UNIVERSITE PAUL SABATIER - TOULOUSE III U.F.R. Sciences de la Vie et de la Terre Ecole Doctorale Biologie Santé Biotechnologies
THESE
Pour obtenir le grade de
Docteur de l’Université Toulouse III
Discipline : Biologie
Mention : Génétique, Epidémiologie, et Santé publique
TITRE :
Enjeux méthodologiques de l’analyse de marqueurs génétiques dans les études d’association de maladies multifactorielles : application à la
polyarthrite rhumatoïde
Présentée et soutenue par
Thomas BARNETCHE
Le 21 Septembre 2007
___________
Directeur de thèse :
Anne Cambon Thomsen ; DR CNRS Toulouse ___________
JURY
Benoît Arveiler, Professeur Bordeaux II - Praticien Hospitalier ; Membre invité Alain Cantagrel, Professeur Toulouse III - Praticien Hospitalier
Arnaud Constantin, Maître de conférences Toulouse III - Praticien Hospitalier François Cornélis, Maître de conférences Paris 7 - Praticien Hospitalier; Rapporteur Thierry Schaeverbeke, Professeur Bordeaux II - Praticien Hospitalier ; Membre invité Gilbert Semana, Professeur Rennes I - Praticien Hospitalier ; Rapporteur
UNIVERSITE PAUL SABATIER - TOULOUSE III U.F.R. Sciences de la Vie et de la Terre Ecole Doctorale Biologie Santé Biotechnologies
THESE
Pour obtenir le grade de
Docteur de l’Université Toulouse III
Discipline : Biologie
Mention : Génétique, Epidémiologie, et Santé publique
TITRE :
Enjeux méthodologiques de l’analyse de marqueurs génétiques dans les études d’association de maladies multifactorielles : application à la
polyarthrite rhumatoïde
Présentée et soutenue par
Thomas BARNETCHE
Le 21 Septembre 2007
___________
Directeur de thèse :
Anne Cambon Thomsen ; DR CNRS Toulouse ___________
JURY
Benoît Arveiler, Professeur Bordeaux II - Praticien Hospitalier ; Membre invité Alain Cantagrel, Professeur Toulouse III - Praticien Hospitalier
Arnaud Constantin, Maître de conférences Toulouse III - Praticien Hospitalier
François Cornélis, Maître de conférences Paris 7 - Praticien Hospitalier; Rapporteur Thierry Schaeverbeke, Professeur Bordeaux II - Praticien Hospitalier ; Membre invité Gilbert Semana, Professeur Rennes I - Praticien Hospitalier ; Rapporteur
Remerciements
Je tiens à adresser mes plus vifs et sincères remerciements
A ma directrice de thèse, le Dr Anne Cambon-Thomsen, pour ses qualités humaines, son sens de l’écoute et la grandeur de ses connaissances. Ses cinq années de travail à vos côtés m’ont permis de constituer une base solide pour mon futur professionnel.
A mes rapporteurs et membres du jury, pour avoir accepté de juger mon travail de thèse.
A Pierre-Antoine Gourraud, pour sa disponibilité et sa patience sans faille. Tes compétences et ta générosité dans le travail constituent pour moi une référence.
A l’ensemble des membres de l’équipe 4, au premier rang desquels Arnaud et Emmanuelle, qu’ils trouvent l’expression de ma reconnaissance et de mon affection.
Avec le professeur Dehais, je tiens à remercier tout le service de rhumatologie du CHU de Bordeaux de m’avoir accueilli. Avec une mention spéciale à : Marie-Jeanne, Nancy, Sophie, Elodie, Camille, Chantal, Marie-Elise, Lorna, Patricia et l’ensemble des infirmières et aides-soignantes.
Au Professeur Schaeverbeke, pour son précieux concours à l’élaboration de ce travail, pour ses compétences et pour la confiance qu’il me témoigne au quotidien.
A l’ensemble de l’équipe GHDC, et notamment Christophe, Fred, Caroline, Ingrid, Delphine, Céline, Virginie, Michelle, Isabelle, Cyril… Je promets d’améliorer mon maniement de la pipette et des cartes de tarot…
Au Professeur Arveiler, pour son écoute, ses précieux conseils et son humour… En espérant pouvoir continuer à partager nos différentes passions, tant sur le plan professionnel, que personnel.
A tous mes amis, Alexandre, Emilie, Stéphane, Baptiste, Julien-Pierre & Carole, Mathieu & Morgane, Cédric & Audrey, Mathieu & Agathe, Washington & Pam… Vous avez chacun à votre manière permis l’aboutissement de ce travail et je vous en serai éternellement reconnaissant.
A toute ma famille, et notamment mes parents et mon frère, qui m’ont toujours suivi et soutenu pendant ces longues années d’étude et que je sais fiers aujourd’hui.
A Solène… Il me faudrait beaucoup plus que ces quelques lignes pour te remercier et te décrire la grandeur de mes sentiments pour toi. Je te promets que je finirai l’année plus calme et reposé que je ne l’ai commencée.
TABLE DES MATIERES
PREAMBULE... 10
INTRODUCTION... 16
A- Distinction entre les maladies monogéniques et les maladies multifactorielles : implications en termes de composante génétique ... 17
B- Principales méthodologies existantes pour l’étude génétique de maladies multifactorielles. ... 22
1. Les stratégies d’approche des études génétiques ... 22
a. Etudes d’agrégation familiale ... 22
b. L’héritabilité : Etudes de jumeaux... 23
c. Le « whole genome scan »... 24
d. L’approche « gène-candidat »... 25
2. Les stratégies d’étude en génétique ... 26
a. La méthode des lod-scores ... 26
b. La méthode des paires de germains atteints... 26
c. Les études d’association allélique ... 28
d. Une méthode « combinée » : le Transmission Disequilibrium Test (TDT) .. 30
3. Les grandes familles de marqueurs utilisés en génétique ... 31
a. Les marqueurs microsatellites ... 31
b. Les marqueurs SNPs ... 33
c. Les marqueurs RFLP... 34
C- Présentation de la polyarthrite rhumatoïde ... 36
1. Définition... 36
2. Critères diagnostiques... 36
3. Epidémiologie générale... 37
4. Facteurs prédisposants ... 38
a. Des facteurs endocriniens ... 38
b. L’hypothèse infectieuse... 39
c. Autres facteurs individuels et environnementaux... 39
5. Epidémiologie génétique de la PR ... 40
D- Objectifs ... 46
PATIENTS ET METHODES ... 48
A- Etude de susceptibilité/sévérité de la PR : utilisation de marqueurs microsatellites de la région HLA ... 49
1. La cohorte RMP... 49
2. L’échantillon de donneurs de moelle osseuse ... 50
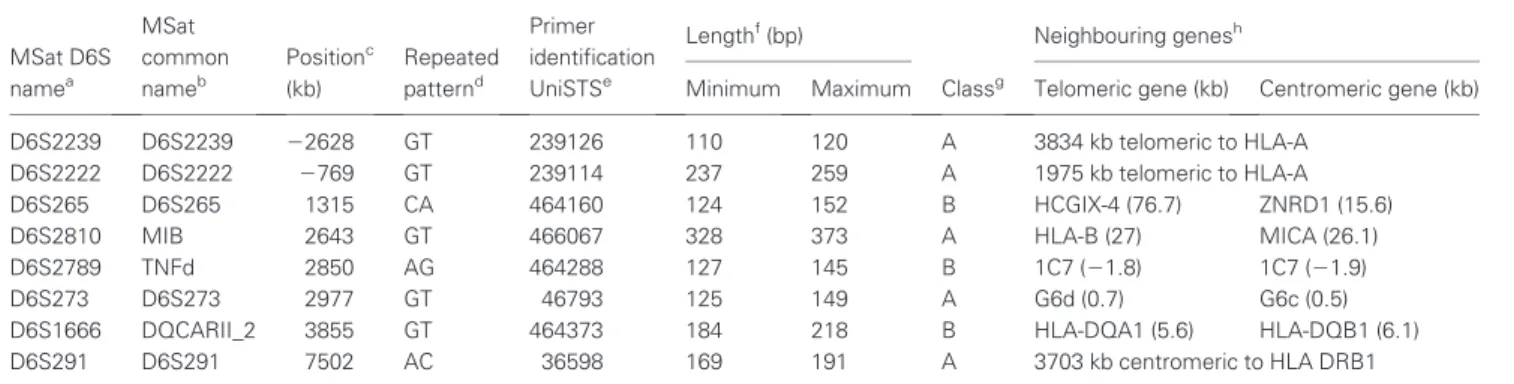
3. Typage HLA et stratégie de choix des marqueurs microsatellites ... 51
a. Le typage HLA ... 51
b. Le choix des marqueurs microsatellites ... 51
4. Analyse statistique ... 54
b. La multiplicité des tests ... 56
c. Principes méthodologiques du test de permutation... 59
B- Analyse des données PR du 13ème Workshop International d’Histocompatibilité : pertinence d’une nouvelle classification des allèles « épitope partagé » ... 62
1. Description des échantillons du 13ème Workshop International d’histocompatibilité (IHWG) ... 62
2. Sélection des échantillons d’intérêt pour la réalisation de la méta-analyse .... 63
3. Principes de la nouvelle classification des allèles codant pour l’épitope partagé ... 64
4. Principes méthodologiques de la réalisation d’une méta-analyse ... 66
RESULTATS ... 71
A- Principes méthodologiques de l’étude de maladies multifactorielles en génétique épidémiologique ... 73
B- Utilisation de marqueurs microsatellites de la région HLA dans l’étude de la susceptibilité à la PR ... 96
C- Etude de la nouvelle classification des allèles HLA-DRB1 en termes de sévérité et de susceptibilité à la PR ... 121
DISCUSSION ... 152
A- Evolution des méthodes d’étude des maladies multifactorielles : les études d’association pangénomiques... 153
B- Stratégie de choix de marqueurs dans les études d’association en génétique ... 161
C- HLA-DRB1 : un marqueur à l’épreuve de la méta-analyse ... 166
D- Quelle peut-être la place de la génétique dans la prévention et la prise en charge des maladies complexes ? ... 171
CONCLUSION ... 179
REFERENCES BIBLIOGRAPHIQUES ... 184
ABSTRACT ... 198
TABLE DES ILLUSTRATIONS
Figure 1 : Exemple d’arbre généalogique pour une maladie à hérédité complexe (p.21)
Tableau 1 : Exemples de maladies multifactorielles avec leurs facteurs génétiques de susceptibilité correspondants
(p.21)
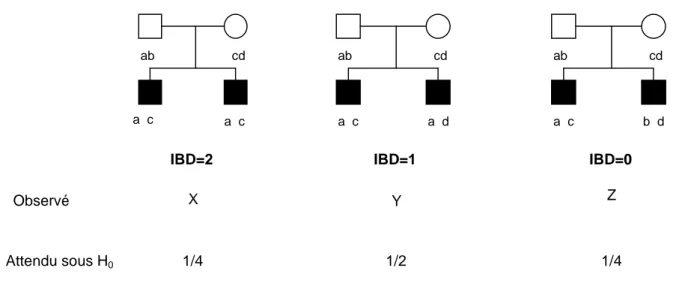
Figure 2 : Les états IBD pour une paire de germains atteints (p.27)
Tableau 2 : Critères de classification d’une PR selon l’ACR (p.37)
Figure 3 : Carte de la région HLA et des séquences microsatellites sélectionnées (p.52)
Tableau 3 : Composition des échantillons sélectionnés (p.64)
Figure 4 : Extraite de « Genetic Epidemiology and Public Health: hope, hype and future prospects » de Georges Dovey Smith et al. Lancet 2005;366:1484-98 (p.183)
L’épidémiologie se définit comme la science des déterminants des événements de santé dans les populations, et de leur répartition. L’épidémiologie génétique se rapproche de l’épidémiologie traditionnelle puisqu’elle concentre son étude sur les déterminants familiaux des maladies, et en particulier génétiques, ainsi que sur l’analyse des effets de facteurs génétiques et non génétiques, et de leurs interactions.
L’épidémiologie génétique est une discipline hybride avec une histoire relativement courte de moins de 50 ans. Neel et Schull, dans leur ouvrage datant de 1954 (1), sont les premiers à faire mention de la notion « d’épidémiologie génétique », en suggérant que les deux disciplines devraient considérer leurs stratégies respectives pour améliorer l’étude de maladies communes telles que les maladies cardio-vaculaires, l’asthme ou le cancer, maladies multifactorielles résultant de l’interaction de facteurs génétiques et environnementaux. Cette nouvelle discipline émergea lentement sur les vingt années qui suivirent, pour connaître un développement important depuis une quinzaine d’années.
L’épidémiologie, considérée comme la science de la Santé Publique, est la « science-mère » la plus ancienne des deux disciplines considérées dans ce propos. Elle tire ses racines de l’étude de paramètres vitaux et/ou de mécanismes de transmission des maladies, durant la « révolution bactériologique » du 19ème siècle. L’épidémiologie, qui tire son propos de la mise en place d’études d’observation, a permis le développement de nombreuses méthodes statistiques pour réaliser des tests d’hypothèses et déterminer des inférences causales (2).
La génétique, qui se définit comme la science de l’hérédité, a trouvé son origine avec les travaux de Gregor Mendel, un moine autrichien dont les expériences d’hybridation chez les plantes ont conduit à mettre en évidence le paradigme sous
-jacent fondamental pour la génétique moderne (3). La valeur réelle des travaux de
Mendel ne fut complètement appréciée qu’à partir du début du 20ème siècle, lorsque
des biologistes cellulaires démontrèrent que les chromosomes des cellules eucaryotiques (et les gènes qu’ils portent) suivent des modèles de ségrégation durant la méiose, ce qui déclencha de nombreuses recherches (4).
La génétique épidémiologie a commencé à émerger lorsque les généticiens et les épidémiologistes ont réalisé que l’étiologie de la majorité des maladies chroniques ne pouvait être comprise de manière complète sans considérer une action conjointe de facteurs génétiques et environnementaux, notamment dans le cadre des maladies multifactorielles. Alors que les chercheurs des deux disciplines commençaient à travailler de concert, les différences de terminologie constituèrent un obstacle à la compréhension des outils respectifs de chacun, et à la mise en place d’études conjointes. Par exemple, les généticiens ont mis du temps à reconnaître qu’une population à structure hétérogène (i.e., une population est composée de sous-groupes génétiques distincts) pouvait entraîner la mise en évidence d’associations saugrenues entre un marqueur génétique et une maladie. Le terme de « population stratifiée » fut défini pour décrire ce phénomène d’hétérogénéité à l’intérieur d’une population de cas et de témoins, alors que les épidémiologistes utilisaient le terme de facteur de confusion pour désigner de tels phénomènes. Ainsi les 2 disciplines divergent dans leur mode d’expression et n’ont pas exploité de façon optimale leur complémentarité. Donc, il n’est pas surprenant aujourd’hui que des confusions persistent sur la façon d’interpréter certaines associations statistiques entre un marqueur génétique et une maladie.
Il existe toute une « gamme de la causalité » entre les maladies monogéniques ou « mendéliennes » (où les gènes constituent le seul facteur de prédisposition), et les
maladies complexes où certains gènes ne déterminent la maladie que sous l’effet d’une exposition à un facteur environnemental spécifique (par exemple le déficit congénital en glucose-6-déshydrogénase qui cause une anémie hémolytique quand le sujet est exposé à des traitements anti-paludéens). Même si les maladies « mendéliennes » restent pour chacune d’elles un événement rare, elles constituent un fardeau cumulatif important en terme de Santé Publique. De plus, certaines de ces maladies peuvent être présentes en grand nombre dans certaines populations et ainsi devenir une priorité dans les programmes de Santé Publique (ex : La beta-thalassémie dans certaines populations du bassin Méditerranéen). La génétique médicale, dont l’intérêt s’est initialement porté sur les maladies monogéniques (généralement rares), s’est régulièrement trouvée éloignée de l’épidémiologie dont le sujet d’étude favori reste les maladies communes (à majorité multifactorielles). Mais lorsque des variants géniques furent identifiés comme facteur de risque, les deux disciplines furent dans l’obligation de considérer l’impact en termes de Santé Publique des facteurs de risque génétique.
King et al (5) ont défini trois questions essentielles en génétique épidémiologique : 1) Existe-t-il une composante familiale dans la maladie étudiée ? 2) Est-ce que cette agrégation familiale est compatible avec un mécanisme génétique ou non? et 3) Quel est le modèle de transmission de la pathologie ? Ces questions peuvent être développées de manière plus précise, et pour chaque question il convient de mettre en place des stratégies d’analyse adéquates.
En ce début du 21ème siècle, la recherche de gènes pouvant contribuer à l’étiologie
de maladies complexes prend une part prépondérante dans les activités de recherche et de développement de la génétique épidémiologique. Depuis que l’intervention de nombreux gènes a été mise en évidence pour un certain nombre de
maladies, il devient primordial de gérer la difficulté de prise en compte de l’hétérogénéité étiologique pouvant exister entre des populations de patients différentes, ou des groupes de familles distincts. Adapter des principes épidémiologiques à des études génétiques classiques pour prendre en considération l’inconsistance des résultats soulève encore d’autres défis méthodologiques, puisque les chercheurs se doivent de considérer les différents patrimoines génétiques des différentes populations, sources d’hétérogénéité dans les résultats.
L’épidémiologie et les statistiques, de par leur rigueur méthodologique, constituent une base de développement très solide au niveau génétique, permettant notamment de mieux appréhender le traitement en masse de données telles que la génétique en produit aujourd’hui. Ceci avec pour objectif final une meilleure exploitation de l’ensemble des informations générées par des équipes de chercheurs, ou mises à leur disposition, dans le but notamment de permettre l’identification de nouvelles cibles thérapeutiques par l’élucidation des mécanismes génétiques de pathologies communes.
C’est dans ce contexte de recherche pluridisciplinaire que ce manuscrit de thèse se place. L’objectif principal de ce travail de thèse est de montrer quel peut être l’apport de l’utilisation de méthodes d’épidémiologie génétique dans l’étude de la physiopathologie d’une maladie multifactorielle, en l’occurrence la polyarthrite rhumatoïde.
Il s’agira tout d’abord de présenter le contexte méthodologique, en détaillant la nature exacte des méthodes permettant l’investigation du rôle d’un variant génétique dans l’étiologie d’une maladie complexe. Puis nous verrons comment cette
méthodologie d’analyse a notamment trouvé son application dans l’étude de la polyarthrite rhumatoïde.
Dans un deuxième temps, nous tenterons d’analyser, en fonction des différents types de marqueurs génétiques, quelles peuvent être les stratégies de choix de ces marqueurs afin d’assurer la cohérence de l’information génétique avec le modèle biologique étudié et sa prise en considération dans la physiopathologie.
Enfin, nous tenterons de voir quels peuvent être des exemples de solution à une prise en compte pertinente de l’hétérogénéité de l’information génétique, et ainsi permettre une interprétation pertinente des résultats.
Ce travail a été mené dans le but de participer à la compréhension des mécanismes complexes intervenant dans la pathogénie de la polyarthrite rhumatoïde, et à une meilleure appréciation de la part respective des différents facteurs impliqués. Ces analyses permettront également de souligner la nécessité de prendre en compte une variabilité génétique (identification de sujets à risque) dans le développement et l’élaboration de stratégies diagnostiques et thérapeutiques.
Cette introduction va poser les bases des études des maladies multifactorielles en génétique, en exploitant notamment comme exemple le modèle d’une maladie multifactorielle : la polyarthrite rhumatoïde (PR).
Une petite précision s’avère néanmoins nécessaire avant de commencer : bien qu’étant conscient des discussions sur le sujet, j’ai pris le parti dans ce manuscrit de définir les maladies complexes (ou multifactorielles) et les maladies communes comme des synonymes, en considérant que les maladies les plus communes sont le plus souvent complexes, et/ou que les maladies complexes sont également les plus communes dans la population (6).
A- Distinction entre les maladies monogéniques et les
maladies multifactorielles : implications en termes de
composante génétique
Au cours des vingt-cinq dernières années, plus d’un millier de gènes impliqués des maladies monogéniques ont été localisées et un grand nombre d’entre eux ont été clonés. Ces maladies, autrement qualifiées de « mendéliennes », se situent à une extrémité du « spectre » et sont purement héréditaires. Les exemples les plus connus de ce genre d’affections sont l’hémophilie, la chorée de Huntington ou encore la dystrophie musculaire de Duchenne. Ces maladies reposent sur la présence d’une mutation, dominante ou récessive, sur un chromosome sexuel ou non. A l’autre extrémité du spectre des maladies, nous trouvons les pathologies que l’on pourrait qualifier de « purement environnementales », telle que la fracture d’une jambe d’un skieur après une chute. Entre ces deux extrêmes, se situe la très grande majorité des maladies humaines qui résultent d’interactions entre des facteurs environnementaux et le génome d’un individu. Cependant il faut nuancer cette
classification car de nombreuses maladies mendéliennes laissent place aussi à des facteurs environnementaux influençant par exemple l’âge d’apparition ou la gravité des symptômes et des événements a priori non concernés par la génétique peuvent recéler une composante génétique ignorée, par exemple rendant la survenue d’une fracture plus ou moins probable dans la cadre d’un traumatisme.
Le développement rapide des technologies moléculaires en génétique, se traduisant notamment par une augmentation notable des publications scientifiques du domaine, a conduit à une augmentation rapide et significative des connaissances, à la fois sur l’étiologie des maladies et sur le fonctionnement du vivant. De nombreux phénomènes comme le contrôle de la respiration, la régulation de la tension artérielle, l’extraction des nutriments de l’alimentation quotidienne, ou encore le système de surveillance immunitaire font appel à des mécanismes complexes, d’ordre qualitatif aussi bien que quantitatifs. Tous les systèmes sollicités sont typiquement sous contrôle génétique et répondent par des mécanismes de feed-back à des stimuli environnementaux. Le fonctionnement de ces systèmes implique des mécanismes de redondance et de compensation pour en pallier les éventuelles défaillances. L’architecture des mécanismes vitaux peut être assimilée à un système hiérarchique, permettant d’aller du génotype au phénotype, celui-ci pouvant être constitué par des signes cliniques qui définissent la maladie.
A cette complexité physiologique de chaque individu, dont les gènes définissent ultimement, de concert avec l’environnement, les phénotypes observés, se surajoute la dimension populationnelle. La population humaine est de grande taille et a subi un certain nombre de divisions ou de stratifications au cours de son évolution, ainsi qu’une exposition à des milieux environnementaux variés. Ainsi des variants génétiques, intervenant chacun dans un mécanisme physiologique, peuvent varier
en fréquence dans différentes sous-populations de la population humaine. Certains de ces variants ont un effet délétère (possiblement dans certaines sous-populations uniquement), car ils co-existent avec d’autres gènes de prédisposition pour l’affection étudiée, ou car ils sont exposés à des facteurs environnementaux qui révèlent leur possible effet menant à un trait pathologique (7-9). A côté de processus aléatoires ou stochastiques qui influencent la présence ou non d’un gène à l’intérieur d’une population et sa fréquence, d’autres facteurs interviennent comme les migrations, les changements environnementaux, qui sont autant d’effets difficilement identifiables de manière exacte.
La complexité de la physiologie humaine et celle des structures génétiques des populations humaines sous-tendent les difficultés méthodologiques de l’étude des maladies multifactorielles.
Dans le cadre des maladies à hérédité complexe, la physiopathologie implique la présence simultanée de nombreuses variations génétiques affectant des gènes différents et ayant chacune un effet modeste. Ce concept n’est pas strictement superposable à celui de maladie polygénique. Les modèles polygéniques utilisés en analyse de ségrégation impliquent en effet une contribution strictement additive de plusieurs gènes, au détriment d’interactions épistatiques plus complexes. Pour cette raison, il est donc préférable de parler de déterminisme « multigénique », plutôt que « polygénique ». Chacun de ces variants génétiques, considérés isolément, n’est ni indispensable ni suffisant pour entraîner la maladie. Au contraire, celle-ci n’apparaît que lorsqu’un certain seuil de susceptibilité est dépassé et ce seuil, dans un modèle général, peut être atteint par l’action conjointe de facteurs génétiques (résultant de
diverses combinaisons alléliques entre différents loci) et de facteurs
conférant un risque pour la maladie peut donc ne s’avérer que très légèrement augmentée dans un groupe de malades comparé à un groupe de témoins. De plus, lorsque l’on considère la co-ségrégation de tel allèle particulier et de la maladie au sein d’une généalogie, le type de transmission est très différent de celui observé dans le cas des maladies mendéliennes. Alors que dans celles-ci, la co-ségrégation entre le génotype pathologique et la maladie est parfaite ou du moins très significative, il n’en va pas de même en ce qui concerne les facteurs de risque. Il existe une part des individus portant le génotype « à risque » au locus considéré, et non atteints (cela correspond au concept de pénétrance incomplète de ce génotype) et, d’autre part, des individus non porteurs de ce génotype qui sont atteints (il s’agit alors de phénocopies). En conséquence, dans le cas des maladies à hérédité complexe, la correspondance génotype/phénotype est, pour un gène donné, très complexe. Dés lors, il n’est pas étonnant que si peu de facteurs de risque génétiques aient été identifiés, comparativement à l’extraordinaire moisson de mutants rares identifiés dans les maladies mendéliennes. Le tableau ci-après, qui ne se veut pas exhaustif, présente quelques exemples de loci impliqués dans la physiopathologie de maladies multifactorielles. A partir de ces quelques exemples, nous pouvons constater que le panel est vaste (cardiovasculaire, neurologique…).
Ainsi, l’identification des gènes associés aux maladies complexes demande d’adopter des stratégies appropriées, aussi bien en termes de recrutement des sujets de recherche qu’en termes de méthodologies utilisées (12).
Figure 1 : exemple d’arbre généalogique génétique pour une maladie à hérédité complexe
En analysant cet arbre généalogique on ne peut pas mettre en évidence une transmission des caractères suivant les lois de Mendel. Il s'agit d'un profil de transmission complexe dans lequel la variation des gènes seule ne peut pas expliquer la maladie.
Tableau 1 : exemples de maladies multifactorielles avec leurs facteurs génétiques de susceptibilité correspondants
Maladie Gènes
Hypertension artèrielle gène ACE2 (chromosome X) Maladie d’Alzheimer Apolipoprotéine E (19q) Diabète insulino-dépendant HLA-DR3 et HLA-DR4 (6p) Diabète non insulino-dépendant Calpaïne 10 (2q)
Thrombose veineuse Mutation du facteur V de Leiden (1q) Hypertryglycéridémie Lipoprotéine lipase (8q)
Sclérose en plaques HLA-DR2 (6p)
Spondylarthrite ankylosante HLA-B27 (6p)
Maladie coeliaque HLA-DQ2 et HLA-DQ8 (6p)
Dans ce manuscrit, le propos sera centré sur une maladie auto-immune : la polyarthrite rhumatoïde dont la région génomique d’intérêt est la région HLA.
B- Principales méthodologies existantes pour l’étude
génétique de maladies multifactorielles.
1. Les stratégies d’approche des études génétiques
Ces différentes stratégies d’approche vont varier suivant l’angle d’étude choisi. S’il s’agit d’observer la maladie et sa distribution, les chercheurs réalisent des études d’agrégation familiale et/ou des études de jumeaux. Si, en revanche, il s’agit d’identifier des marqueurs génétiques pouvant être liés et/ou associés à la maladie, deux grandes stratégies co-existent : le « whole genome scan » et la stratégie « gène-candidat ».
a. Etudes d’agrégation familiale
L’objectif initial de beaucoup d’études épidémiologiques est d’obtenir une estimation de la mesure d’agrégation familiale de la maladie étudiée. Le degré d’atteinte familiale pour un trait dichotomique peut être exprimé par la fraction λS, assimilable
au risque relatif de développer la maladie chez les apparentés du premier degré d’une personne atteinte de polyarthrite rhumatoïde par exemple. Le proposant (ou « proband » en anglais) correspond à la première personne chez qui on détecte cliniquement l’affection génétique d’intérêt. S peut alors représenter les parents, un frère ou une sœur, ou tout autre personne ayant un lien de parenté avec le patient.
Lorsque λS =1, il n’existe aucune agrégation familiale. En général, une valeur élevée
pour λS va suggérer l’intervention d’une composante génétique, bien qu’une
estimation élevée de λS puisse aussi être le reflet de facteurs environnementaux
Même si classiquement cette méthode d’analyse reste la plus utilisée et la plus connue, il existe beaucoup d’autres approches qui permettent d’estimer l’agrégation familiale d’une maladie d’intérêt (6).
b. L’héritabilité : Etudes de jumeaux
Les études portant sur la contribution relative des gènes et des facteurs environnementaux sont souvent mises en place avant les études d’association ou de liaison directement centrées sur l’ADN. L’objectif est d’estimer la proportion de la variance phénotypique totale d’un trait provenant de l’action de l’ensemble des facteurs génétiques. Cette proportion est souvent dénommée « héritabilité », est
notée h2 et exprimée en pourcentage.
Dans le cadre des maladies multifactorielles, l’hypothèse de départ des études est que les effets génétiques sont la résultante d’un grand nombre de loci, chacun avec un effet faible, présentant des effets additifs sur l’expression du phénotype. Plusieurs approches existent pour estimer cette héritabilité : des analyses de régression, des analyses de variance ou de maximum de vraisemblance (6, 12), mais ce sont les études étudiant les taux de concordance entre jumeaux qui présentent l’un des schémas d’utilisation le plus simple (13).
Le principe de base des études de jumeaux est que les facteurs environnementaux partagés sont les mêmes, que ce soit pour des jumeaux monozygotes (MZ) ou dizygotes (DZ).
Ces études sont réalisées dans le but de discriminer l'influence des gènes de celle de l'environnement. Si le taux de concordance entre les jumeaux monozygotes et celui entre les jumeaux dizygotes sont égaux, c’est à dire s’il n’y a pas de différence dans les taux de concordance entre ces deux types de jumeaux cela veut dire que le
fait d’avoir soit 50% des gènes en commun, soit 100% des gènes en commun, ne fait pas de différence sur l’incidence de la maladie. Il n’y a donc pas de facteur causal génétique. Au contraire, s’il existe une grande différence des taux de concordance chez les monozygotes et les dizygotes, elle reflète un poids important des facteurs génétiques. Cela ne veut pas dire que cela permet de les identifier encore, mais cela permet de quantifier le poids des influences génétiques dans une maladie : plus cette différence est grande, plus cette influence est grande.
c. Le « whole genome scan »
Le genome scan est un moyen de réaliser une approche au hasard des régions candidates dans une maladie. C’est une exploration génomique au hasard qui consiste à étudier des marqueurs répartis tout au long du génome. Dans cette stratégie, le but est de rechercher une région génétique contenant un gène inconnu ou non, associée à la maladie ou à un trait quantitatif lié à la maladie (ex : la masse grasse pour l’obésité).
Si les loci marqueurs présentent une liaison, il est alors possible de déterminer une région où peut être localisé un gène important dans le mécanisme de la maladie étudiée. De même, il est possible d’isoler des loci génétiques associés à un trait quantitatif ou QTL (Quantitative Trait Locus). Si une indication de liaison est retrouvée avec un ou plusieurs marqueurs, il est alors indispensable de confirmer ces résultats dans un autre échantillon de familles, puis si la liaison s’avère confirmée, de rechercher le gène mis en cause par clonage positionnel. Cette stratégie permet de cibler de plus en plus précisément la localisation du gène et enfin de le définir.
L’approche au hasard permet de progresser par exclusion successive de régions génomiques (cartographie d'exclusion). Le but n'est pas de trouver directement le gène responsable de la maladie mais de rechercher sa localisation la plus probable entre des marqueurs dont l'emplacement précis est connu sur la carte génétique.
d. L’approche « gène-candidat »
Elle cherche à mettre en cause ou au contraire à exclure certains gènes connus, codant pour des protéines pouvant être impliquées dans la physiopathologie de la maladie d’intérêt.
Dans le cas des maladies multifactorielles, il n'existe pas à proprement parler de réel gène candidat. Le grand nombre de gènes potentiellement impliqués rend compte de l'absence de "piste" privilégiée. Si on prend l’exemple de l’obésité à nouveau, un gène-candidat pourrait par exemple intervenir dans le mécanisme de satiété, de prise alimentaire, dans l'insulino-résistance, dans la réponse lipogénique ou lipolytique du tissu adipeux, dans le métabolisme lipidique ou glucidique, dans la dépense d'énergie, dans l'absorption intestinale des nutriments ou encore jouer un rôle au niveau de la régulation du métabolisme énergétique.
Ces deux dernières techniques d’approche, que sont le « genome scan » et le « gène-candidat » nécessitent la mise en place de stratégies d’études spécifiques à l’information génétique mise à la disposition du chercheur, ainsi qu’à son objectif final. Ainsi des analyses de liaison (Lod-score ou méthode des paires de germains atteints) seront réalisées à partir de familles, pour tenter de retrouver un lien physique existant entre un marqueur et la maladie. Et des études d’association (cohorte ou cas-témoins) seront pratiquées à partir d’informations individuelles ou
populationnelles pour tenter d’établir des corrélations entre un marqueur et une maladie.
2. Les stratégies d’étude en génétique
a. La méthode des lod-scores
Grâce à la méthode des lod-scores, un grand nombre de gènes impliqués dans des maladies mendéliennes ont pu être cartographiés.
La méthode des lod-scores est une méthode d’analyse de liaison basée sur l’exploitation de données familiales sur plusieurs générations. Ce score, défini par Morton (14) comme le logarithme de l’odds, est basé sur le calcul de la fraction de recombinaison θ (ou sur la distance en cM) entre un marqueur m et un gène de la maladie. Cela signifie donc que le lod-score va varier suivant la valeur de θ considérée. Un score largement positif est synonyme de liaison génétique (ou co-ségrégation), et inversement un score fortement négatif signifie une absence de liaison. Classiquement, un score de 3 est considéré comme une évidence de liaison,
score correspondant à p=10-4. (15)
Cette méthode d’analyse de liaison requiert la connaissance du mode de transmission au locus « maladie » et la spécification de paramètres concernant cette transmission (fréquence de l’allèle pathologique, pénétrances des divers génotypes).
b. La méthode des paires de germains atteints
Proposée par Penrose dès 1935, cette méthode est également une analyse de liaison. Elle consiste à compter, chez des paires de germains atteints, le nombre d’allèles transmis du marqueur qu’ils partagent (16, 17). Si le locus analysé est proche d’un locus impliqué dans la maladie, les paires de germains qui se
ressemblent pour le phénotype étudié doivent présenter un excès de concordance pour le marqueur génétique. Cette méthode permet donc de tester simultanément la transmission conjointe d’une maladie, et d’un ou deux marqueurs génétiques dans les couples de germains atteints. Aucune hypothèse a priori n’est formulée sur le mode de transmission de la maladie, puisque seuls les germains atteints sont analysés. Il est important de noter que, contrairement à la méthode du lod score, la méthode présentée ici ne nécessite pas la connaissance du modèle génétique sous-jacent à la maladie (méthodes « model-free »). C’est une méthode non paramétrique. Pour les marqueurs complètement informatifs, il existe 4 allèles ou haplotypes parentaux différents a, b, c, d et la variable IBD (identity by descent) représente le nombre d’allèles présents conjointement chez les deux germains (0, 1 ou 2).
Figure 2 : les états IBD pour une paire de germains atteints
Sous l’hypothèse d’indépendance complète entre la maladie et le marqueur, les probabilités des valeurs théoriques sont P(IBD=2)=1/4 pour 2 allèles partagés entre germains, P(IBD=1)=1/2 pour un seul allèle en commun et P(IBD=0)=1/4 pour
ab cd ab cd ab cd
a c a c a c a d a c b d
IBD=2 IBD=1 IBD=0
Observé X Y Z
l’absence d’allèle en commun. Il est alors possible de tester l’hypothèse de l’indépendance entre le marqueur et la maladie par un test comparant distribution théorique et distribution observée.
La méthode des paires de germains atteints représente une stratégie de faible puissance car, contrairement à l’analyse de liaison sur des familles étendues, elle n’étudie qu’un seul type d’apparentement. Il est donc difficile de séparer statistiquement la concordance des allèles entre germains de la probabilité de ségrégation au hasard du marqueur et de la maladie. La méthode nécessite donc l’étude d’un grand nombre de familles présentant au moins deux cas dans la même génération. D’autres paramètres entrent en jeu dans les valeurs d’IBD observées :
valeur du taux de recombinaison θ entre le locus du marqueur et celui de la maladie,
polymorphisme du marqueur, contribution du locus à la maladie.
c. Les études d’association allélique
La recherche d’associations, entre des marqueurs génétiques et des maladies, est une des voies possibles pour identifier des gènes de susceptibilité aux maladies. La technique d’étude est relativement simple : on compare la fréquence d’un marqueur génétique chez les sujets atteints et chez les témoins (étude cas-témoins). L’analyse statistique est fondée sur le tableau de contingence où figurent, pour les cas et les témoins, les fréquences de chaque génotype ou de chaque allèle au locus considéré. Le marqueur utilisé peut être un polymorphisme sans relation causale avec la pathologie ou bien un polymorphisme d’un gène candidat. Un résultat positif (différence de distribution allélique entre les cas et les témoins) suggère que le marqueur est soit directement impliqué, soit en déséquilibre de liaison avec une ou plusieurs variation(s) génétique(s) causale(s).
Un déséquilibre de liaison entre deux loci est défini par l’existence d’une combinaison d’allèles à ces loci plus fréquente que celle attendue sous l’hypothèse d’indépendance. Deux marqueurs génétiques en déséquilibre de liaison sont en général très proches sur le même chromosome (18). Il est important de souligner que la mise en évidence d’une association n’est pas synonyme de relation de causalité, qui implique en général la démonstration cellulaire et biochimique du mécanisme d’action de la variation étudiée. Si la population témoin est suffisamment importante et représentative, il est possible de calculer le risque relatif de présenter la pathologie en fonction de l’état homozygote ou hétérozygote de l’allèle. Dans une étude cas-témoins, on va remplacer le risque relatif par l’odds-ratio (OR). Il faut cependant prendre garde aux faux positifs qui découlent d’un biais de sélection des cas et des témoins, dont la sélection peut être inappropriée parce qu’ils diffèrent par l’âge, la répartition des sexes et des groupes ethniques, ou la présence d’une autre pathologie associée au critère de choix - autant de paramètres susceptibles d’entraîner des différences de fréquences génotypiques non attribuables à la pathologie.
Les études d’association sont particulièrement intéressantes lorsqu’il existe plusieurs loci impliqués et que l’effet quantitatif de chaque allèle délétère est trop faible pour être facilement analysé par une étude de liaison familiale. La probabilité de détecter une association dépend de l’intensité du déséquilibre de liaison entre le marqueur et la variation causale, de la fréquence de la variation dans la population, de l’effet observable de la variation (sa pénétrance) sur la pathologie, et enfin de la distance génétique entre les marqueurs et le locus impliqué dans la maladie lorsqu’on ne se réfère pas directement au gène candidat. Dans ces conditions, un résultat négatif ne permet pas d’exclure le rôle d’un locus. Il se peut en effet qu’il ne soit pas en
déséquilibre de liaison avec une variation morbide, même très proche. De même, une mutation à effet biologique fort et plus généralement une variation génétique, mais rarement représentée dans la population, peut aboutir à un test statistique négatif et passer inaperçue.
d. Une méthode « combinée » : le Transmission Disequilibrium Test (TDT)
Cette dernière méthode, communément appelée TDT (19), prend en compte à la fois l’information sur la liaison génétique apportée par l’analyse de la ségrégation à l’échelle familiale, et celle du déséquilibre gamétique apportée par l’étude de la liaison marqueur-maladie à l’échelle de la population. Il a d’abord été décrit dans le cadre de l’étude des relations entre le gène de l’insuline et le diabète insulinodépendant.
Ce test considère des trios constitués de l’enfant atteint et de ses deux parents. Il peut-être pratiqué sur des familles comportant au moins un parent hétérozygote pour le marqueur analysé. Le test compare la fréquence avec laquelle l’une ou l’autre forme allélique du marqueur est transmise aux sujets atteints. Si le résultat du test est significatif, on pourra conclure à l’existence d’une association ainsi que d’une liaison génétique entre le marqueur et la maladie.
Cette méthode est en fait une méthode qui permet la recherche simultanée d’un déséquilibre de liaison et d’une liaison génétique, dans le cas d’une association connue.
Un inconvénient du TDT est qu’il rejette effectivement de l’information génotypique, en raison de sa dépendance envers l’exigence du statut hétérozygote des parents.
Ceci ayant pour conséquence une perte de puissance statistique pour détecter de réelles associations alléliques.
De plus, dans sa forme originelle, le TDT requiert de disposer de l’information concernant les génotypes des parents, information qui n’est pas accessible de manière systématique dans le cas de maladies se déclarant à un âge avancé de la vie, par exemple. Des adaptations de cette méthode ont été proposées notamment pour permettre la prise en compte de données parentales manquantes ou la considération alternative d’informations apportées par d’autres apparentés (20). Schématiquement soit on connaît un gène et ses variants que l’on étudie, soit on cherche un marqueur génétique qui permettra de donner des indications sur des gènes proches. Dans ce dernier cas il est important que les caractéristiques du marqueur apportent de l’information de façon optimale sans apporter de facteurs de confusion avec l’effet du gène lui-même.
3. Les grandes familles de marqueurs utilisés en génétique
Un « bon » marqueur doit être : polymorphe, codominant (l'individu hétérozygote peut être distingué car il présente simultanément les caractères de ses parents homozygotes), insensible au milieu, non épistatique, multiallélique.
Nous allons ici présenter les principaux types de marqueurs utilisés en génétique épidémiologique.
a. Les marqueurs microsatellites
Les microsatellites appartiennent à la famille des séquences répétées au sein du génome plus particulièrement ce que l’on appelle les VNTR (Variable Number of Tandem Repeat) ou encore les HVR (Highly Variable Repeat). Ils sont constitués de
motifs nucléotidiques associés en tandem 10 à 50 fois. Ce motif est souvent un dinucléotide : C-A ; mais il existe de nombreux motifs tri, tétra, penta, voire hexa nucléotidiques ou plus complexes. Un allèle de microsatellites est un nombre de répétitions d’un motif simple. Les allèles sont de longueurs différentes, c’est un cas de polymorphisme de longueur.
La technique de réaction de polymérisation en chaîne (PCR) permet d’augmenter de façon sélective la quantité d’un fragment d’ADN au sein d’un échantillon. Des techniques de séparation en fonction de la taille ou la séquence directe permettent de mettre en évidence des variations de longueur pour un même microsatellite qui constituent autant «d’allèles» très utilisés dans les études de liaison et la cartographie génétique. Dans les années 90, c’est le polymorphisme des microsatellites qui a retenu l’attention des chercheurs. De tels marqueurs sont ubiquitaires dans les génomes des vertébrés. Ils existent en particulier dans le génome humain. Au sein de celui-ci on en trouve plusieurs centaines dans la région HLA sur le chromosome 6 qui nous intéressera particulièrement dans ce travail. Les microsatellites, bien que plus denses aux niveaux centromérique et télomérique, sont bien répartis dans l’ensemble du génome. On estime en effet qu’il y a dans la région HLA un microsatellite tous les 30kb.
Ce sont des marqueurs pertinents dans les études de cartographies de gènes impliqués dans une pathologie, et dans la génétique des populations, ceci étant dû à leur haut degré de polymorphisme et leur densité importante dans le génome. (21) Les études d’évolution moléculaire ont démontré, sur de courtes périodes de temps, le très important taux de mutation de ce type de séquence. (22-24)
Le phénomène de déséquilibre de liaison est observé entre les allèles de marqueurs microsatellites sur les haplotypes HLA dits « ancestraux ». On parle dans ce cas d’haplotype étendu. Les haplotypes ancestraux sont le siège de déséquilibres de liaison très fort entre leurs allèles. La notion de déséquilibre de liaison entre marqueurs microsatellites et gènes HLA ouvre de nouvelles perspectives en matière d’étude de la région HLA. En effet un allèle de microsatellite est techniquement plus facilement détectable qu’un allèle de gène HLA, pour un moindre coût.
b. Les marqueurs SNPs
Les SNPs (Single Nucleotide Polymorphisms) constituent la forme la plus abondante de variations génétiques dans le génome humain. Ils représentent plus de 90% de toutes les différences entre individus. C'est un type de polymorphisme bi-allélique ponctuel de l'ADN au niveau d’une paire de bases.
Dans deux génomes humains tirés au hasard, 99,9% de la séquence d'ADN est identique. Les 0,1% restants contiennent des variations de séquence dont le type le plus commun est le polymorphisme pour un nucléotide (SNP). Les SNPs sont stables, très abondants et distribués uniformément dans tout le génome. Ces variations peuvent être des marqueurs de la diversité entre populations ou individus, être associées à des différences de susceptibilité à des maladies, de sévérité des maladies et/ou à la réponse individuelle aux médicaments.
De nombreux SNPs ont été mis en évidence lors de l'étude de sujets sains et malades portant des allèles différents d'un gène donné. Il existe aujourd'hui de nombreuses méthodes pour mettre en évidence des différences d'un nucléotide. Une des plus performante actuellement et qui paraît offrir de grandes espérances est l'usage des puces à ADN.
c. Les marqueurs RFLPs
Ce sont des variations individuelles de la séquence d’ADN révélées par des modifications de la carte de restriction. Ils génèrent des polymorphismes de longueur: la taille des fragments d’ADN varie après traitement par une enzyme de restriction d’où le nom de RFLP (Restriction Fragment Lenght Polymorphism).
Les RFLPs sont le plus fréquemment bialléliques. Ils correspondent à des mutations ponctuelles abolissant ou créant un site de restriction.
Les RFLPs ont été utilisés pour établir les premières cartes génétiques chez l’homme, cartes dites de première génération. Ils ont contribué à la localisation de gènes responsables de maladies génétiques telles que la myopathie de Duchenne (1982), la chorée de Huntington (1983) ou la mucoviscidose (1985).
Les enzymes de restriction, isolées de bactéries, ont la particularité de couper l’ADN en des endroits précis et en fragments de tailles variables (polymorphes). Un RLFP est un fragment d’ADN génomique. Utilisé comme sonde sur de l’ADN génomique humain digéré par une enzyme de restriction, il détecte des fragments dont la taille varie (polymorphisme) au sein d’une population d’individus. Ces variations pour un même locus sont dues soit à la répartition des sites de restriction (coupure), soit à la présence de minisatellites (séquences répétitives d’ADN) dans la région reconnue par la sonde.
Les approches méthodologiques pour permettre de quantifier et de caractériser l’intervention de facteurs génétiques dans la physiopathologie d’une maladie multifactorielle sont nombreuses et en constante évolution. De plus en plus d’études se concentrent sur l’étude des interactions gène-gène, ainsi que sur les interactions gène-environnement, plutôt que de considérer simplement l’intervention d’un facteur génétique unique. De plus, de nouvelles technologies comme les profils d’expression
géniques vont permettre d’identifier les effets fonctionnels de plusieurs gènes simultanément (25). Toutes ces informations glanées sur les mécanismes physiopathologiques des maladies complexes sont utilisées dans le cadre de la recherche thérapeutique, de la prévention des maladies avec comme objectif final une amélioration de la prise en charge des patients affectés par ces maladies multifactorielles, comme la polyarthrite rhumatoïde.
C- Présentation de la polyarthrite rhumatoïde
1. Définition
Dans la majorité des cas, la polyarthrite rhumatoïde (PR) se présente comme un rhumatisme inflammatoire chronique caractérisé par :
- Topographiquement, des arthrites des articulations des membres. L’atteinte initiale des articulations distales est presque constante, de même que l’extension secondaire à d’autres articulations, avec une tendance nette à la symétrie des localisations. Les articulations sacro-iliaques et le rachis dorso-lombaire sont respectés, mais la localisation au rachis cervical est fréquente. - Morphologiquement, des synovites prolifératives (« pannus synovial »). Elles
sont agressives, provoquant des érosions osseuses et une dégradation des cartilages de conjugaison. La destruction progressive des structures articulaires conduit aux déformations caractéristiques de la maladie.
- Biologiquement, des désordres auto-immuns. Les facteurs rhumatoïdes (immunoglobulines anti-IgG) et les anticorps anti-protéines citrullinées, constituent ainsi des marqueurs sériques de la maladie, utiles au diagnostic et au pronostic, mais inconstants.
Dans un nombre faible de cas, la PR peut se présenter comme une maladie systémique, du fait de l’importance des manifestations extra-articulaires associées aux arthrites, notamment en cas de vascularite rhumatoïde.
2. Critères diagnostiques
Afin d’homogénéiser les études sur la PR, l’American College of Rheumatology (ACR) a défini une série de critères de classification de la PR (26). Ces critères de classification sont présentés dans le tableau ci-aprés.
Tableau 2 : critères de classification d’une PR selon l’ACR.
Critères ACR de classification d’une polyarthrite rhumatoïde
-Dérouillage matinal péri-articulaire et articulaire durant au moins une heure avant l'amélioration maximale.*
- Au moins trois régions articulaires doivent avoir présenté simultanément une tuméfaction des tissus mous ou la présence de liquide (pas uniquement d'excroissance osseuse) observée par un médecin. Les 7 régions concernées sont les inter-phalangiennes proximales (IPP),
métacarpo-phalangiennes (MCP), métatarso-métacarpo-phalangiennes, poignets, coudes, genoux, et les chevilles.*
- Au moins une région tuméfiée parmi les 3 suivantes : IPP, MCP, ou poignets.*
- Atteinte simultanée de la même région articulaire (voir ci-dessus) bilatérale (l'atteinte bilatérale des IPP, MCP, ou MTP est acceptable sans symétrie absolue).*
*ces critères doivent être présents depuis au moins 6 semaines
- Nodules sous cutanés sur excroissance osseuse ou sur de larges surfaces ou juxtra-articulaires, observés par un médecin.
- Démonstration de taux sérique de facteur rhumatoïde anormal par toute méthode dont les résultats se sont avérés positifs chez moins de 5% des témoins normaux. - Signes radiographiques caractéristiques de la PR (en postéro antérieur de la main et du poignet) qui doivent révéler des érosions ou une décalcification osseuse non équivoque au niveau ou au voisinage des articulations atteintes (les seuls signes d'arthrose ne sont pas pris en compte). Pour que le diagnostic de PR soit confirmé, le patient doit présenter au moins 4 critères parmi les 7 suivants
Référence : Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria
for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315-24.
3. Epidémiologie générale
La PR est la plus fréquente des arthrites rhumatismales chroniques, mais sa fréquence est variable suivant les populations. Dans la population française, sa prévalence serait de l’ordre de 0.3% (27), avec des variations inter-régionales observées allant de 0.14% au Nord à 0.66% au Sud (28).
Cette maladie peut être observée dans les deux sexes et à tout âge, mais il est désormais bien établi que la femme est atteinte entre deux et cinq fois plus souvent que l’homme, et particulièrement avec un début de la maladie plus fréquent entre 30 et 50 ans (29-31).
4. Facteurs prédisposants
Différents facteurs environnementaux ont été évoqués dans la PR parmi lesquels on peut retenir l’existence de facteurs hormonaux, de facteurs infectieux ou de facteurs individuels diverses (32).
a. Des facteurs endocriniens
L’intervention de facteurs hormonaux dans le développement de la PR est suspectée sur la base de plusieurs observations cliniques : la prédominance de la maladie chez la femme, la fréquence de survenue de la maladie en période préménopausique, la possible rémission de la maladie lors de la grossesse (33, 34), l’effet protecteur des contraceptifs oraux dont la mise sur le marché coïncide avec une baisse de l’incidence observée ces dernières décennies (35).
Toutes ces considérations cliniques sont le reflet d’interactions fonctionnelles entre système endocrinien et système immunitaire. Les hormones sexuelles sont impliquées dans la réponse immune, notamment de par le rôle des oestrogènes comme modulateur de l’immunité humorale, ainsi que des androgènes et de la progestérone comme suppresseurs naturels de l’immunité (36-38). Une modification de l’expression de ces hormones, ou une altération de leurs fonctions, entraîne un déséquilibre dans le complexe interactionnel hormono-immunitaire, et peut ainsi contribuer à l’étiologie de la PR (37).
Taneja et al. ont récemment développé un modèle de souris transgénique, exprimant l’allèle de susceptibilité HLA-DRB1*0401 à la PR, et ont retrouvé un ratio de 3 souris femelles atteintes pour une souris mâle, redémontrant ainsi la prédisposition féminine à développer la maladie (39).
b. L’hypothèse infectieuse
Nous avons précédemment vu que l’incidence de la PR avait diminué depuis plusieurs décennies, cette baisse a également été observée chez l’homme, ce qui tendrait à supposer qu’il existe d’autres facteurs intervenant dans la physiopathologie de la PR. L’amélioration de la prévention des maladies infectieuses et l’évolution des thérapeutiques disponibles pourraient expliquer en partie cette décroissance. Cependant aucun épisode épidémique n’a été observé (40).
Plusieurs agents infectieux ont été mis en cause dans la pathogénie de la PR. Certains rétrovirus, comme HTLV1 dans une population martiniquaise (41), ou le parvovirus humain B19 (42, 43) ont été retrouvés dans les tissus (articulations et moelle osseuse) de patients atteints de PR. D’autres agents comme Escherichia coli, ou le virus Epstein-Barr ont également été suspectés.
Il ne semble donc pas exister un agent infectieux, bactérien ou viral, unique mais plusieurs agents infectieux non spécifiques qui interviendraient en tant que facteurs déclenchants de la maladie.
c. Autres facteurs individuels et environnementaux
Hormis les facteurs infectieux et hormonaux présentés ci-dessus, plusieurs autres facteurs de risque notamment environnementaux ont été évoqués dans la polyarthrite rhumatoïde.
Le rôle du tabac a fait l’objet de plusieurs études, dont les résultats sont contradictoires (44-46). Il semblerait que les patients fumeurs atteints de PR présenteraient significativement plus de forme sévère de la maladie, avec une augmentation significative des taux de facteur rhumatoïde (47-49). Le tabac pourrait
aussi agir sur les hormones comme anti-oestrogène ou directement sur le système immunitaire (45).
Par ailleurs différents facteurs de risque ont également été étudiés comme : la consommation d’alcool (44), le rôle du stress (50), la consommation de café (51), ou encore le possible rôle protecteur de certains régimes alimentaires comme la régime méditerranéen (52, 53).
Enfin, plusieurs études ont démontré l’existence d’interactions entre des facteurs environnementaux et des facteurs de susceptibilité génétiques. Klareskog et al. font ainsi la revue des études menées à partir d’une cohorte de patients et de témoins suédois, qui ont notamment permis de montrer l’augmentation significative du risque de développer une PR pour les patients porteurs de deux copies d’allèles codant pour l’épitope partagé, fumeurs et présentant des anticorps anti-CCP positifs (54).
5. Epidémiologie génétique de la PR
La PR est une maladie complexe, multifactorielle, mettant vraisemblablement en jeu des interactions de facteurs génétiques et de facteurs environnementaux.
L’existence d’une composante familiale de la maladie a été envisagée devant les résultats des études d’agrégation familiale : la prévalence de la PR chez les apparentés du 1er degré d’un patient touché par la maladie est de 2 à 20%, alors qu’elle est estimé entre 0.2 et 1% dans la population générale (55-57). En reprenant les données de la littérature, Seldin et al. ont estimé que la fraction (définie précédemment) serait située entre 1,2 et 17 (58). Les études de concordance familiale sont difficilement comparables car elles peuvent être basées sur des modes de recrutement différents des patients, avec des critères diagnostiques variables
d’une étude à l’autre (utilisation ou non des critères de l’ACR 1988). Cependant, dans la plupart des études, les résultats observés restent discordants avec des estimations du risque variant de 2 à 15 (59, 60). Le risque relatif d’apparition de la maladie chez l’apparenté du 1er degré apparaît tout de même plus faible dans le cadre de la PR que pour d’autres maladies multifactorielles comme le diabète insulino-dépendant (risque relatif à environ 15) ou la sclérose en plaque (risque relatif à environ 20).
La part des facteurs génétiques dans la PR a été estimée à partir des études du taux de concordance de la maladie chez des jumeaux (proportion de seconds jumeaux atteints). A partir des résultats de différentes études, nous pouvons penser que ce taux de concordance est compris entre 12 et 30% chez les jumeaux monozygotes,
alors qu’il serait de l’ordre de 3 à 10% chez les jumeaux dizygotes (61-63). Le taux
de concordance pour des jumeaux monozygotes atteints par la PR est donc approximativement quatre fois plus important que pour des jumeaux dizygotes, ceci indiquant une héritabilité comprise entre 40% et 60% (63). Ces études de jumeaux fournissent une limite supérieure concernant la contribution génétique à la PR. Les études incluant des patients atteints d’une forme moins sévère de la maladie ont mis en évidence des taux de concordance entre monozygotes moins importants. Ceci est très probablement dû à des facteurs génétiques intervenant dans le déterminisme de la PR, et qui influencent non seulement la susceptibilité, mais aussi la sévérité, de la maladie (64).
En 1976, Peter Stastny rapporte que les réactions mixtes lymphocytaires (réaction MLR) entre des lymphocytes de patients atteints de PR sont moins souvent
activatrices que celles mettant en jeu des lymphocytes de témoins sains (65). Stastny émet alors l’hypothèse que la plupart des patients atteints de PR partagent un déterminant antigénique commun, codé par un gène de la région HLA, prédisposant à la maladie, nommé HLA-Dw4. Avec le développement des méthodes de typage sérologique des molécules HLA, Stastny montrera plus tard que l’antigène HLA-DRw4 défini sérologiquement est exprimé chez environ 70% des patients atteints par la PR (66).
A la fin des années 1980, alors que l’avènement des techniques de biologie moléculaire a permis de séquencer le locus HLA-DRB1, Gregersen et al. remarquent que les allèles HLA DRw4 (*0401), DRw14 (*404), DRw15 (*0405) et DR1 (ensemble des allèles *01 sauf *0103) partagent la même séquence d’acides aminés dans la troisième région hypervariable (67). Gregersen avance alors l’hypothèse de l’épitope partagé comme explication à l’association entre la région de classe II du Complexe Majeur d’Histocompatibilité (CMH) et la susceptibilité à la PR. Cette hypothèse attribue l’association entre la région HLA-DR et la susceptibilité à la PR aux seuls allèles HLA-DRB1 codant pour l’épitope partagé. Cet épitope partagé correspond à une séquence particulière d’acides aminés (séquence RAA) en position 72-74 de la
3ème région hypervariable du 1er domaine de la chaîne β des molécules HLA-DR (67).
Pratiquement vingt ans après, le rôle exact de l’épitope partagé dans la susceptibilité à la PR n’a pas été clairement établi (68). Le gène HLA-DRB1 n’explique qu’une partie du risque familial global (69). Près de 40% de la population générale porte un des allèles HLA-DRB1 de prédisposition à la PR, contre plus de 70% des personnes malades. De plus la proportion de la susceptibilité à la PR conférée par la région HLA sur l’ensemble de la composante génétique est estimée à moins de 50%, voire moins de 30% (58, 70, 71).
Les différentes études de criblage du génome réalisées ont permis de confirmer les premiers résultats de Stastny, en confirmant l’importance de l’implication de la région HLA. Ces études ont notamment été menées sur des populations française, anglaise et nord-américaine (72-81). Toutes ces études ont notamment suggéré l’existence d’autres loci impliqués dans la physiopathologie de la PR, mais sans identifier de manière formelle un nouveau de gène susceptibilité à la maladie indépendant de HLA-DRB1. L’une des raisons principales de cette disparité dans les résultats observés est l’hétérogénéité, tant sur le plan clinique que génétique, qui existe entre les populations étudiées.
Nous pouvons tout de même noter plusieurs éléments communs à certaines études. Tout d’abord, toutes les études retrouvent les scores de liaison les plus élevés pour la région HLA, confirmant ainsi les résultats princeps de Stastny et le rôle primordial joué par cette partie du génome dans la susceptibilité aux maladies auto-immunes, comme la PR. Une étude menée sur une population japonaise en 1998 (82) a néanmoins mis en évidence des scores de liaison très élevés pour trois régions chromosomiques (D1S253/214, D8S556 and DXS1232), alors que dans le même temps le lod-score de la région HLA était infèrieur à 3 (82).
D’autres régions chromosomiques semblent également contribuer à l’étiologie de la maladie puisqu’elles sont retrouvées dans la plupart des études de liaison réalisées : le bras long du chromosome 6 (6q), le bras court du chromosome 12 (12p) ou encore la bras court du chromosome 16 (16p) (83). Deux méta-analyses récentes (76, 84) confirment ces résultats, et mettent également en évidence le possible rôle joué par d’autres régions du génome (chromosome 1, 3, 4…). Néanmoins aucun nouveau locus de susceptibilité à la maladie, indépendant de HLA-DRB1, n’a pu être clairement identifié. John et al. ont suggéré dans une étude récente que certaines de
ces régions génomiques (notamment 6q et 16p) pouvaient interagir avec la région HLA dans le processus physiopathologique de la maladie (78). Au travers de ces études de liaison réalisées dans le cadre de plusieurs maladies multifactorielles, il apparaît également que certains loci sont communs à différentes maladies auto-immunes comme la PR, le lupus érythémateux disséminé, le diabète insulino-dépendant ou le vitiligo, suggérant la participation de facteurs génétiques de susceptibilité communs (73).
Les études d’association réalisées sur des gènes candidats ont suggéré l’existence d’autres facteurs génétiques de susceptibilité, tout ceci allant dans le sens que l’ensemble de la susceptibilité à la PR conférée par la région HLA ne revient vraisemblablement pas au seul locus HLA-DRB1.
Plusieurs loci ont été étudiés :
1) les allèles HLA-DM, HLA-DP ou HLA-DQ (85-89) qui n’ont pas montré d’association indépendante de HLA-DRB1,
2) le gène PADI4, codant pour une des enzymes « peptidyl-arginine déiminases » impliqués dans la citrullination des résidus arginine dont l’association est bien documentée sur des populations asiatiques (90-92). Plusieurs études de réplication sur des populations européennes se sont révélées négatives (93, 94). Plus récemment, une méta-analyse (95) et une étude avec une population de grande taille (96) ont démontré que ce gène était également retrouvé comme facteur génétique de susceptibilité à la PR dans des populations caucasoïdes.
3) SCL22A4 (gène codant pour un transporteur cationique) et RUNX1 (qui participe à la régulation de l’expression de gènes inclus dans la réponse immunitaire) dont