Ability of nitrones of various structures to control the radical polymerization of styrene mediated by in situ formed nitroxides
Texte intégral
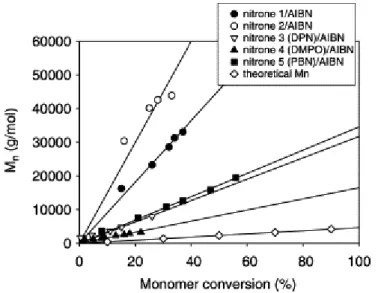
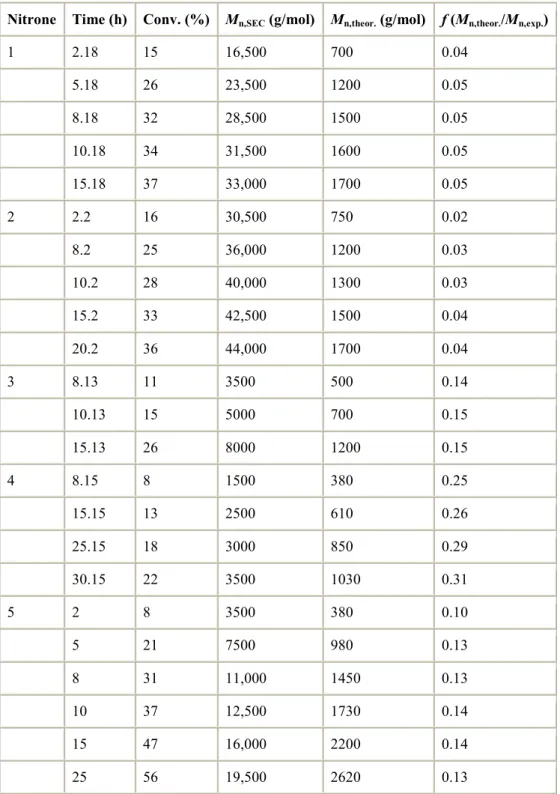
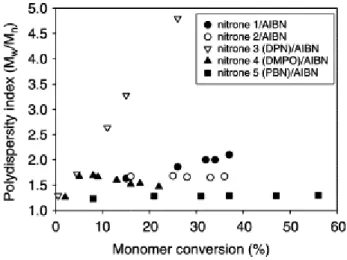
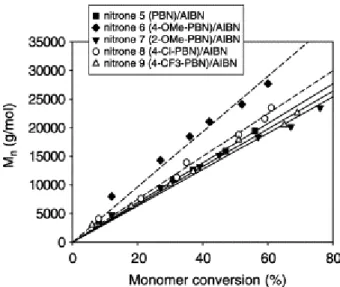
Figure
![Fig. 2. Time dependence of ln ([M] 0 /[M]) for the bulk radical polymerization of styrene at 110 °C [Pre-reaction of nitrone and AIBN in toluene at 85 °C for 4 h; [nitrone 1]=0.19 M and [AIBN]=9.7×10 −2 M (nitrone 1/AIBN:](https://thumb-eu.123doks.com/thumbv2/123doknet/5461944.129218/6.892.103.452.490.746/dependence-radical-polymerization-styrene-reaction-nitrone-nitrone-nitrone.webp)
![Fig. 6. Time dependence of ln ([M] 0 /[M]) for the radical polymerization of styrene at 110 °C in toluene (styrene/toluene: 1/1; v/v) and in bulk, in the presence of PBN [Pre-reaction of nitrone and AIBN in toluene at 85 °C for 4 h; [nitrone]=0.19 M and](https://thumb-eu.123doks.com/thumbv2/123doknet/5461944.129218/10.892.117.801.101.280/dependence-radical-polymerization-toluene-presence-reaction-nitrone-toluene.webp)

![Fig. 8. SEC chromatograms for the bulk polymerization of styrene at 110 °C in the presence of PBN [Pre- [Pre-reaction of PBN and AIBN in toluene at 85 °C for 4 h; [PBN]=0.19 M and [AIBN]=9.7×10 −2 M (PBN/AIBN:](https://thumb-eu.123doks.com/thumbv2/123doknet/5461944.129218/12.892.110.581.94.585/chromatograms-polymerization-styrene-presence-reaction-aibn-toluene-aibn.webp)
Documents relatifs
Characterisation of the free radical chemistry in physicochemical conditions of the digestive tract.. Philippe Gatellier, Anne Meynier, Claire Dufour, Catherine Caris-Veyrat,
Il ressort par ailleurs de l’article 374 du Code civil que l’exercice de l’autorité parentale demeure en principe conjoint en cas de séparation des père et mère et que le
Essentially represented by atom-transfer radical polymerization (ATRP), reversible addition-fragmentation chain transfer (RAFT) polymerization and nitroxide-mediated
In the Brassicaceae studies of compatibility in SC Arabidopsis thali- ana have identified an oleosin and LTPs as important for the initial stages of the pollen – pistil interaction
Die Resultate der Studie zeigen, dass trotz einem erhöhten Risiko zu psychischen Folgen eines Einsatzes Rettungshelfer Zufriedenheit und Sinn in ihrer Arbeit finden können und
In the Falck case, the far-sighted family champion of change Alberto Falck—with crucial support of the external CEO Achille Colombo—was able to de-escalate the family business
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
l’utilisation d’un remède autre que le médicament, le mélange de miel et citron était le remède le plus utilisé, ce remède était efficace dans 75% des cas, le
