induit par
des cytokines et d’autres molécules de costimulation.65
Les lymphocytes B assurent la réponse immunitaire humorale grâce à leur capacité de sécréter un récepteur spécifique d’antigènes (B Cell antigen Receptor – BCR), aussi connu sous le nom d’immunoglobuline (Ig) ou d’anticorps, qui va reconnaitre et neutraliser les pathogènes extracellulaires.1 Il existe cinq classes
différentes d’immunoglobulines – IgG, IgM, IgA, IgE et IgD – qui possèdent des activités biologiques qui leur sont propres. Les trois rôles principaux des anticorps sont de reconnaitre leur antigène spécifique, d’activer le système du complément et de recruter ainsi que d’activer les autres cellules du système immunitaire.66
2. Le système immunitaire en conditions pathologiques
Normalement, le système immunitaire répond de manière appropriée et ne cause pas de dommages collatéraux à l’organisme : il y a reconnaissance et élimination de ce qui est nocif, puis les mécanismes de défense reviennent en état de veille jusqu’à la prochaine infection.67 Il arrive cependant que le système immunitaire de certains individus soit déréglé ou affaibli. C’est le cas des réactions allergiques, des immunodéficiences, des maladies immunes et des maladies auto-inflammatoires. Les réactions allergiques ou d’hypersensibilités sont caractérisées par une réponse immunitaire disproportionnée face à un antigène inoffensif.68 Les immunodéficiences se définissent par un déficit du système immunitaire : faible production d’anticorps et/ou de leucocytes ou encore présence d’un désordre au niveau de la phagocytose ou du complément, ce qui résulte en une incapacité de lutter efficacement contre les agents infectieux.69,70 Finalement, d’autres maladies, comme les maladies auto-immunes et auto-inflammatoires, sont caractérisées par un désordre du système immunitaire où il y a destruction des tissus sains de l’organisme et/ou présence d’une inflammation excessive et prolongée contre ces derniers, ce qui mènent à des dommages importants.71
2.1 Maladies auto-immunes vs auto-inflammatoires
Les maladies auto-immunes et auto-inflammatoires diffèrent les unes des autres en raison des mécanismes physiopathologiques impliqués. De façon générale, les maladies auto-immunes sont issues d’un dysfonctionnement du système immunitaire adaptatif et sont caractérisées par la présence d’auto-anticorps et/ou de lymphocytes T autoréactifs. Les maladies auto-inflammatoires, quant à elles, découlent d’un dysfonctionnement du système immunitaire inné et vont donc se distinguer par la présence de cellules hyperactives de l’immunité innée, i.e. dont la sécrétion en cytokines et en médiateurs pro-inflammatoires est accrue, ainsi que par l’absence d’auto-anticorps et/ou de lymphocytes T autoréactifs.71 Dans les deux cas, ces maladies pourront être spécifiques, i.e. pour un organe donné, ou systémiques. Cependant, bien qu’à priori il existe une séparation nosologique
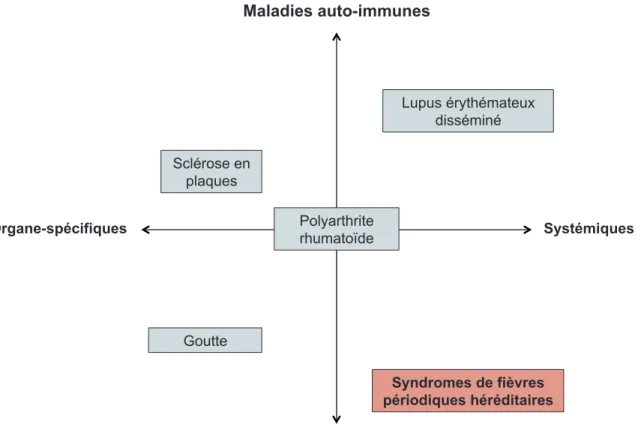
entre ces deux entités, ces maladies sont plutôt réparties au sein d’un spectre s’étendant des maladies auto-immunes aux maladies auto-inflammatoires et des maladies organe-spécifiques aux maladies systémiques (Figure 2).72
Inspiré de Park et al. (2012)72
Figure 2 : Spectre des maladies auto-immunes et auto-inflammatoires.
Le spectre s’étend des maladies auto-immunes aux maladies auto-inflammatoires (axe vertical de haut en bas) et des maladies organe-spécifiques aux maladies systémiques (axe horizontal de gauche à droite). La sclérose en plaques et le lupus érythémateux disséminé sont des exemples de maladies auto-immunes, alors que la goutte et les syndromes de fièvres périodiques héréditaires sont des exemples de maladies auto-inflammatoires. Finalement, d’autres maladies comme la polyarthrite rhumatoïde, par exemple, sont à la croisée des chemins entre ces différentes affections inflammatoires.
La sclérose en plaques, caractérisée par la destruction de la myéline, est un exemple de maladie auto-immune organe-spécifique73, alors que le lupus
Maladies auto-immunes
Maladies auto-inflammatoires
Organe-spécifiques Polyarthrite Systémiques
rhumatoïde Lupus érythémateux disséminé Sclérose en plaques Goutte Syndromes de fièvres périodiques héréditaires
érythémateux disséminé (Systemic Lupus Erythematosus – SLE) est l’exemple parfait d’une maladie auto-immune systémique puisqu’elle affecte plusieurs organes à la fois comme la peau, les articulations et les reins.74 Dans les deux cas,
des auto-anticorps et/ou des lymphocytes T autoréactifs sont identifiés chez les individus atteints. Du côté des maladies auto-inflammatoires, des mécanismes physiopathologiques différents sont impliqués. La goutte, caractérisée par une accumulation de cristaux d’acide urique dans les organes périphériques, notamment le gros orteil, est désormais considérée comme une maladie auto-inflammatoire organe-spécifique puisqu’il a été démontré que ces cristaux activaient de manière excessive les voies de signalisation du système immunitaire inné et la production de cytokines pro-inflammatoires.75 Il existe aussi des maladies auto-inflammatoires systémiques; c’est le cas de plusieurs syndromes auto-inflammatoires dont les fièvres périodiques héréditaires. Finalement, bien qu’il soit possible de classer certaines de ces maladies, il est évident aujourd’hui que de nombreuses pathologies sont un mélange de composantes immunes et auto-inflammatoires. C’est le cas de la RA qui se retrouve au centre du spectre de ces affections inflammatoires; une maladie caractérisée par une inflammation chronique de la membrane synoviale et la destruction des articulations, impliquant aussi bien des lymphocytes T et B que des macrophages et des PMN.76
2.2 Les syndromes auto-inflammatoires, aussi appelés cytokinopathies
Les syndromes auto-inflammatoires sont un groupe de maladies génétiques rares qui découlent de mutations dans différents gènes de régulation du système immunitaire inné. Comme la plupart de ces mutations mènent à une hyperactivation des cellules immunitaires ainsi qu’à une hypersécrétion de cytokines pro-inflammatoires, ces maladies sont également désignées comme étant des cytokinopathies.77 Les symptômes les plus souvent associés à ces
maladies sont les fièvres récurrentes, les éruptions cutanées, les douleurs abdominales ainsi que les douleurs musculaires et/ou articulaires.78
2.2.1 Historique et classification des syndromes auto-inflammatoires
Bien que ces maladies soient observées depuis longtemps, les syndromes de fièvres périodiques héréditaires n'ont été caractérisés qu'au cours des vingt dernières années, en grande partie grâce au raffinement des protocoles qui permettent d’exclure les causes microbiennes.79 La fièvre méditerranéenne
familiale (Familial Mediterranean Fever – FMF) fut le premier syndrome à être caractérisé en 1997 à la suite de l’identification du gène muté responsable de la maladie : le gène MEFV qui code pour la protéine pyrine.80,81 Le syndrome périodique associé au récepteur du TNF (TNF Receptor Associated Periodic
Syndrome – TRAPS) a été caractérisé en 1999 à la suite de la découverte de
mutations dans le gène TNFRSF1A codant pour le TNFRI. Le terme auto-inflammation a alors été proposé par McDermott et ses collaborateurs afin de regrouper tous les syndromes caractérisés par des épisodes récurrents d’inflammation systémique qui se produisaient en absence d’infection et de manifestations auto-immunes.82
Depuis, la découverte de nombreuses mutations dans différents gènes du système immunitaire inné a permis la caractérisation de plus d’une vingtaine de syndromes auto-inflammatoires qui étaient considérés comme des maladies orphelines.83
Parmi ceux-ci, on retrouve notamment les fièvres périodiques héréditaires, composées des syndromes FMF, TRAPS et Hyper immunoglobulinémie D avec fièvres périodiques (Hyper Immunoglobulinemia D Syndrome with periodic fever – HIDS), les syndromes périodiques associés à la cryopyrine (Cryopyrin-Associated
Periodic Syndromes – CAPS), composés du syndrome auto-inflammatoire familial
associé au froid (Familial Cold Autoinflammatory Syndrome – FCAS), du syndrome Muckle-Wells (Muckle-Wells Syndrome – MWS) et du syndrome inflammatoire multi-systémique débutant à l’enfance (Neonatal Onset Multisystem Inflammatory
Disease – NOMID), le syndrome de Blau, le syndrome d’arthrite pyogène associé
au pyoderma gangrenosum et à l’acné (Pyogenic Arthritis, Pyoderma
gangrenosum and Acne – PAPA), le syndrome associé à la déficience de
Antagonist – DIRA), le syndrome associé à la déficience de l’antagoniste du
récepteur de l’IL-36 (Deficiency of Interleukin Thirty-six Receptor Antagonist – DITRA) et le syndrome de dermatose neutrophilique atypique chronique avec lipodystrophie et température élevée (Chronic Atypical Neutrophilic Dermatosis
with Lipodystrophy and Elevated temperature – CANDLE) (Tableau I).22,72
Finalement, les syndromes auto-inflammatoires se distinguent les uns des autres selon la nature des cytokines dérégulées et les composantes de la réponse immunitaire innée qui sont affectées par les mutations à l’origine de ces maladies (Tableau I).22,72
Tableau I : Liste partielle des syndromes auto-inflammatoires et leurs principales caractéristiques.
Syndrome Gène muté Protéine associée Cytokines dérégulées
FMF MEFV MEFV/Pyrine IL-1β
TRAPS TNFRSF1A TNFRI IL-1, TNF
HIDS MVK Mévalonate kinase IL-1β
CAPS NLRP3 NLRP3/NALP3/Cryopyrine IL-1β
Blau NOD2 NOD2 IL-1, TNF, IL-6
PAPA PSTPIP1 CD2BP1 IL-1β, TNF
DIRA IL1RN IL-1RA IL-1α et β
DITRA IL36RN IL-36RA IL-36α, β et γ
CANDLE PSMB8 S.-u. β5i du protéasome IFN et IL-6
Inspiré de Almeida de Jesus et al. (2015)22 et de Park et al. (2012)72
Blau : Syndrome de Blau; CANDLE : Syndrome de dermatose neutrophilique atypique chronique avec lipodystrophie et température élevée; CAPS : Syndromes périodiques associés à la cryopyrine; DIRA : Syndrome associé à la déficience de l’antagoniste du récepteur de l’IL-1; DITRA : Syndrome associé à la déficience de l’antagoniste du récepteur de l’IL-36; FMF : Fièvre méditerranéenne familiale; HIDS : Hyper immunoglobulinémie D avec fièvres périodiques; PAPA : Syndrome d’arthrite pyogène associé au pyoderma gangrenosum et à l’acné; S.-u. : Sous-unité; TRAPS : Syndrome périodique associé au récepteur du TNF
2.2.2 Quatre grands mécanismes physiopathologiques impliqués dans les maladies auto-inflammatoires
Les mutations à l’origine de ces maladies peuvent soit : 1) rendre les PRR intracellulaires hyperactifs, 2) conduire à l’accumulation de molécules à l’intérieur de la cellule comme des substrats d’enzymes ou des protéines incorrectement repliées ou défectueuses, ce qui induit un stress et l’activation subséquente des PRR, 3) provoquer la perte de régulateurs négatifs de l’inflammation, ce qui mène à un déséquilibre entre les cytokines pro-inflammatoires et les antagonistes de ces cytokines, ou 4) affecter les molécules de signalisation et leurs récepteurs de surface, ce qui mène également à une dérégulation des fonctions cellulaires.22
2.2.2.1 Hyperréactivité intrinsèque des PRR
Des mutations de gain de fonction dans les gènes codant pour des PRR intracellulaires peuvent mener à leur hyperactivation ainsi qu’à une production excessive ou continue de médiateurs pro-inflammatoires.22 Certaines des protéines affectées par ces mutations sont des protéines qui constituent les inflammasomes : la pyrine causant le syndrome FMF, NLRP3/NALP3 causant les syndromes CAPS84, et NLRC4/IPAF qui est à l’origine d’un autre type de syndrome auto-inflammatoire où il y a activation excessive des macrophages, appelé syndrome NLRC4-MAS (NLRC4-Macrophage Activation Syndrome).85 D’autres
protéines ne composant pas les inflammasomes peuvent aussi être affectées, comme c’est le cas dans le syndrome de Blau, où le récepteur NOD2, un constituant du signalosome (complexe protéique impliqué dans la régulation de la dégradation des protéines) NOD1/NOD2, est en cause.84
2.2.2.1.1 La fièvre méditerranéenne familiale – FMF
La FMF est le syndrome auto-inflammatoire le plus répandu à travers le monde.86 En général, la maladie se manifeste avant l’âge de 20 ans et se caractérise par des crises inflammatoires récurrentes qui durent, en moyenne, de 12 à 72 heures et qui reviennent à chaque semaine, mois ou quelque fois par année, en
intermittence avec des périodes peu symptomatiques ou complètement asymptomatiques. Les symptômes les plus couramment observés sont les fièvres récurrentes, les péritonites généralisées, les douleurs abdominales, la constipation et l’arthrite. Les péricardites, les méningites aseptiques et les éruptions cutanées de type érythème érysipéloïde au niveau des membres inférieurs sont aussi des symptômes observés chez certains patients, bien qu’ils soient plus rares.84 Les patients souffrant d’inflammation chronique ont également un risque élevé de développer, à long terme, une amyloïdose systémique pouvant mener à l’insuffisance rénale et à la mort. L’amyloïdose est une pathologie qui se caractérise par l’accumulation de protéines insolubles dans un tissu ou un organe, ce qui mène éventuellement à leur dysfonctionnement.87 Finalement, bien que le gène muté à l’origine de la maladie soit identifié (MEFV codant pour la pyrine), les mécanismes physiopathologiques demeurent encore mal compris. Il existe deux théories : 1) la pyrine activerait directement la caspase-1, indépendamment de l’inflammasome NLRP3/NALP3 (dans ce cas-ci on parlera alors de l’inflammasome pyrine), 2) la pyrine mutée perdrait son effet inhibiteur sur l’inflammasome NLRP3/NALP3, entraînant l’hyperactivation de ce dernier et l’hypersécrétion subséquente d’IL-1β.88
2.2.2.1.2 Les syndromes périodiques associés à la cryopyrine – CAPS
La prévalence de chacun des syndromes CAPS, incluant FCAS, MWS et NOMID, est estimée à une à deux personnes sur 1 million.22 Tous les patients atteints vont,
entre autres, souffrir d’épisodes récurrents de fièvres, d’arthralgie, de conjonctivites et d’urticaire neutrophilique. Certains patients MWS et NOMID peuvent également développer une perte d’audition permanente ainsi qu’une méningite aseptique chronique pouvant mener à diverses atteintes du système nerveux central (SNC) : atrophie cérébrale, déficience cognitive, atrophie des nerfs optiques et/ou perte de la vision. Il a été montré que des mutations de gain de fonction dans le gène NLRP3 qui code pour le récepteur NLRP3/NALP3, aussi appelée cryopyrine, permettent l'oligomérisation spontanée et l'activation excessive de l'inflammasome NLRP3/NALP3. Basé sur ce mécanisme
physiopathologique, la cellule n’a plus besoin du deuxième signal (abordé dans la section 1.1.4.1) pour induire une sécrétion rapide et maximale d’IL-1β.89
2.2.2.1.3 Le syndrome de Blau
Le syndrome de Blau est une maladie rare qui affecte moins d’une personne sur un million. Les principaux symptômes observés sont l’uvéite granulomateuse, menant à une perte visuelle progressive chez 40% des patients; une polyarthrite chronique qui se développe principalement au niveau des chevilles, des genoux et des poignets; et une éruption cutanée qui se caractérise par la présence de papules rougeâtres sur le visage et le tronc.90 La maladie découle de mutations de gain de fonction dans le gène NOD2 qui code pour le récepteur NOD2.91 En condition physiologique, ce récepteur s’oligomérise à la suite de la reconnaissance de différents PAMP intracellulaires comme le muramyl dipeptide (MDP), un constituant du peptidoglycane de la paroi des bactéries positives et gram-négatives, ce qui permet le recrutement de la kinase RIP-2 (Receptor-Interacting
Protein-2 Kinase – RIP-2K) et l’activation des voies de signalisation NFκB et
MAPK.92 Chez les patients atteints, les mutations dans le gène NOD2 entrainent une oligomérisation spontanée du récepteur, ce qui permet l’activation de la voie NFκB en absence d’agents bactériens et l’hypersécrétion subséquente des cytokines pro-inflammatoires à l’origine des symptômes, notamment l’IL-1β, le TNF et l’IL-6.93
2.2.2.2 Stress intracellulaire menant à l’inflammation
Des mutations autosomiques récessives de perte de fonction ou causant de l’haploinsuffisance dans les gènes codant pour des molécules ou des enzymes essentielles au maintien de l’homéostasie cellulaire peuvent entrainer l'accumulation de facteurs de stress intracellulaires qui stimulent l'activation des PRR intracellulaires et la production de médiateurs pro-inflammatoires.22
2.2.2.2.1 Le syndrome périodique associé au récepteur du TNF – TRAPS
Le TRAPS est la seconde maladie auto-inflammatoire la plus répandue.94 Les
premiers symptômes de la maladie peuvent se manifester à tout moment de la vie (dès 2 semaines de vie jusqu’à la cinquantaine) mais dans la majorité des cas, ils apparaissent avant l’âge de 20 ans.95 Les patients atteints souffrent de crises
inflammatoires récurrentes qui se caractérisent par des fièvres qui peuvent durer en moyenne 21 jours et revenir à chaque 5-6 semaines, des douleurs abdominales et/ou thoraciques sévères, des éruptions cutanées, souvent maculaires, érythémateuses ou urticariennes, des manifestations oculaires telles que des conjonctivites et des uvéites, et des douleurs musculaires et/ou articulaires, appelées respectivement myalgies et arthralgies. Les patients qui ne sont pas traités adéquatement ont également un risque élevé de développer, à long terme, une amyloïdose rénale et hépatique.96 Plus de 100 mutations dans le gène
TNFRSF1A ont été identifiées à ce jour, la plupart affectant le domaine
extracellulaire du récepteur et menant à un défaut dans la structure et le repliement de la protéine.97 Les mutants TNFRI s’accumulent dans le réticulum endoplasmique et induisent un stress intracellulaire important menant à l’activation spontanée des voies NFκB et MAPK ainsi qu’à la transcription subséquente de médiateurs pro-inflammatoires, notamment l’IL-1 et le TNF.98 De plus, les cellules des patients atteints produisent de façon exagérée des ROS mitochondriaux qui maintiennent l’activation des voies d’activation menant à la sécrétion de cytokines pro-inflammatoires.99
2.2.2.2.2 Le syndrome d’hyper IgD avec fièvres périodiques – HIDS
Les patients atteints du HIDS souffrent de crises inflammatoires récurrentes qui durent en moyenne 3-7 jours et qui se caractérisent par un niveau élevé d’IgD dans le sérum, des fièvres récurrentes, des douleurs abdominales, des arthralgies, et diverses éruptions cutanées, principalement maculopapulaires, urticariennes et purpuriques. Les cas les plus sévères peuvent aussi présenter des atteintes au niveau du SNC comme un retard psychomoteur ou de la dysmorphie faciale.100 Les mutations dans le gène MVK causent une diminution de l’activité enzymatique