Université de Montréal
Évolution à fine échelle des sites d’épissage des introns dans les gènes des oomycètes
par
Steven Sêton Bocco
Département de biochimie et médecine moléculaire Faculté de médecine
Mémoire présenté à la Faculté des études supérieures en vue de l’obtention du grade de Maître ès sciences (M.Sc.)
en bio-informatique
Université de Montréal Faculté des études supérieures
Ce mémoire intitulé:
Évolution à fine échelle des sites d’épissage des introns dans les gènes des oomycètes
présenté par: Steven Sêton Bocco
a été évalué par un jury composé des personnes suivantes: Sylvie Hamel, président-rapporteur
Miklós Cs˝urös, directeur de recherche Simon Joly, membre du jury
RÉSUMÉ
Les introns sont des portions de gènes transcrites dans l’ARN messager, mais retirées pendant l’épissage avant la synthèse des produits du gène. Chez les eucaryotes, on rencontre les introns splicéosomaux, qui sont retirés de l’ARN messager par des splicéosomes.
Les introns permettent plusieurs processus importants, tels que l’épissage alternatif, la dé-gradation des ARNs messagers non-sens, et l’encodage d’ARNs fonctionnels. Leurs rôles nous interrogent sur l’influence de la sélection naturelle sur leur évolution. Nous nous intéressons aux mutations qui peuvent modifier les produits d’un gène en changeant les sites d’épissage des introns. Ces mutations peuvent influencer le fonctionnement d’un organisme, et constituent donc un sujet d’étude intéressant, mais il n’existe actuellement pas de logiciels permettant de les étudier convenablement. Le but de notre projet était donc de concevoir une méthode pour détecter et analyser les changements des sites d’épissage des introns splicéosomaux.
Nous avons finalement développé une méthode qui repère les évènements évolutifs qui affectent les introns splicéosomaux dans un jeu d’espèces données. La méthode a été exécutée sur un ensemble d’espèces d’oomycètes. Plusieurs évènements détectés ont changé les sites d’épissage et les protéines, mais de nombreux évènements trouvés ont modifié les introns sans affecter les produits des gènes.
Il manque à notre méthode une étape finale d’analyse approfondie des données récoltées. Cependant, la méthode actuelle est facilement reproductible et automatise l’analyse des génomes pour la détection des évènements. Les fichiers produits peuvent ensuite être analysés dans chaque étude pour répondre à des questions spécifiques.
ABSTRACT
Introns are portions of genes transcribed into messenger RNA, but removed during RNA splicing. In eukaryotes, they are called spliceosomal introns as they are removed by spliceosomes.
Introns allow many important processes such as alternative splicing, nonsense-mediated decay and functional-RNA coding. These roles leads to the question of the influence of natural selection on evolution of introns. We focus on mutations that are able to change gene products by modifing introns splice sites. These mutations seems to be an interesting topic as they can affect proteins, but there is currently no software to study them properly. The aim of our project was to design a method to detect and analyze changes in splice sites of spliceosomal introns.
We finally developed a method that locates the evolutionary events on splice sites of spliceo-somal introns in a given species set. The method was performed on a set of oomycetes. Several detected events change splice sites and proteins, but there is also many events that seems to modify introns without affecting gene products.
Our method lacks a final step for thorough analysis of the collected events. However, the current method is easily reusable and automates genome analysis for the detection of events. The resulting files can then be analyzed in each study to answer specific questions.
TABLE DES MATIÈRES
RÉSUMÉ . . . iii
ABSTRACT . . . iv
TABLE DES MATIÈRES . . . v
LISTE DES TABLEAUX . . . ix
LISTE DES FIGURES . . . x
LISTE DES SIGLES . . . xi
REMERCIEMENTS . . . xii
CHAPITRE 1 : INTRODUCTION . . . 1
1.1 Définition et propriétés des introns . . . 1
1.1.1 Définition . . . 1
1.1.2 Phases d’introns . . . 1
1.1.3 Classes d’introns . . . 2
1.1.4 Structure des introns splicéosomaux . . . 2
1.2 Rôles et fonctions des introns . . . 3
1.2.1 Épissage alternatif . . . 4
1.2.2 Dégradation des ARNs messagers non-sens . . . 6
1.2.3 Encodage d’ARNs fonctionnels . . . 7
1.3 Évolution des introns . . . 8
1.3.1 Origine des introns splicéosomaux . . . 8
1.3.2 Mécanismes d’évolution des introns . . . 9
1.3.2.1 Évolution des introns par mutations introniques globales . . . 10
1.4 Outils bio-informatiques disponibles pour l’étude de l’évolution des introns . . 14
1.5 Notre projet . . . 15
1.5.1 Objectifs . . . 15
1.5.2 Hypothèse . . . 16
1.5.3 Aboutissement . . . 16
CHAPITRE 2 : COLLECTE DES DONNÉES ET TRAITEMENTS PRÉLIMINAIRES 18 2.1 Choix des espèces à étudier . . . 18
2.1.1 Théorie . . . 18
2.1.2 Application . . . 18
2.2 Utilisation des protéines à la place des gènes dans notre méthode . . . 19
2.3 Construction des familles de protéines . . . 21
2.3.1 Théorie . . . 21
2.3.1.1 OrthoMCL . . . 22
2.3.2 Application . . . 22
2.3.2.1 Paramètres utilisés pour l’exécution d’OrthoMCL . . . 22
2.3.2.2 Résultats obtenus . . . 23
2.3.2.3 Distributions des familles selon le nombre d’espèces et le nombre de séquences . . . 24
2.3.2.4 Génération des fichiers FASTA des familles de protéines . . . 26
2.4 Alignement des familles de protéines . . . 26
2.5 Utilisation des familles de protéines strictement orthologues dans la suite de l’étude 27 2.5.1 Théorie . . . 27
2.5.2 Application . . . 28
2.6 Construction de l’arbre phylogénétique des espèces . . . 29
2.6.1 Théorie . . . 29
2.6.1.1 Sélection des familles à utiliser pour construire l’arbre . . . . 29
2.6.1.2 Concaténation des familles sélectionnées . . . 31
2.6.1.3 Conversion du fichier concaténé au format PHYLIP . . . 31
2.6.1.4 Construction proprement dite de l’arbre phylogénétique des
espèces . . . 31
2.6.2 Application . . . 32
2.7 Extraction des positions des introns à partir des annotations des génomes . . . . 36
2.7.1 Théorie . . . 36
2.7.1.1 Utilisation des fichiers d’annotations des génomes . . . 36
2.7.1.2 Gestion des annotations des introns dans les familles stricte-ment orthologues en cas d’épissage alternatif . . . 37
2.7.1.3 Programmes d’extraction et format de sortie . . . 38
2.7.2 Application . . . 40
2.7.2.1 Analyse statistique des longueurs des introns . . . 40
2.8 Génération de fichiers FASTA personnalisés rassemblant les alignements des familles et les positions des introns . . . 41
CHAPITRE 3 : DÉTECTION DES MUTATIONS INTRONIQUES LOCALES . . 43
3.1 Reconstruction et alignement des séquences ancestrales . . . 44
3.1.1 Projection des introns sur les protéines . . . 45
3.1.2 Détermination des homologies entre caractères . . . 46
3.1.2.1 Stratégie utilisée . . . 46
3.1.2.2 Pertinence de la stratégie . . . 48
3.1.3 Détermination des caractères ancestraux dans chaque groupe de carac-tères homologues . . . 49
3.1.3.1 Algorithme de Fitch adapté . . . 50
3.1.3.2 L’étape ascendante de l’algorithme de Fitch . . . 50
3.1.3.3 L’étape descendante de l’algorithme de Fitch . . . 51
3.1.3.4 Modification de l’algorithme de Fitch pour le choix d’un caractère 52 3.1.3.5 Pseudocode général utilisé pour l’algorithme de Fitch . . . . 54
3.1.4 Assemblage et vérification des séquences reconstruites . . . 58
3.1.4.1 Réajustement des extrémités des séquences reconstruites . . 58
3.1.6 Application . . . 62
3.2 Déduction des évènements évolutifs impliquant les introns . . . 64
3.2.1 Hypothèse de travail . . . 64
3.2.1.1 Observation en faveur de notre hypothèse . . . 66
3.2.2 Algorithme . . . 67
3.2.2.1 Extension des zones autour des introns . . . 68
3.2.2.2 Typage des évènements évolutifs autour des introns . . . 74
3.2.3 Implémentation de l’algorithme . . . 75
3.2.4 Application . . . 77
3.2.4.1 Pertinence des évènements détectés . . . 77
3.2.4.2 Identification des types d’évènements . . . 79
3.2.4.3 Évènements de conservation d’introns . . . 81
3.2.4.4 Évènements d’insertions et de suppressions complètes d’introns 81 3.2.4.5 Fichier-rapport sur les nombres d’évènements par branche de l’arbre des espèces . . . 84
3.2.4.6 Tendances de gains d’introns . . . 85
3.2.4.7 Tendances de créations d’introns . . . 86
3.2.4.8 Tendances de déplacement des sites d’épissage vers le côté 3’ des gènes . . . 88 CHAPITRE 4 : CONCLUSION . . . 93 4.1 Perspectives . . . 93 4.2 Résumé et disponibilité . . . 95 BIBLIOGRAPHIE . . . 97 viii
LISTE DES TABLEAUX
2.I Liste des espèces étudiées et origine des données collectées pour chaque espèce. . . 20 2.II Paramètre d’exécution de RAxML pour l’inférence de l’arbre
phylogéné-tique des espèces étudiées. . . 33 2.III Statistiques sur les longueurs des introns. . . 42 3.I Dénombrement des fenêtres d’alignements de longueur 10 détectées dans
les familles de protéines et définies en fonction de la présence de trous et d’introns. . . 67 3.II Types de colonnes possibles dans un alignement simple mettant en évidence
les introns, et traitement de ces colonnes pendant la détection des zones d’évolutions entourant les introns. . . 70 3.III Catégorisation des colonnes prises en compte pendant la détection des
zones d’évolution entourant les introns. . . 71 3.IV Effet des colonnes d’alignement prises en compte sur l’extension des zones
d’évolution autour des introns. . . 73 3.V Exemple de typage d’un évènement réel détecté dans une famille de
pro-téines orthologues. . . 75 3.VI Information sur quelques types d’évènements reconnaissables. . . 82 3.VII Exemples d’évènements détectés dans les familles de protéines orthologues
LISTE DES FIGURES
1.1 Structure générique d’un intron splicéosomal. . . 4 1.2 Structure consensus des sites d’épissage de la majorité des introns
splicéo-somaux observés chez les eucaryotes. . . 4 2.1 Nombre de familles de protéines en fonction du nombre de séquences
contenues dans les familles. . . 25 2.2 Nombre de familles de protéines en fonction du nombre d’espèces qui
apparaissent dans les familles. . . 25 2.3 Arbres phylogénétiques obtenus pour la première et la seconde exécution
de RAxML. . . 34 2.4 Arbre phylogénétiue d’oomycètes publié en 2012 dans la littérature [45]. 35 3.1 Exemple d’alignement de protéines avec mise en évidence des introns en
phase 2 dans un codon. . . 47 3.2 Exemple de réajustement des extrémités des séquences ancestrales de la
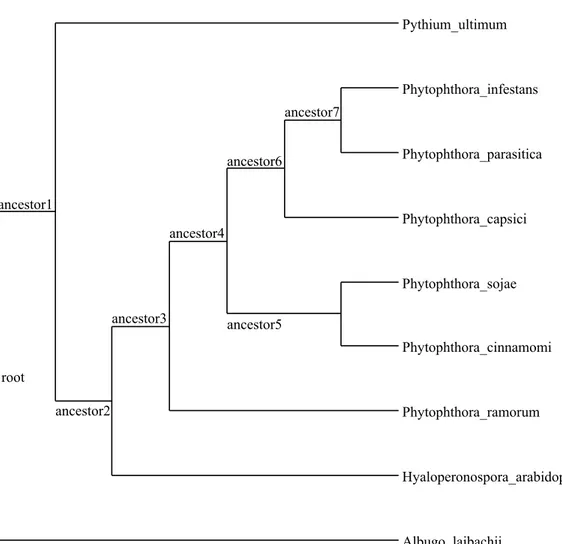
famille oomycetes4897 pour empêcher la prédiction d’introns à l’extérieur des séquences. . . 61 3.3 Topologie de l’arbre des espèces montrant les noms attribués aux noeuds
internes. . . 63 3.4 Exemples de contenus des fichiers de sortie pour la détection des
évène-ments évolutifs. . . 77 3.5 Nombre de gains et de pertes d’introns comptés sur chaque branche de
l’arbre des espèces. . . 87 3.6 Nombre de créations d’introns, et de conversions d’introns en exons,
comp-tées sur chaque branche de l’arbre des espèces. . . 89 3.7 Nombre de déplacements de sites d’épissage vers les extrémités des gènes,
comptés sur chaque branche de l’arbre. . . 92 4.1 Algorithme général de la méthode. . . 96
LISTE DES SIGLES
albu Albugo laibachii AMF Aligned Marked Fasta
EJC Exon-exon Junction Complex hyal Hyaloperonospora arabidopsidis
ILS Incomplete Lineage Sorting
LECA Last Eukaryotic Common Ancestor PCA Parent-Children Alignments
phca Phytophthora capsici phci Phytophthora cinnamomi phin Phytophthora infestans phpa Phytophthora parasitica
phra Phytophthora ramorum phso Phytophthora sojae
REMERCIEMENTS
Un grand merci à mon directeur de recherche M Miklós Cs˝urös, mon parrain M Simon Joly, mes parents, ma famille, mes amis, et toutes les personnes qui m’ont de près ou de loin aidé à finir cette maîtrise !
CHAPITRE 1
INTRODUCTION
1.1 Définition et propriétés des introns 1.1.1 Définition
À la lumière des connaissances actuelles, on peut définir un gène comme étant une suite de séquences localisée sur un ADN et contenant les codes de fabrication pour une ou plusieurs molécules nécessaires au fonctionnement d’un organisme [20, 21]. Les molécules encodées dans les gènes sont des protéines ou des ARNs, à la base du fonctionnement des êtres vivants.
Il arrive cependant qu’un gène ne contienne pas exclusivement les codes de fabrication de ses produits. Dans de nombreux gènes, la séquence codante est en réalité découpée en plusieurs pièces, appelées exons, entrecoupées de séquences qui ne codent pas pour les produits de ces gènes. Ces séquences dites non-codantes sont appelées introns. Pour synthétiser les produits d’un gène, ce dernier est transcrit en ARN messager sans perte d’information. Les introns sont ensuite retirés de cet ARN au cours d’une phase appelée épissage. L’ARN messager mature finalement obtenu ne contient que les exons, et est utilisé soit pour fabriquer des protéines, soit en tant qu’ARN fonctionnel dans la cellule.
Les introns sont observés dans de nombreux gènes chez tous les eucaryotes, mais on en trouve aussi chez les procaryotes [53].
1.1.2 Phases d’introns
Dans les gènes qui codent pour des protéines, la séquence codante est une suite de triplets de nucléotides, appelés codons, qui codent chacun pour un acide aminé précis des protéines encodées dans ce gène. Comme les introns fragmentent la séquence codante en plusieurs exons, on peut les retrouver entre deux codons, ou à l’intérieur d’un codon. La phase d’un intron désigne
ainsi la situation relative de l’intron par rapport aux codons du gène [17]. On peut classer les introns dans 3 phases :
• Les introns en phase 0 (ou en phase 3, selon l’appellation choisie) sont les introns situés entre deux codons (donc après le 3e nucléotide d’un codon).
• Les introns en phase 1 sont les introns situés après le 1ernucléotide d’un codon.
• Les introns en phase 2 sont les introns situés après le 2èmenucléotide d’un codon.
1.1.3 Classes d’introns
Il existe différentes classes d’introns, en fonction du processus d’épissage qui les élimine. Certains, comme les introns du groupe I et II, sont capables de s’auto-épisser grâce à un replie-ment dépendant de leur structure. D’autres, appelés « introns splicéosomaux » (« spliceosomal introns»), sont identifiés par des signaux particuliers qui permettent leur épissage au moyen d’un complexe ribonucléoprotéique appelé splicéosome [44, 53]. Les introns splicéosomaux sont rencontrés spécifiquement chez les eucaryotes, et constituent le sujet d’étude de notre projet.
1.1.4 Structure des introns splicéosomaux
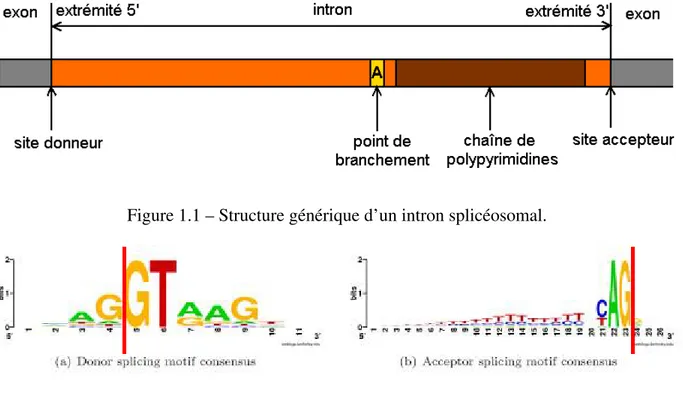
Les introns splicéosomaux sont reconnus par le splicéosome grâce à leur structure caractéris-tique [44] présentée dans la figure 1.1. La structure d’un intron splicéosomal est composée de 4 sites particuliers :
• Le site donneur : il représente la frontière entre l’extrémité 5’ de l’intron (en amont) et l’exon qui le précède.
• Le site accepteur : il s’agit de la frontière entre l’extrémité 3’ de l’intron (en aval) et l’exon qui le suit.
• Le point de branchement : c’est une molécule d’adénine située dans l’intron, plus proche du site accepteur que du site donneur, et qui interagit avec le site donneur lorsque l’intron est retiré de l’ARN messager.
• La chaîne de polypyrimidines : c’est une suite de pyrimidines (cytosine et thymine dans l’ADN, cytosine et uracile dans l’ARN) située avant le site accepteur et rencontrée dans la majorité des introns splicéosomaux.
Les sites donneur et accepteur sont appelés sites d’épissage, et représentent les frontières de l’intron. Un site d’épissage est composé de 2 parties : une partie exonique située dans l’exon voisin, et une partie intronique située dans l’intron. Dans le site donneur, la partie exonique précède la partie intronique, tandis que dans le site accepteur, c’est la partie intronique qu’on rencontre avant la partie exonique. Lorsqu’un intron est enlevé de l’ARN messager au cours de l’épissage, il subsiste dans l’ARN la partie exonique du site donneur de l’intron et la partie exonique du site accepteur de l’intron. Ces deux parties mises bout à bout forment une séquence reconnaissable appelée site de proto-épissage.
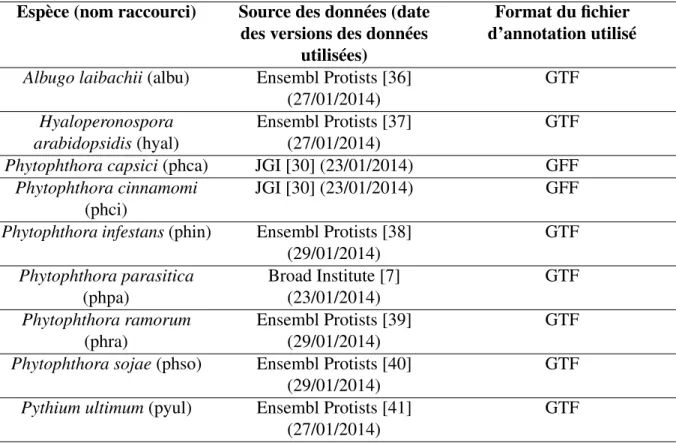
En fonction de la séquence de nucléotides des sites d’épissage, on distingue deux principales structures d’introns, chacune reconnue par un splicéosome particulier. La structure la plus souvent rencontrée est représentée dans la figure 1.2. Dans cette structure, le site donneur a pour séquence-consensus « AG|GT », avec « AG » comme partie exonique et « GT » comme partie intronique. Dans le site accepteur, la partie intronique a pour séquence-consensus « AG », mais la partie exonique est plus variable [44].
D’autres variations de structure sont parfois rencontrées chez certains eucaryotes. Par exemples, certains eucaryotes unicellulaires n’ont pas de chaînes de polypyrimidines dans leurs introns [44].
1.2 Rôles et fonctions des introns
Comme les introns sont retirés de l’ARN messager pendant son épissage, on pourrait penser qu’ils sont inutiles pour les cellules. Cependant, de nombreuses recherches montrent que les introns peuvent jouer plusieurs rôles dans le fonctionnement des cellules et dans la diversification des produits des gènes. Ils permettent notamment l’épissage alternatif et la dégradation des ARNs messagers non-sens, et peuvent même coder pour certains ARNs actifs dans les cellules.
Figure 1.1 – Structure générique d’un intron splicéosomal.
Figure 1.2 – Structure consensus des sites d’épissage donneur et accepteur de la majorité des introns splicéosomaux observés chez les eucaryotes. Les lignes verticales rouges représentent les frontières entre l’intron et les exons voisins. La figure provient de la référence [44].
1.2.1 Épissage alternatif
Lorsqu’un gène est exprimé, il est transcrit en ARN messager qui subit un épissage au cours duquel les introns sont retirés, et les exons restants sont alors assemblés dans l’ARN messager mature. Cependant, pour certains gènes, et selon les besoins de la cellule, tous les exons en provenance du gène ne sont pas forcément insérés dans l’ARN messager mature, et les exons retenus ne sont pas obligatoirement assemblés dans l’ordre dans lequel ils apparaissent sur l’ADN. Ce processus de sélection et de mise en ordre des exons d’un gène dans l’ARN messager est appelé épissage alternatif.
L’épissage alternatif rend donc possible la création de plusieurs ARNs messagers matures différents à partir d’un même gène de départ, qui seront convertis en molécules (protéines ou ARNs fonctionnels selon le gène) également différentes. Grâce à ce processus, un seul gène peut ainsi coder pour plusieurs molécules différentes, ce qui augmente la diversité des molécules synthétisées par un organisme sans que le nombre ou la longueur des gènes varient
significativement. Un exemple d’épissage alternatif est donné par la drosophile, dont le gène « dsx » code pour deux protéines impliquées dans la différenciation sexuelle de la mouche. Une des deux protéines est exprimée à un stade précoce du développement de la mouche, et, selon la protéine, l’organisme deviendra soit un mâle, soit une femelle [27].
Il existe plusieurs modes d’épissage alternatif [4, 10], parmi lesquels on peut citer :
• La sélection d’un exon. Un exon du gène peut être retenu ou ignoré dans l’ARN épissé. • La sélection mutuellement exclusive entre deux exons. L’un ou l’autre est présent dans
l’ARN après épissage, mais jamais les deux simultanément.
• La sélection alternative d’un site d’épissage en 5’ ou en 3’. La frontière de l’intron enlevé est modifiée, et la longueur de l’exon retenu change donc elle aussi, selon la portion d’exon qui a été enlevée avec l’intron retiré.
• La rétention d’intron. Une portion génique ayant les caractéristiques typiques d’un intron peut être retenue dans l’ARN messager épissé au lieu d’être enlevée.
L’épissage alternatif joue donc un rôle important dans la diversification des produits des gènes, et notamment des protéines. Certaines études estiment que, chez l’Homme, 60 % de tous les gènes [4], ou 95 % des gènes qui contiennent des introns [33], subissent l’épissage alternatif.
Lorsque des mutations se produisent au niveau des sites d’épissage, elles peuvent affecter les possibilités d’épissage alternatif d’un gène, et même engendrer des maladies génétiques. Ainsi, d’après López-Bigas et al. [26], 62 % des mutations causant des maladies chez l’Homme affecteraient les sites d’épissage plutôt que les séquences codantes elles-mêmes. Lim KH et al. [25] soutiennent quant à eux qu’un tiers des maladies causées par des mutations génétiques seraient liées à des problèmes d’épissage.
Les introns, sans lesquels l’épissage alternatif n’existerait pas, sont ainsi impliqués dans des maladies d’ordre génétique et dans la diversité des protéines fabriquées par un organisme.
1.2.2 Dégradation des ARNs messagers non-sens
Les gènes qui codent pour des protéines sont convertis en ARNs messagers matures qui contiennent une suite de codons, chaque codon correspondant à un acide aminé de la protéine à synthétiser. Pour fabriquer la protéine, des ribosomes lisent l’ARN messager mature à partir d’un codon de départ qui indique le premier acide aminé de la protéine, et s’arrêtent lorsqu’ils rencontrent un codon qui indique la fin de la protéine, et qui ne correspond lui-même à aucun acide aminé. Le codon d’arrêt est appelé codon STOP, et se trouve donc dans un exon. On peut rencontrer 3 codons STOP dans l’ADN des êtres vivants : UAA, UAG et UGA.
Si le gène contient des introns, on s’attend donc à ce que le codon STOP soit situé dans le tout dernier exon présent dans l’ARN messager mature. Cependant, à la suite par exemple de mutations ponctuelles, il peut arriver que des codons STOP apparaissent dans d’autres exons du gène en amont du dernier codon STOP. Il s’agit de codons STOP prématurés. Si les ribosomes rencontrent un tel codon, ils produiront une protéine tronquée, inutilisable par la cellule ou susceptible de perturber gravement son fonctionnement. Des ARNs messagers contenant des codons STOP prématurés doivent donc être rapidement repérés et détruits, afin qu’ils ne soient pas réutilisés pour fabriquer d’autres protéines dysfonctionnelles. Ces ARNs sont appelés ARNs messagers non-sens, et leur dégradation est un mécanisme de régulation important observé chez tous les eucaryotes [9].
On sait maintenant que le découpage des gènes en exons, du fait de la présence des introns, est impliqué dans un mode de détection des ARNs messagers non-sens chez les eucaryotes, notamment chez les mammifères [3, 9]. En effet, la présence d’introns permet l’existence de jonctions exon repérables dans l’ARN messager mature. Un complexe de jonction exon-exon appelé EJC (« exon-exon-exon-exon junction complex »), se fixe en amont de chaque jonction exon-exon-exon-exon à une distance d’environ 20 à 24 nucléotides de la jonction. L’ARN messager mature et les EJCs qui se sont associés à lui forment un complexe qui est ensuite lu par les ribosomes. Au cours de la traduction, les ribosomes enlèvent les EJCs qu’ils rencontrent, et arrêtent la traduction au premier codon STOP lu.
Puisque le premier codon STOP rencontré devrait normalement se trouver dans le dernier exon, il devrait être situé après la dernière frontière exon-exon dans l’ARN messager, donc après l’EJC le plus proche de la région terminale de l’ARN messager. Ainsi, si les ribosomes ont lu l’ARN jusqu’à rencontrer ce codon STOP, tous les EJCs auront été retirés. Mais si un codon STOP prématuré est rencontré plus tôt par les ribosomes, avant qu’ils n’arrivent au dernier exon, alors il restera au moins un EJC rattaché à l’ARN messager. Or un complexe ARN messager - EJC peut être repéré par la machinerie de dégradation des ARNs messagers non-sens. Celle-ci est alors mobilisée autour de l’ARN non-sens ainsi détecté, et se charge de supprimer rapidement l’ARN afin qu’il ne soit pas réutilisé pour synthétiser de nouvelles protéines tronquées.
Dans les gènes qui ne contiennent pas d’introns, mais qui comporteraient des codons STOP prématurés, un tel mode de détection des ARNs messagers non-sens est impossible, car aucune jonction exon-exon n’est disponible pour la liaison des EJCs. Les introns peuvent ainsi jouer un rôle crucial dans les divers mécanismes qui assurent le bon fonctionnement d’une cellule.
1.2.3 Encodage d’ARNs fonctionnels
On appelle ARN non-codant un ARN qui ne code pas pour une protéine. Il existe une grande variété d’ARNs qui sont non-codants mais qui jouent des rôles importants dans le fonctionnement des cellules. On connaît notamment les petits ARNs du nucléole (snoRNA), les micro-ARNs (miRNA), et les petits ARNs interférents (siRNA), qui sont connus pour réguler l’expression des gènes [5].
On sait désormais que de nombreux introns du génome humain contiennent les codes d’une grande variété d’ARNs non-codants régulateurs utilisés dans les cellules [5, 42]. Les introns qui codent pour ces ARNs sont donc directement impliqués dans le contrôle de la production de nombreuses protéines.
1.3 Évolution des introns
La présence d’introns dans de nombreuses espèces nous incite à nous demander s’ils étaient déjà présents dans des espèces ancestrales, voire dans des organismes situés très hauts dans l’arbre du vivant. En outre, puisqu’ils peuvent influencer le fonctionnement des organismes, on peut supposer que les introns constituent une source intéressante de variation pour la sélection naturelle. Les études menées sur l’évolution des introns cherchent donc aussi à déterminer s’ils sont effectivement soumis à des pressions sélectives pendant l’évolution des diverses lignées du vivant. Plusieurs études ont déjà été menées pour tenter d’apporter des réponses à ces interrogations.
1.3.1 Origine des introns splicéosomaux
La présence d’introns splicéosomaux dans toutes les lignées d’eucaryotes suggère que leur apparition est ancienne, et pose la question de leurs origines. Deux théories ont été proposées pour expliquer l’origine des introns splicéosomaux : la théorie des introns tôt et la théorie des introns tard [44].
La théorie des « introns tôt » propose que tous les introns des eucaryotes proviennent d’un ancêtre procaryote, commun aussi bien aux eucaryotes qu’aux procaryotes actuels. Les différences observées au niveau des introns chez les espèces eucaryotes actuelles proviendraient donc essentiellement de pertes d’introns spécifiques dans chaque lignée. Cette théorie implique que les procaryotes actuels, qui partageaient le même ancêtre, ont progressivement perdu presque tous leurs introns au cours de l’évolution de leurs génomes. Des versions plus récentes de la théorie admettent la possibilité que de nouveaux introns soient apparus chez certains eucaryotes au fil du temps, s’ajoutant aux introns ancestraux.
La théorie des « introns tard » quant à elle propose plutôt que les introns splicéosomaux sont apparus exclusivement chez les eucaryotes, via des gains d’introns qui se sont produits pendant leur évolution. Selon cette théorie, les procaryotes n’auraient donc jamais eu d’introns splicéosomaux ni de splicéosomes.
Les études récentes tendent à montrer que les ancêtres des eucaryotes, incluant le dernier ancêtre commun à tous les eucaryotes (« LECA »), possédaient des gènes riches en introns [44]. Les lignées d’eucaryotes auraient donc évolué en combinant des évènements de pertes et de gains d’introns, avec des pertes se produisant plus fréquemment que des gains.
Ces études posent alors la question de l’origine des introns de LECA. Des comparaisons des régions terminales des introns splicéosomaux et de certains introns auto-épissables du groupe II suggèrent que ces deux classes d’introns sont apparentées. Les introns splicéosomaux auraient ainsi évolué à partir des introns auto-épissables du groupe II, qu’on retrouve principalement chez certains procaryotes [44]. Une théorie, associant cette observation et les connaissances sur l’origine des eucaryotes, propose que les introns splicéosomaux dérivent d’introns du groupe II qui se seraient trouvés chez un procaryote absorbé au cours d’une endosymbiose par un autre procaryote, au cours de la formation des premières cellules eucaryotes [44]. Les introns du groupe II auraient alors migré vers le génome de l’hôte, tout en subissant des modifications qui ont rendu leur épissage dépendant d’un splicéosome.
À l’heure actuelle, la théorie des introns « tôt » est donc la plus soutenue pour expliquer l’origine des introns des eucaryotes.
1.3.2 Mécanismes d’évolution des introns
Les théories relatives à l’origine des introns considèrent que des gains et des pertes d’introns dans les espèces peuvent se produire au fil du temps. Ces hypothèses suggèrent que les introns changent au cours de l’évolution des espèces, ce qui est soutenu par les observations faites dans les génomes connus, car le nombre et la longueur des introns varient significativement d’une espèce à une autre [44]. On peut donc se demander si les introns sont soumis à des pressions sélectives qui guident leur évolution, ou s’ils évoluent de façon totalement aléatoire.
Pour répondre à cette question, il est nécessaire d’identifier les différents mécanismes d’évolu-tion des introns, afin de vérifier si certains mécanismes sont privilégiés par rapport à d’autres en fonction de conditions observées, et donc déterminer si l’évolution des introns n’est pas aléatoire.
Plusieurs modèles d’évolution ont été proposés pour expliquer les changements observés au niveau des introns dans les gènes. Certains modèles, tels que l’évolution par mutations ponctuelles de nucléotides, sont évidents et faciles à soutenir. D’autres modèles, tels que l’évolution par transposition d’introns, manquent d’études approfondies et ne sont donc pas suffisamment soutenus [53].
Les modèles proposés nous permettent cependant de définir plusieurs mutations introniques potentielles. Nous appelons mutation intronique une mutation qui insère, supprime ou modifie un intron dans un gène. À partir des modèles d’évolution proposés, et en se servant du gène comme repère, nous pouvons distinguer 2 catégories de mutations introniques :
• Les mutations introniques génomiques, qui sont dues à l’interaction du gène avec d’autres séquences issues du génome et situées à l’extérieur de ce gène. On peut les qualifier de mutations introniques globales, car elles se produisent à l’échelle du génome.
• Les mutations introniques intragéniques, qui sont dues exclusivement à des mutations ponctuelles (changement de nucléotides) à l’intérieur du gène. On peut les qualifier de mutations introniques locales, car elles se produisent à l’échelle du gène seul.
Le but de notre projet est d’étudier les mutations introniques locales des introns splicéosomaux.
1.3.2.1 Évolution des introns par mutations introniques globales
Les mutations introniques globales se produisent lorsqu’un gène interagit avec une autre séquence nucléotidique issue du génome. Ces interactions génomiques peuvent conduire à des insertions ou des suppressions complètes d’introns dans le gène [53]. Les mutations introniques globales se manifestent donc par des pertes ou des gains d’introns.
Les pertes d’introns peuvent se produire via une recombinaison génomique [53]. Un ARN messager mature, ne contenant donc aucun intron, peut être converti en une séquence d’ADN complémentaire via une transcription inverse. Cette séquence peut alors être impliquée dans une
recombinaison génomique avec le gène initial, au cours de laquelle les introns situés dans le gène seront perdus. Des introns entiers peuvent ainsi être supprimés sans affecter les exons voisins.
Les gains d’introns peuvent se produire via diverses interactions [53] :
• La transposition d’intron. Un intron retiré d’un ARN messager au cours de l’épissage peut s’insérer dans un autre ARN messager, du même gène ou d’un gène différent. Cet ARN messager peut être converti par transcription inverse en un ADN complémentaire, qui contient donc un nouvel intron. Une recombinaison génomique peut ensuite avoir lieu avec le gène de l’ARN messager rétro-transcrit, au cours de laquelle le nouvel intron est inséré dans le gène.
• Le transfert d’intron entre deux gènes paralogues. Deux gènes sont paralogues s’ils pro-viennent d’une duplication d’un gène ancestral dans un même génome. Au cours de l’évolution, des introns peuvent apparaître dans un des gènes sans apparaître dans l’autre, ou apparaître dans les deux gènes mais pas forcément aux mêmes positions. Une recom-binaison entre deux gènes paralogues après une certaine période d’évolution peut alors permettre le passage de nouveaux introns d’un gène à un autre.
• L’insertion d’un transposon. Un transposon (séquence d’ADN mobile dans le génome) s’insère dans un gène et est progressivement converti en intron au fil de l’évolution. • L’auto-épissage d’introns du groupe II. Comme nous l’avons vu, ce modèle est suggéré pour
expliquer l’origine des introns splicéosomaux. Il propose que des introns auto-épissables du groupe II, libérés par l’épissage de gènes d’organelles acquises par endosymbiose avec des procaryotes, s’insèrent dans le génome de la cellule hôte puis se transforment progressivement en introns splicéosomaux, c’est-à-dire en introns reconnus et épissés par un splicéosome de la cellule eucaryote hôte.
1.3.2.2 Évolution des introns par mutations introniques locales
Les mutations introniques locales se produisent lorsqu’un gène est modifié par des mutations ponctuelles. Les mutations ponctuelles désignent les mutations des nucléotides du gène, telles
que les insertions, les suppressions, les substitutions ou les duplications de nucléotides ou de petits blocs de nucléotides à l’intérieur du gène. Les mutations ponctuelles peuvent avoir divers effets sur les introns, selon l’endroit où elles se produisent.
Par exemple, dans un exon, l’apparition de 2 nouveaux sites d’épissage donneur et accepteur peut délimiter une portion de l’exon qui évoluera ensuite vers un intron. Une duplication géno-mique en tandem sur un site de proto-épissage situé dans l’exon peut aussi conduire à la naissance d’un nouvel intron. En effet, chez les eucaryotes, la séquence AGGT est un site de proto-épissage potentiel. L’intron se trouve entre AG et GT, mais une majorité d’introns contient également GT à leur extrémité 5’ et AG à leur extrémité 3’. Si un tel site est dupliqué dans un exon, on obtient une sous-séquence de la forme AGGT...AGGT. Un nouvel intron peut alors apparaître à partir de la séquence GT...AG, flanqué d’AG en amont et GT en aval, ce qui redonne le site de proto-épissage AGGT [53].
Si les mutations ponctuelles se produisent dans un intron, elles peuvent modifier sa longueur (par insertion ou suppression de nucléotides à l’intérieur de l’intron), ou rendre son épissage impossible (par destruction de sa structure en dénaturant ses sites d’épissage, son point de branchement, ou sa chaîne de polypyrimidines) et le convertir ainsi en exon. Les mutations qui affectent les sites d’épissage peuvent avoir des conséquences plus variées, telles que la modification des frontières de l’intron. La suppression de tout ou une partie de l’intron est également possible suite à divers phénomènes évolutifs (par exemple des suppressions ponctuelles accumulées, ou une suppression segmentale). Dans le cas d’une suppression partielle du début ou de la fin d’un intron, qui supprime donc un de ses sites d’épissage, la partie restante de l’intron peut subsister et modifier les exons voisins en fusionnant avec eux [53].
Cas particulier des glissements d’introns
Certaines observations faites dans des alignements de gènes orthologues montrent des introns, qui semblent apparentés, mais dont les positions varient de quelques nucléotides d’un gène à un autre. Ces observations suggèrent un autre type de mutation intronique potentielle appelée glissement d’introns, qui désigne le déplacement d’un intron dans son gène.
Plusieurs glissements d’introns pourraient n’être qu’apparents, et pourraient représenter, par exemple, des insertions parallèles d’introns dans les gènes d’une même famille à des positions proches. Cependant, Tarrio et al. [48] ont proposé une théorie basée sur l’évolution d’un même intron grâce à l’épissage alternatif. Selon cette théorie, de nouveaux sites d’épissage peuvent apparaître près des sites d’épissage initiaux d’un intron, ce qui permet un épissage alternatif. Cependant, les anciens sites d’épissage peuvent disparaître suite à des mutations, si bien que les nouveaux sites les remplacent définitivement pour l’épissage de cet intron, ce qui provoque un changement de position de ce dernier, et donc une observation qui laisse supposer un glissement d’intron. Des études plus approfondies sont nécessaires pour déterminer si les glissements d’introns sont de véritables mutations introniques, et quelle est leur nature (globale ou locale).
1.3.2.3 Impact des mutations introniques sur les produits des gènes
La conséquence d’une mutation intronique globale est l’insertion ou la suppression d’un intron complet. Ainsi, plusieurs dizaines de nucléotides peuvent apparaître ou disparaître dans le gène, ce qui modifie significativement sa longueur et son apparence sur l’ADN. Cependant, ce sont des introns entiers qui sont gagnés ou perdus, sans que les exons qui les entourent soient forcément affectés. Ainsi, même si le gène change beaucoup, ses produits peuvent rester identiques à la suite de mutations introniques globales. Les mutations introniques globales peuvent ainsi avoir un effet négligeable, voire nul, sur le fonctionnement de l’organisme, puisqu’elles ne modifient pas forcément les protéines qu’il utilise.
Les mutations introniques locales, quant à elles, affectent des portions de nucléotides dans les gènes. Seuls quelques nucléotides sont changés, supprimés ou insérés dans un gène. Les modifications sont donc mineures sur la longueur et l’apparence du gène. Cependant, si ces mutations affectent les sites d’épissage des introns, elles peuvent convertir tout ou une partie d’un intron en exon, ou tout ou une partie d’un exon en intron. Ainsi, même si le gène change très peu, ses produits peuvent être significativement modifiés. Les mutations introniques locales peuvent donc avoir un effet important, voire majeur, sur un organisme, en modifiant les protéines nécessaires à son fonctionnement. Il serait donc intéressant d’étudier ces mutations afin de
déterminer leur fréquence et leur impact réel sur l’évolution des introns et des espèces.
1.4 Outils bio-informatiques disponibles pour l’étude de l’évolution des introns
Les mutations introniques locales pourraient influencer la production des protéines chez les eucaryotes lorsqu’elles affectent les sites d’épissage des introns. L’étude de l’évolution des sites d’épissage peut donc présenter un intérêt important pour la compréhension de l’évolution des gènes, de leurs produits et des organismes. En outre, l’étude des mutations introniques peut fournir de nombreuses informations sur les pressions sélectives qui s’exercent sur les introns.
Pour faciliter l’étude des mutations introniques, il serait convenable de disposer d’un logiciel adapté. Cependant, les outils bio-informatiques actuellement disponibles qui permettent l’étude des introns ne proposent pas d’options pour analyser efficacement les mutations introniques.
Par exemple, le logiciel « CIWOG » [52] (« Common Introns Within Orthologous Genes ») permet d’étudier l’évolution des introns splicéosomaux en comparant leur positionnement et leur longueur dans des gènes homologues. Il peut également associer à chaque intron le splicéosome qui l’épisse parmi les 2 splicéosomes qu’on retrouve chez les eucaryotes, et déterminer ainsi les classes des introns splicéosomaux. Avec ce logiciel, il est donc possible d’analyser l’évolution des changements de longueurs, de positions et de classes des introns splicéosomaux, et récolter des statistiques sur ces évolutions. Mais aucune option ne permet d’identifier les mutations introniques responsables des changements observés. On ne peut donc pas utiliser cet outil pour étudier précisément les mutations introniques locales. On ne peut pas non plus chercher des corrélations entre des critères potentiellement sélectifs et les changements observés au niveau des introns.
Un autre logiciel, appelé « MALIN » [12] (« MAximum Likelihood analysis of INtron »), est disponible pour étudier les introns dans les familles de gènes. Il propose diverses fonctionnalités, telles que :
• L’étude comparative et statistique des positions et des phases des introns dans des aligne-ments de protéines.
• La prédiction des positions des introns présents chez le gène ancestral. • La prédiction et l’analyse statistique des pertes et des gains d’introns.
• L’inférence de l’histoire évolutive de sites d’introns individuels (apparition, disparition ou déplacement d’un intron donné).
Cependant, en dépit des options proposées, MALIN ne se focalise que sur l’étude des pertes et des gains d’introns. On ne peut donc pas étudier efficacement d’autres évènements tels que les glissements d’introns. De plus, MALIN n’identifie pas la cause des pertes et des gains, et ne permet donc pas de déterminer si elles sont dues à des mutations introniques globales ou à des mutations introniques locales. Aussi, les évolutions qui changent les sites d’épissage des introns, sans entraîner leur disparition ou leur apparition complètes, ne peuvent pas être prédites ni analysées. Cet outil ne permet donc pas non plus une étude complète et approfondie de l’évolution des sites d’épissage.
1.5 Notre projet 1.5.1 Objectifs
Le but de notre projet était de proposer une méthode qui permettrait d’étudier les mutations introniques locales chez les eucaryotes, et plus particulièrement l’évolution des sites d’épissage susceptibles de modifier les produits des gènes. La méthode devait être une alternative généralisée et plus efficace que les logiciels actuellement disponibles, capable d’analyser non seulement les gains et les pertes d’introns, mais aussi l’ensemble des mutations introniques locales qui surviennent dans les génomes des eucaryotes. À partir de cette méthode, nous devions pouvoir répondre à plusieurs questions relatives aux mutations introniques locales pour un ensemble d’espèces données. Par exemple :
• Évaluer leur importance relative par rapport aux mutations globales. Parmi les introns étudiés, combien ont évolué via des mutations locales ?
• Évaluer la fréquence et l’importance des mutations introniques locales. Combien d’allon-gements, de raccourcissements, d’apparitions ou de disparitions complètes, combien dans chaque direction (5’ ou 3’) ou dans les deux directions en même temps ?
• Chercher des corrélations entre la fréquence des évènements évolutifs et des pressions sélectives (par exemple les fonctions des gènes). Certaines évolutions locales surviennent-elles plus souvent que d’autres selon le rôle de la protéine dans la cellule ?
1.5.2 Hypothèse
Pour développer notre méthode, nous avons émis l’hypothèse selon laquelle l’évolution des sites d’épissage des introns pourrait être inférée à partir d’alignements entre génomes et d’ana-lyses comparatives locales dans les gènes, en considérant des génomes assez proches pour être correctement alignés, mais suffisamment éloignés pour que des changements soient visibles. À partir des alignements des familles de gènes, nous pourrions détecter et quantifier les mutations introniques locales qui affectent les sites d’épissage des génomes étudiés.
1.5.3 Aboutissement
Nous avons développé une méthode bio-informatique qui permet, à l’heure actuelle, de détecter les évènements évolutifs qui changent les sites d’épissage dans un ensemble de génomes données, et de les sauvegarder dans un ensemble de fichiers texte qui constituent la base de données des évènements collectés.
Les évènements sont repérés en cherchant des mutations introniques locales, mais plusieurs mutations mises en évidence, telles que les pertes et les gains d’introns, peuvent être des mutations introniques globales, et notre méthode actuelle ne permet pas encore d’identifier clairement la catégorie (locale ou globale) des mutations introniques détectées. Elle ne propose pas non plus d’outils permettant d’analyser l’ensemble des évènements identifiés pour répondre aux questions
d’intérêt sur les pressions sélectives qui s’exercent sur les introns. La méthode est donc perfectible, et de nombreuses options et améliorations seront prochainement ajoutées pour affiner la détection des mutations et l’extraction d’informations pertinentes à partir de la collection des mutations détectées.
Cependant, la méthode automatise tout le processus d’identification et de collecte des muta-tions introniques. Les chercheurs peuvent donc s’en servir pour rassembler plus facilement les évènements évolutifs des introns, puis développer plus rapidement des requêtes à exécuter sur la base de données obtenue pour répondre à leurs questions de recherche.
Nous avons développé notre méthode en étudiant une famille de champignons unicellulaires parasites, les oomycètes. Les résultats obtenus montrent plusieurs mutations introniques locales reconnaissables détectées dans de nombreuses familles de protéines orthologues. Cependant, les mutations les plus nombreuses sont les pertes et les gains d’introns, qui peuvent être causés par des mutations introniques globales. L’ensemble des mutations détectées suggère des tendances de gains d’introns et de déplacement des sites d’épissage vers le côté 3’ des gènes, au cours de l’évolution des oomycètes étudiés. Ces tendances sont des hypothèses, qui pourront être analysés plus précisément avec les prochaines versions de notre méthode.
Dans les chapitres suivants, nous décrivons les étapes de la méthode, les paramètres, les avantages, les inconvénients et les améliorations possibles pour chaque étape, et nous présentons les résultats obtenus pour la famille des oomycètes. Nous expliquons enfin comment récupérer les codes nécessaires pour réutiliser notre méthode dans d’autres projets.
CHAPITRE 2
COLLECTE DES DONNÉES ET TRAITEMENTS PRÉLIMINAIRES
2.1 Choix des espèces à étudier 2.1.1 Théorie
Notre hypothèse de travail requiert que les données en entrée proviennent d’espèces proches (pour faciliter l’identification des séquences homologues) mais suffisamment différentes pour que des variations soient observables au niveau des gènes et des protéines. Il faut donc choisir un ensemble d’espèces en fonction de ce critère. De plus, l’étude de l’évolution des introns nécessitera la construction d’arbres phylogénétiques, et donc la présence d’un outgroup dans l’ensemble des espèces choisies. Un outgroup est un ensemble d’espèces témoins qui permettent de repérer la racine de l’arbre phylogénétique reconstruit.
Une manière simple de choisir est de prendre des espèces qui sont considérées comme apparte-nant à un même taxon, et de former un outgroup avec quelques espèces prises à l’extérieur de ce taxon, mais proches de ce taxon.
2.1.2 Application
Pour développer notre méthode, nous avons travaillé avec le taxon des oomycètes. Ce sont des organismes unicellulaires semblables à des champignons, dont plusieurs espèces sont des parasites. Certains oomycètes sont bien connus pour provoquer d’importantes maladies chez les plantes cultivées par l’Homme. L’espèce Phytophthora infestans, par exemple, est l’oomycète responsable du mildiou de la pomme de terre, une maladie capable de détruire des récoltes entières [31].
Au travers d’études antérieures menées sur ces parasites, nous savions que les oomycètes constituaient un bon candidat d’étude. Des génomes complets et déjà annotés de plusieurs espèces de ce taxon sont déjà disponibles et facilement accessibles. De plus, les espèces annotées sont
suffisamment proches pour faciliter la détection des homologies entre les gènes. Les oomycètes constituent également un sujet d’étude intéressant, car on peut se demander si leurs introns ont évolué en fonction d’adaptations à leurs hôtes ou aux différents environnements qu’ils colonisent.
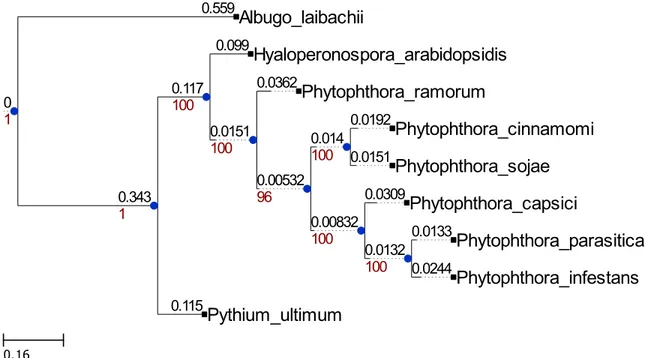
Nous avons constitué un ensemble de 9 espèces d’oomycètes, qui comporte 6 espèces du genre Phytophthora, et 1 espèce dans chacun des genres Pythium, Hyaloperonospora et Albugo. Nous avons choisi l’espèce Albugo laibachii comme outgroup en analysant un arbre phylogénétique d’oomycètes disponible dans la littérature [34]. Les données collectées pour ces espèces incluaient les séquences des gènes, des transcrits et des protéines, et les annotations de leurs génomes, ainsi que les séquences de contigs ou de chromosomes pour certaines espèces. Toutes les données proviennent de 3 bases de données génomiques accessibles sur l’Internet :
• La base de données Ensembl Protists [23]. • La base de données de Broad Institute [6].
• La base de données du Joint Genome Institute (JGI) [30].
Le tableau 2.I présente la liste des espèces, l’origine des données pour chacune d’elles, et la date à laquelle ces données ont été récoltées.
2.2 Utilisation des protéines à la place des gènes dans notre méthode
Le développement de notre méthode nécessite de comparer des séquences entre elles pour identifier des séquences homologues et des composants homologues dans les séquences. Pour effectuer ces comparaisons, nous devrons réaliser des alignements de séquences, à partir desquels nous déduirons des informations indispensables à l’étude de l’évolution des introns. Il faut donc déterminer quel type de séquences nous comptons utiliser parmi les différents types disponibles, à savoir les protéines, les gènes, et les dérivés des gènes tels que les transcrits (qu’on peut considérer comme les gènes avec quelques portions de séquences enlevées, comme les introns).
Tableau 2.I – Liste des espèces étudiées et origine des données collectées pour chaque espèce. Espèce (nom raccourci) Source des données (date
des versions des données utilisées)
Format du fichier d’annotation utilisé Albugo laibachii(albu) Ensembl Protists [36]
(27/01/2014) GTF Hyaloperonospora arabidopsidis(hyal) Ensembl Protists [37] (27/01/2014) GTF Phytophthora capsici(phca) JGI [30] (23/01/2014) GFF
Phytophthora cinnamomi (phci)
JGI [30] (23/01/2014) GFF
Phytophthora infestans(phin) Ensembl Protists [38] (29/01/2014) GTF Phytophthora parasitica (phpa) Broad Institute [7] (23/01/2014) GTF Phytophthora ramorum (phra) Ensembl Protists [39] (29/01/2014) GTF Phytophthora sojae(phso) Ensembl Protists [40]
(29/01/2014)
GTF Pythium ultimum(pyul) Ensembl Protists [41]
(27/01/2014)
GTF
Le code génétique, qui représente la table de correspondance entre les codons et les acides aminés, contient 64 codons, dont 3 sont généralement des codons STOP, et 61 codent effective-ment pour une vingtaine d’acides aminés [32]. Puisqu’il y a plus de codons que d’acides aminés, plusieurs codons différents codent pour un même acide aminé. De ce fait, les gènes peuvent accumuler plusieurs mutations sans que les protéines (séquences d’acides aminés) pour lesquelles ils codent soient obligatoirement modifiées. Les gènes peuvent donc évoluer plus rapidement que les protéines. Or une évolution rapide peut rendre trop différentes des séquences géniques qui sont apparentées, et donc fausser les alignements de ces séquences et les déductions à faire sur leurs homologies. Pour cette raison, nous avons utilisé les protéines à la place des gènes pour le développement de notre méthode, car elles évoluent moins vite que les gènes, et conservent donc plus d’homologies.
L’utilisation des protéines présente aussi un autre avantage. En effet, elles sont décrites avec un alphabet de 20 états (20 acides aminés), alors que les gènes et les transcrits sont des séquences nucléotidiques décrites avec un alphabet de seulement 4 états (4 nucléotides). Le risque de considérer comme homologues deux états qui ne le sont pas est donc beaucoup plus élevé si nous comparons les gènes, alors que les protéines offrent une plus grande diversité d’états, donc une probabilité un peu plus faible que deux états supposés homologues ne le soient pas réellement.
Travailler avec des protéines présente quelques complications lorsqu’il s’agit d’étudier des introns, car ces derniers sont des séquences nucléotidiques qui sont plus faciles à localiser dans des gènes. Nous avons cependant géré ces problèmes en créant des programmes et des notations adaptés, présentés dans les prochaines sections, et qui permettent de positionner et de mettre en évidence les introns d’un gène directement sur ses protéines.
2.3 Construction des familles de protéines 2.3.1 Théorie
Notre projet a pour but d’étudier les mutations introniques locales, qui se produisent à l’intérieur d’un gène. Pour le faire, il nous semble convenable d’étudier l’évolution du gène proprement dit,
donc en l’occurrence des protéines synthétisées par ce gène. L’étude de l’évolution des protéines nécessite de repérer les homologies entre les protéines, et d’en déduire les groupes de protéines homologues, qu’on appelle communément familles de protéines. Nous pourrons ensuite étudier l’évolution des introns qui sont présents dans chaque famille de protéines déterminée.
2.3.1.1 OrthoMCL
Pour construire les familles de protéines, nous avons utilisé OrthoMCL [24]. Il s’agit d’un ensemble de scripts écrits dans le langage de programmation Perl et qui doivent être exécutés dans une suite précise d’étapes, conjointement avec d’autres logiciels, pour construire les familles de protéines. Le site de téléchargement d’OrthoMCL fournit un guide d’utilisation détaillé pour l’exécution de ces étapes [18]. Les autres logiciels utilisés sont MySQL [14] (pour gérer une base de données), et BLAST [2] (pour comparer et aligner les protéines).
OrthoMCL analyse d’abord les protéines disponibles et retient uniquement les protéines qu’il juge de bonne qualité. Ses critères pour filtrer les protéines sont basés sur leurs longueurs (les protéines trop courtes sont ignorées) et sur le pourcentage de codons STOP qu’elles contiennent (les protéines contenant trop de codons stop prématurés sont ignorées). Le logiciel BLAST est ensuite exécuté pour comparer toutes les protéines entre elles et récolter les informations sur leurs similarités. OrthoMCL utilise enfin ces informations pour générer les familles de protéines. Il produit un fichier texte dans lequel chaque ligne décrit une famille en lui donnant un identifiant unique et en listant les identifiants des protéines de cette famille.
2.3.2 Application
2.3.2.1 Paramètres utilisés pour l’exécution d’OrthoMCL
Tous les scripts d’OrthoMCL ont été exécutés avec les valeurs par défaut chaque fois que c’était possible. La version de BLAST que nous avons utilisée est la version 2.2.29 qui fournit les programmes « BLASTP » (pour comparer les protéines) et « makeblastdb » (pour manipuler des bases de données compréhensibles pour BLAST). BLASTP a également été exécuté avec ses options par défaut, sauf pour le paramètre qui contrôle le format des fichiers de sortie, car
OrthoMCL requiert un format particulier pour bien fonctionner. Le paramètre « e-value » de BLASTP a donc lui aussi été laissé à sa valeur par défaut (10), bien que OrthoMCL recommande une valeur différente (10-5). Puisque notre but est avant tout de développer une méthode, nous
considérons que l’e-value est un paramètre global qui peut être ajustée si nécessaire à chaque exécution de la méthode. Nous avons donc travaillé avec les familles de protéines produites selon les valeurs par défaut de BLASTP.
2.3.2.2 Résultats obtenus
Notre jeu de données contenait un total de 162 566 protéines réparties dans nos 9 espèces. 162 564 protéines ont été retenues par OrthoMCL, et 2 protéines ont été ignorées : la protéine « HpaP802526 » de l’espèce Hyaloperonospora arabidopsidis, qui ne contient que 6 acides aminés, et la protéine « CCA28415 » de l’espèce Albugo laibachii, qui ne contient que 10 acides aminés.
BLASTP, au cours de son exécution, a ignoré 2 autres protéines suite à des erreurs qu’il a trouvées dans leurs séquences : la protéine « 109327 » de l’espèce Phytophthora capsici (200 acides aminés), et la protéine « 92918 » de l’espèce Phytophthora cinnamomi (109 acides aminés). BLASTP a recommandé de vérifier ces protéines ou les paramètres de filtrage des protéines de bonne qualité. Les protéines concernées ne contenaient aucun caractère atypique, si bien que ces erreurs nous semblent provenir des paramètres de filtrage d’OrthoMCL, ou d’erreurs que nous n’identifions pas encore. Nous avons cependant poursuivi le processus avec les protéines restantes, car les paramètres de filtrage d’OrthoMCL peuvent ici aussi être considérés comme des paramètres de la méthode.
Au total, OrthoMCL a généré 18 955 familles de protéines, contenant 134 154 protéines, soit 82,52 % des protéines de bonne qualité. On constate donc que 28 410 protéines de bonne qualité n’ont pas été classées par OrthoMCL dans des familles. Même si le nombre reste relativement faible par rapport au nombre total de protéines initialement disponibles, nous ne comprenons pas encore pourquoi autant de protéines n’ont pu être rangées dans des familles. Nous pensons que l’utilisation de valeurs appropriées pour les paramètres de filtrage des protéines et pour BLASTP
pourrait modifier la proportion de protéines classées et le nombre de familles déterminées, mais nous devrons exécuter à nouveau l’étape OrthoMCL pour déterminer comment les familles construites varieront en fonction des ajustements de ces paramètres.
2.3.2.3 Distributions des familles selon le nombre d’espèces et le nombre de séquences Nous avons écrit un programme JAVA qui produit un ensemble de statistiques sur les familles de protéines obtenues, à partir desquelles nous pouvons collecter diverses informations, notamment par rapport au nombre de séquences et au nombre d’espèces dans les familles.
La figure 2.1 présente, pour chaque nombre de séquences de 0 à 40 compté dans les familles, le nombre de familles qui contiennent chacune ce nombre séquences parmi les 18 955 familles déterminées. Les nombres de séquences supérieurs à 40 sont groupés afin d’être tous affichés sur la figure. Ils vont de 41 à 618 séquences. On remarque notamment que 4 163 familles (soit 21,96 % des familles disponibles) contiennent seulement 2 séquences, tandis que 108 familles contiennent plus de 40 séquences, avec 1 famille contenant jusqu’à 618 séquences. Il est très probable que de nombreuses familles soient en fait des faux positifs, notamment celles qui contiennent de si grands nombres de séquences, et plusieurs parmi celles qui en contiennent seulement 2.
La figure 2.2 présente, pour chaque nombre d’espèces de notre jeu de données, le nombre de familles dans lesquelles apparaissent ce nombre d’espèces parmi les 18 955 familles déterminées. La figure décrit donc le nombre de familles dans lesquelles apparaissent un certain nombre d’espèces. On constate notamment qu’une seule espèce apparaît dans 3 948 familles (soit 20,83 % des familles disponibles), tandis que les 9 espèces étudiées apparaissent dans 3 604 familles (soit 19,01 % des familles disponibles). Puisque beaucoup de familles contiennent plus de 9 séquences (comme le montre la figure 2.1), donc plus de séquences que d’espèces étudiées, il semble donc que de nombreuses familles contiennent des protéines issues de gènes paralogues (c’est-à-dire des gènes issus de la duplication d’un gène ancestral). De plus, au moins 1 espèce n’apparaît pas dans 15 351 familles, soit 80,99 % des familles disponibles. Il semble donc y avoir eu beaucoup de disparitions de gènes dans les différentes espèces au cours de leur évolution.
4163 1663 1177 1109 1267 1666 2117 2731 1173 411 254 177 143 118 110 98 71 52 57 32 27 29 13 24 15 18 17 16 16 8 18 12 11 5 8 4 9 3 5 108 0 1000 2000 3000 4000 5000 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 (>) Nombre de séquences Nombre de f amilles
Figure 2.1 – Nombre de familles de protéines en fonction du nombre de séquences contenues dans les familles. « (>) » désigne les nombres de séquences trouvés supérieurs à 40, qui vont de 41 à 618 séquences.
3948
2334
1225
957 1020
1350
2035
2482
3604
0 1000 2000 3000 4000 1 2 3 4 5 6 7 8 9 Nombre d'espèces Nombre de f amillesFigure 2.2 – Nombre de familles de protéines en fonction du nombre d’espèces qui apparaissent dans les familles.
2.3.2.4 Génération des fichiers FASTA des familles de protéines
OrthoMCL fournit une description des familles de protéines déterminées, en donnant pour chacune d’elles un identifiant et la liste des noms des protéines qu’elle contient. Nous n’avons donc pas directement les séquences des protéines, qui sont nécessaires à la poursuite de nos analyses.
Nous devons donc générer nous-même pour chaque famille un fichier qui rassemble les séquences de ses protéines. Pour le faire, nous avons écrit un programme JAVA capable de générer ces fichiers au format FASTA. Le programme prend en entrée le fichier de familles fourni par OrthoMCL, un dossier de fichiers contenant les séquences des protéines des espèces étudiées, et le nom d’un dossier de sortie qui contiendra les fichiers FASTA représentatifs des familles. Les fichiers FASTA des 18 955 familles de protéines ont ainsi été générés.
2.4 Alignement des familles de protéines
À partir des familles de protéines déterminées, nous devrons construire des arbres phylogéné-tiques, et identifier les homologies entre les introns qui apparaissent dans chaque famille. Pour réaliser ces étapes, nous devons d’abord aligner les familles de protéines, pour déterminer les homologies entre les acides aminés à l’intérieur de chaque famille.
Pour aligner les familles, nous avons utilisé le logiciel « MUSCLE » [15] avec l’option «-maxiters 1000» qui assure que le logiciel analyse suffisamment chaque famille afin de maximiser la qualité de l’alignement généré. Comme le nombre de familles est grand, et que certaines contiennent beaucoup de séquences, nous avons écrit un script PHP exécutable en ligne de commande, qui permet de lancer parallèlement plusieurs lots d’alignements. On peut donc exploiter plusieurs machines en même temps pour aligner tous les groupes en peu de temps, au lieu d’aligner les 18 955 groupes sur un seul ordinateur, ce qui prendrait beaucoup de temps.
2.5 Utilisation des familles de protéines strictement orthologues dans la suite de l’étude 2.5.1 Théorie
Pour étudier l’évolution des introns dans chaque famille de gènes, il faut disposer d’un arbre phylogénétique pour guider l’ordre d’analyse des changements observables. Deux types d’arbres phylogénétiques peuvent être obtenus à partir des données disponibles :
• Un unique arbre des espèces, qui décrit l’évolution des 9 espèces.
• Un arbre des protéines pour chaque famille, qui décrit l’évolution des protéines spécifiques de cette famille.
Pour une étude efficace et précise, il faudrait idéalement inférer les arbres des protéines des familles. Cependant, comme notre objectif est de développer et de tester une méthode, nous avons décidé de générer uniquement l’arbre des espèces, et de l’utiliser pour étudier spécifiquement les familles de protéines strictement orthologues, en faisant l’hypothèse que l’arbre des espèces serait identique à l’arbre des protéines pour ces familles. Nous appelons famille strictement orthologue une famille de protéines qui contient exactement 1 protéine pour chaque espèce du jeu de données, donc 9 protéines pour les 9 espèces que nous étudions. Dans une telle famille, toutes les espèces sont représentées et chaque espèce est associée à une seule protéine, ce qui permet d’utiliser l’arbre des espèces pour analyser les protéines exactement comme si on analysait les espèces.
L’évolution des protéines dans une famille strictement orthologue ne suit pas forcément le même schéma que l’évolution des espèces. En effet, certains phénomènes évolutifs, tels que l’ « incomplete lineage sorting » (ILS) et le flux de gènes, peuvent influencer des familles de gènes spécifiques (et donc leurs protéines) et les faire évoluer différemment des espèces. Ces phénomènes sont notamment identifiés dans les génomes de certains primates, dont l’Homme, le Chimpanzé et le Gorille [43].
• L’ILS est un mécanisme de ségrégation de polymorphismes ancestraux, à la suite duquel l’arbre d’une famille de gènes peut être différent de l’arbre des espèces [43]. Lorsqu’une
espèce ancestrale possède plusieurs allèles d’un même gène, chacun de ses descendants peut retenir un seul allèle et éliminer les autres sous l’effet de la dérive génétique ou de la sélection naturelle. Si deux descendants normalement éloignés sélectionnent le même allèle ancestral, alors elles seront considérées comme proches du point de vue de cette famille de gènes. L’arbre des gènes de cette famille ne correspondrait donc pas à l’arbre des espèces auxquelles appartiennent ces gènes.
• Le flux de gènes est le transfert d’allèles ou de gènes entre populations [43]. Si la population d’une espèce reçoit un nouvel allèle pour un gène donné suite à une hybridation avec une population d’une autre espèce éloignée, alors la première population dispose de son allèle initial et du nouvel allèle obtenu. Au cours de l’évolution, le nouvel allèle peut devenir le plus fréquent et s’imposer jusqu’à la disparition de l’ancien allèle. Du point de vue de cette famille de gènes, les deux espèces seront considérées comme très proches (parce qu’elles ont le même allèle), alors qu’elles sont éloignées en réalité. À nouveau, l’arbre des gènes de cette famille ne correspondrait pas à l’arbre des espèces.
L’utilisation de l’arbre des espèces pour étudier les familles strictement orthologues n’est donc pas optimale et ne peut être définitive, mais elle est suffisante pour poursuivre la recherche et le développement des programmes et des algorithmes nécessaires à la méthode, que nous pourrons ensuite réutiliser facilement lorsque nous aurons les vrais arbres des protéines.
2.5.2 Application
Nous avons écrit un programme JAVA qui produit un rapport de statistiques sur les familles de protéines, à partir duquel on peut facilement identifier les familles strictement orthologues. 1 924 familles de protéines sont strictement orthologues sur les 18 955 familles disponibles.
2.6 Construction de l’arbre phylogénétique des espèces 2.6.1 Théorie
2.6.1.1 Sélection des familles à utiliser pour construire l’arbre
Pour construire l’arbre des espèces, il faut sélectionner des familles de protéines strictement orthologues adéquates et les utiliser avec un logiciel de phylogénie.
L’utilisation simultanée de toutes les familles strictement orthologues disponibles est peu recommandable car elles sont trop nombreuses, ce qui ralentirait énormément le logiciel de phylogénie. De plus, puisque les familles sont générées par OrthoMCL, elles ne sont pas encore toutes validées. Il est donc préférable d’opérer une sélection sur les familles, en retenant celles dont les alignements contiennent le plus possible de sites conservés ou similaires et le moins possible de sites instables ou de trous. Pour sélectionner les familles, nous utilisons le logiciel « GBlocks » [8] qui analyse les alignements de séquences.
GBlocks permet de filtrer un alignement pour n’en garder que les meilleures régions à utiliser dans les logiciels de phylogénie. En général il sélectionne des régions bien conservées mais avec quelques sites variables nécessaires pour différencier les espèces. Il prend en entrée des fichiers FASTA et génère en sortie des fichiers FASTA qui ont l’extension « fasta-gb » et qui ne contiennent que les régions qu’il a retenues dans les alignements.
Pendant l’exécution de GBlocks, deux informations ont été collectées pour chaque famille, afin d’aider à les sélectionner :
• Le pourcentage de positions retenues par GBlocks après filtrage. • La longueur de l’alignement après filtrage.


![Figure 2.4 – Arbre phylogénétiue d’oomycètes publié en 2012 dans la littérature [45].](https://thumb-eu.123doks.com/thumbv2/123doknet/2063079.6194/47.918.117.802.121.340/figure-arbre-phylogenetiue-oomycetes-publie-litterature.webp)