Substrate Specificity of Low-Molecular Mass Bacterial DD-Peptidases
Texte intégral
Figure
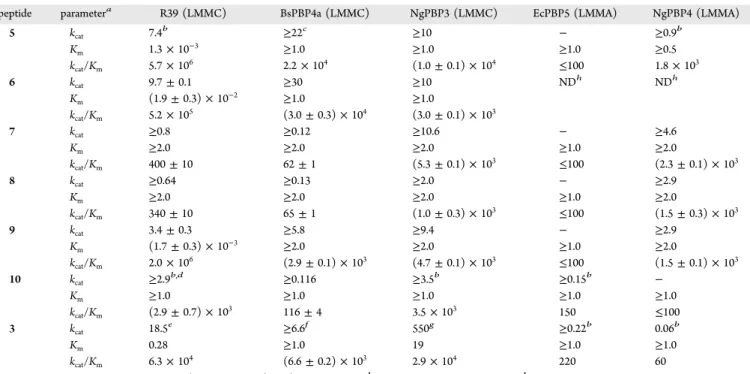

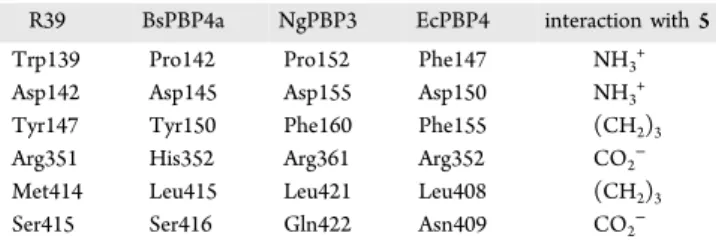
Documents relatifs
the design of farm buildings or structural elements made from steel products should be in accordance with the Chapter on Steel Construction of the Canadian
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
In summary, the network means of communication are critically linked with the neural states dynamical repertoire of the network, which is key since conscious processes are
More generally speaking, if we have a singular curve C, the set of valuation rings of its rational function field is in bijection with the set of points of a non-singular model C ′
Effect of -conjugation on the methyl torsion: The gas phase structures and internal rotation parameters of butadienyl acetate were determined with very high accuracy by
De la combinaison de ces deux facteurs conjoncturels – le rééquilibrage des lignes de production et la mise en conformité avec la nouvelle loi sur le travail tem- poraire – naît
Kramer GD, Pausz C, Herndl GJ (2005) Elemental composition of dissolved organic matter and bacterioplankton production in the Faroe-Shetland Channel (North Atlantic). Deep-Sea
Identi- fication of the L,D-transpeptidases responsible for attachment of the Braun lipoprotein to Escherichia coli peptidoglycan.... coli lipoprotein attachment to peptidoglycan
