Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS École nationale supérieure d'ingénieurs (Poitiers) Institut de chimie des milieux et matériaux de Poitiers - IC2MP
(Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie et microbiologie de l'eau
Présentée par : Odissa Abou Mehrez
Chloration et monochloramination des aminophénols en solution aqueuse
Directeur(s) de Thèse : Bernard Legube, Florence Berne Soutenue le 13 décembre 2013 devant le jury
Jury :
Président Farouk Jaber Professeur, Université Libanaise Rapporteur Farouk Jaber Professeur, Université Libanaise
Rapporteur Pascal Wong Wah Chung Professeur des Universités, Université d'Aix-Marseille Membre Bernard Legube Professeur des Universités, Université de Poitiers Membre Florence Berne Maître de conférences, Université de Poitiers Membre Sylvie Soreau Ingénieur de recherche, EDF R&D, Chatou
Pour citer cette thèse :
Odissa Abou Mehrez. Chloration et monochloramination des aminophénols en solution aqueuse [En ligne]. Thèse Chimie et microbiologie de l'eau. Poitiers : Université de Poitiers, 2013. Disponible sur Internet
Présentée à
L’UNIVERSITE DE POITIERS
ECOLE NATIONALE SUPERIEURE D’INGENIEURS DE POITIERS ECOLE DOCTORALE SCIENCES POUR L’ENVIRONNEMENT GAY LUSSAC
Pour l’obtention du grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS (Diplôme National – Arrêté du 7 août 2006)
SPECIALITE : CHIMIE ET MICROBIOLOGIE DE L’EAU
Par
Odissa ABOU MEHREZ
Maître ès sciencesChloration et monochloramination des aminophénols en
solution aqueuse
Soutenance prévue le 13 Décembre 2013, devant la commission d’examen :
Rapporteurs : M. F. JABER
(Professeur - Université Libanaise) M. P. WONG-WAH-CHUNG
(Professeur - Université d’Aix-Marseille) Examinateur : Mme S. SOREAU
(Ingénieur Recherche - EDF R&D Chatou) Directeurs de thèse : M. B. LEGUBE
(Professeur - Université de Poitiers) Mme F. DOSSIER-BERNE
Ce travail a été effectué à l’Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP) au sein de l’Equipe Eaux, Géochimie organique, Santé (EGS).
Je tiens à remercier notre responsable d’équipe Mme
KARPEL VEL LEITNER Nathalie pour la confiance qu’elle m’a accordée en m’accueillant au sein de l’équipe.
Mes sincères remerciements au CNRS Libanais qui a financé cette thèse. Je remercie en particulier M. TABET Charles pour le soutien qu’il m’a témoigné durant ces trois années.
Je remercie également mon directeur de thèse, M. LEGUBE Bernard, professeur et directeur du PRES Limousin-Poitou-Charentes pour le suivi de mon travail et son temps précieux qu’il m’a offert.
Un grand merci pour ma co-directrice de thèse, Mme DOSSIER-BERNE Florence, Maître de conférences, pour tous ses sacrifices, sa persévérance et sa grande disponibilité. Je suis vraiment reconnaissante qu’elle ait partagé avec moi ses compétences professionnelles.
Je suis très reconnaissante envers M. JABER Farouk, professeur et responsable du Laboratoire d’Analyse des Pesticides et des Polluants Organiques (LAPPO) à la Commission Libanaise de l’Energie Atomique à Beyrouth au CNRS Libanais, pour avoir accepté de rapporter ce travail.
Je souhaite remercier M. WONG-WAH-CHUNG Pascal, professeur au Laboratoire de Chimie de l’Environnement (LCE) de l’Université d’Aix-Marseille, d’avoir accepté de juger ce travail et d’en être rapporteur.
Je tiens à remercier Mme SOREAU Sylvie, Ingénieur Chercheur au département Laboratoire National d’Hydraulique et Environnement (LNHE) de EDF de Chatou, d’avoir accepté d’examiner ce travail.
Je souhaite remercier tous les membres de l’équipe EGS et tout spécialement Mme
DEBORDE Marie et Mme LIU Sylvie pour leur aide en LC/MS ; et M. LABANOWSKI Jérôme pour m’avoir transmis le goût de la recherche lors de mes premiers pas dans l’équipe.
Je ne voudrais pas oublier de remercier ma famille et mes amis pour leur soutien moral.
INTRODUCTION GENERALE……… 1
CHAPITRE I : SYNTHESE BIBLIOGRAPHIQUE I.1- Aminophénols ... 3
I.1.1- Présentation ... 3
I.1.2- Propriétés physiques ... 3
I.1.3- Propriétés chimiques et réactivité ... 4
I.1.4- Utilisations industrielles et domestiques ... 8
I.1.5- Présence dans les eaux ou les sols et risques sanitaires ... 11
I.1.6- Analyse ... 14
I.2- Chlore ... 15
I.2.1- Propriétés physiques du chlore ... 15
I.2.2- Chimie du chlore dans l’eau ... 16
I.2.3- Réactivité du chlore sur les composés minéraux ... 17
I.2.3.1- Oxydation de l’azote ammoniacal ... 18
I.2.3.2- Oxydation des halogénures ... 19
I.2.3.3- Oxydation des nitrites et des sulfites ... 19
I.2.4- Réactivité du chlore sur les composés organiques ... 21
I.2.4.1- Oxydation des composés organiques azotés ... 21
I.2.4.1.1- Amines aliphatiques ... 21
I.2.4.1.2- Aniline ... 24
I.2.4.2- Oxydation des hydroxybenzènes ... 25
I.2.4.2.1- Phénol ... 25
I.2.4.2.2- Résorcinol ... 29
I.3- Monochloramine ... 32
I.3.1- Formation et utilisation des chloramines ... 32
I.3.2- Modélisation cinétique de l’auto-décomposition de la monochloramine ... 34
I.3.3- Réactivité de la monochloramine sur les composés minéraux ... 36
I.3.3.1- Effet des ions nitrite ... 36
I.3.3.2- Effet des ions bromure ... 37
I.3.3.3- Effet des ions carbonate ... 37
I.3.3.4- Effet des ions phosphate ... 38
I.3.3.5- Effet du fer ... 38
I.3.3.6- Effet du cuivre ... 39
I.3.4- Réactivité de la monochloramine sur les composés organiques ... 39
I.3.4.1- Oxydation des composés organiques azotés ... 39
I.3.4.2- Oxydation des hydroxybenzènes ... 42
I.3.4.2.1- Phénol ... 42
II.1. Introduction ... 47
II.2. Matériels et Méthodes ... 48
II.2.1. Préparation des solutions ... 48
II.2.2. Dispositif expérimental ... 49
II.2.3. Méthodes analytiques ... 50
II.2.3.1. Dosage du chlore total ... 50
II.2.3.2. Dosage du chloroforme ... 51
II.2.3.3. Dosage des acides haloacétiques ... 51
II.2.3.4. Dosage des composés extractibles au MTBE ... 52
II.2.3.5. Dosage des composés organohalogénés adsorbables (AOX) ... 53
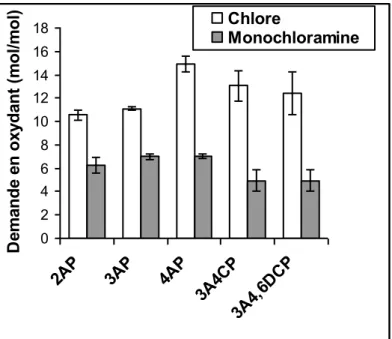
II.3. Demandes en oxydant ... 54
II.3.1. Demandes en chlore et en monochloramine ... 54
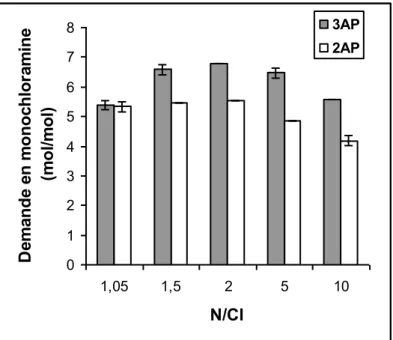
II.3.2. Influence du rapport molaire N/Cl ... 59
II.4. Formation de sous-produits de désinfection ... 61
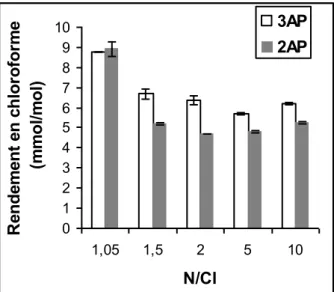
II.4.1. Formation du chloroforme ... 61
II.4.1.1. Détermination du potentiel de formation en chloroforme ... 61
II.4.1.2. Influence du rapport molaire N/Cl ... 65
II.4.2. Formation d’acides haloacétiques ... 67
II.4.3. Formation des composés extractibles au MTBE ... 71
II.4.4. Formation d’AOX ... 73
II.4.4.1. Détermination du potentiel de formation en AOX ... 73
II.4.4.2. Influence du rapport molaire N/Cl ... 75
II.4.4.3. Spéciation des AOX ... 77
II.5. Conclusion………..….81
CHAPITRE III : CINETIQUE DE CHLORATION III.1. Introduction ... 83
III.2. Matériels et Méthodes ... 84
III.2.1. Préparation des solutions ... 84
III.2.2. Dispositif expérimental ... 84
III.2.3. Méthodes analytiques ... 85
III.2.3.1. Dosage de l’oxydant total par la méthode I3- ... 85
III.2.3.2. Détermination des constantes d’acidité des aminophénols par spectrophotométrie ... 85
III.3. Détermination des constantes d’acidité des aminophénols ... 87
III.3.1. Spectres d’absorption UV ... 87
III.3.2. Calcul des constantes d’acidité ... 91
III.4.3. Cinétique de chloration du 2-aminophénol ... 97
III.4.4. Modélisation ... 100
III.4.4.1. Hypothèses des réactions élémentaires ... 100
III.4.4.2. Modélisation de kapp2 en fonction du pH ... 102
III.5. Conclusion ... 110
CHAPITRE IV : CINETIQUE DE MONOCHLORAMINATION IV.1. Introduction ... 111
IV.2. Matériels et Méthodes ... 112
IV.2.1. Préparation des solutions ... 112
IV.2.2. Dispositif expérimental ... 112
IV.2.3. Méthodes analytiques ... 113
IV.2.3.1. Dosage de la monochloramine ... 113
IV.2.3.2. Dosage des aminophénols ... 113
IV.3. Monochloramination en réacteur fermé ... 114
IV.3.1. Consommation en monochloramine ... 114
IV.3.1.1. Expériences à faible taux de monochloramination ... 114
IV.3.1.2. Expériences en excès d’oxydant ... 115
IV.3.2. Suivi du 3-aminophénol et de ses premiers sous-produits ... 116
IV.3.2.1. Suivi de la dégradation du 3AP ... 116
IV.3.2.2. Formation des premiers sous-produits ... 116
IV.3.2.3. Influence du rapport molaire N/Cl ... 120
IV.4. Etude cinétique ... 121
IV.4.1. Détermination des constantes de vitesse ... 121
IV.4.2. Cinétique de monochloramination du 3-aminophénol ... 122
IV.4.2.1. Cinétique à pH = 8,6 ... 122
IV.4.2.2. Influence du rapport molaire N/Cl ... 123
IV.4.2.3. Influence du pH ... 124
IV.4.2.4. Modélisation ... 127
IV.4.2.4.1. Hypothèses des réactions élémentaires ... 127
IV.4.2.4.2. Modélisation de kapp2 en fonction du pH ... 129
IV.4.3. Cinétique de monochloramination du 3-amino-4-chlorophénol ... 133
IV.5. Conclusion ... 135
CHAPITRE V : IDENTIFICATION DES SOUS-PRODUITS V.1. Introduction ... 137
V.2. Matériels et Méthodes ... 138
V.2.3.1. Spectres d’absorption UV ... 139
V.2.3.2. Dosage des AOX ... 139
V.2.3.3. Analyse par HPLC/PDA ... 139
V.2.3.4. Analyse par LC/MS ... 140
V.3. Sous-produits du 3-aminophénol ... 141
V.3.1. Monochloramination du 3-aminophénol ... 141
V.3.1.1. Effet du pH sur la formation des 3-aminochlorophénols ... 141
V.3.1.2. Identification de sous-produits par LC/MS ... 143
V.3.2. Chloration du 3-aminophénol ... 150
V.4. Sous-produits du 2-aminophénol ... 152
V.4.1. Stabilité du 2-aminophénol en milieu aqueux ... 152
V.4.2. Monochloramination du 2-aminophénol ... 155
V.4.2.1. Suivi du 2-aminophénol ... 155
V.4.2.2. Suivi de l’APX formé ... 156
V.4.2.3. Effet de l’oxygène dissous ... 160
V.4.2.4. Suivi des AOX ... 161
V.4.2.5. Identification de sous-produits par LC/MS ... 162
V.4.3. Chloration du 2-aminophénol ... 166
V.5. Conclusion ... 167
CONCLUSION GENERALE………..168
REFERENCES BIBLIOGRAPHIQUES………171
Introduction générale
L’eau est une ressource riche et essentielle à la vie. L’amélioration et la préservation de la qualité de la ressource en eau est une préoccupation constante qui se traduit par l’établissement de normes de plus en plus sévères vis-à-vis des rejets dans le milieu naturel. Néanmoins, l’usage de l’eau pour la consommation humaine ou pour des applications industrielles nécessite la mise en place de filières de traitement permettant de répondre aux exigences de la réglementation.
Parmi les différentes étapes de traitement, la désinfection de l’eau permet de lutter contre les microorganismes pathogènes et donc les maladies d’origine hydrique. Les traitements de désinfection ne sont pas uniquement réservés à l'eau de consommation, mais trouvent également des applications dans les eaux de process, les eaux de circuits de refroidissement ou les eaux récréatives comme les piscines.
La chloration est la méthode de désinfection la plus largement appliquée au monde car le chlore est un désinfectant très efficace. Cependant, en présence de composés organiques, comme la matière organique naturelle de l’eau, le chlore réagit pour former des sous-produits de désinfection indésirables, voire toxiques. Ces sous-produits sont majoritairement halogénés et si les plus connus sont les trihalométhanes (THM), plus de 600 sous-produits ont maintenant été identifiés dans les eaux de distribution chlorées.
Afin de réduire la production de sous-produits de désinfection présentant un risque pour la santé humaine, de nombreux pays anglo-saxons (Etats-Unis, Canada, Australie…) ont adopté la monochloramine (NH2Cl) comme alternative à la désinfection au chlore dans les usines de
production d’eau potable. En France, le traitement des eaux potables par la monochloramine n’est pas autorisé. Cependant, ce biocide est couramment utilisé dans les circuits de refroidissement des centrales nucléaires. Ces circuits semi-fermés sont en effet propices au développement de biofilms et à la prolifération de bactéries pathogènes, ce qui nécessite une désinfection.
La monochloramine est produite à partir de la réaction entre le chlore libre et l’azote ammoniacal. Elle peut être formée in situ (c'est-à-dire par ajout d’ammoniac dans l’eau à traiter suivi d’une chloration) ou préformée (par la production de monochloramine dans un bassin tampon puis utilisation du désinfectant formé).
Moins réactive avec les composés organiques, la monochloramine présente l’avantage par rapport au chlore de permettre le maintien d’un résiduel en oxydant beaucoup plus stable dans les réseaux de distribution et de générer moins de sous-produits indésirables comme les trihalométhanes.
Contrairement au cas du chlore, la réactivité de la monochloramine avec des composés organiques modèles n’a fait l’objet que d’un nombre restreint de travaux. Heasley et al. (1λλλ et 2004) et récemment Cimetière (2009) ont étudié la monochloramination de phénols et de dihydroxybenzènes. Les composés phénoliques sont fréquemment sélectionnés dans les
études d’oxydation en tant que modèles de la structure de la matière organique naturelle ou de micropolluants organiques.
Dans le cadre de ces travaux, les aminophénols ont été choisis comme composés modèles. Les aminophénols sont des intermédiaires dans la fabrication de nombreux produits dans des secteurs industriels variés : industries de teinture (résines, vêtements, fourrures, colorants pour cheveux), chimiques (synthèse des composés hétérocycliques et des pesticides) et pharmaceutiques (fabrication de médicaments).
Ces travaux ont eu pour objectif principal d’étudier la chloration et la monochloramination d’aminophénols. Cette thèse est divisée en cinq chapitres.
Le chapitre I présente une synthèse des données bibliographiques relatives aux aminophénols et aux procédés de chloration et de monochloramination, en portant une attention particulière aux aspects cinétiques et aux sous-produits générés.
Dans le chapitre II, sont exposés les demandes en oxydants de quelques aminophénols et leurs potentiels de formation par chloration et monochloramination de sous-produits tels que les composés organohalogénés adsorbables (AOX), le chloroforme, les acides haloacétiques. Les chapitres III et IV traitent des aspects cinétiques respectivement de la chloration des 2-aminophénol et 3-2-aminophénol et de la monochloramination du 3-2-aminophénol.
Enfin, quelques sous-produits identifiés lors de la monochloramination des 2-aminophénol et 3-aminophénol sont décrits dans le chapitre V.
CHAPITRE I
Chapitre I
Synthèse bibliographique
I.1- Aminophénols ... 3
I.1.1- Présentation ... 3
I.1.2- Propriétés physiques ... 3
I.1.3- Propriétés chimiques et réactivité ... 4
I.1.4- Utilisations industrielles et domestiques ... 8
I.1.5- Présence dans les eaux ou les sols et risques sanitaires ... 11
I.1.6- Analyse ... 14
I.2- Chlore ... 15
I.2.1- Propriétés physiques du chlore ... 15
I.2.2- Chimie du chlore dans l’eau ... 16
I.2.3- Réactivité du chlore sur les composés minéraux ... 17
I.2.3.1- Oxydation de l’azote ammoniacal ... 18
I.2.3.2- Oxydation des halogénures ... 19
I.2.3.3- Oxydation des nitrites et des sulfites ... 19
I.2.4- Réactivité du chlore sur les composés organiques ... 21
I.2.4.1- Oxydation des composés organiques azotés ... 21
I.2.4.1.1- Amines aliphatiques ... 21
I.2.4.1.2- Aniline ... 24
I.2.4.2- Oxydation des hydroxybenzènes ... 25
I.2.4.2.1- Phénol ... 25
I.2.4.2.2- Résorcinol ... 29
I.3- Monochloramine ... 32
I.3.1- Formation et utilisation des chloramines ... 32
I.3.2- Modélisation cinétique de l’auto-décomposition de la monochloramine ... 34
I.3.3- Réactivité de la monochloramine sur les composés minéraux ... 36
I.3.3.1- Effet des ions nitrite ... 36
I.3.3.2- Effet des ions bromure ... 37
I.3.3.3- Effet des ions carbonate ... 37
I.3.3.4- Effet des ions phosphate ... 38
I.3.3.5- Effet du fer ... 38
I.3.3.6- Effet du cuivre ... 39
I.3.4- Réactivité de la monochloramine sur les composés organiques ... 39
I.3.4.1- Oxydation des composés organiques azotés ... 39
I.3.4.2- Oxydation des hydroxybenzènes ... 42
I.3.4.2.1- Phénol ... 42
Chapitre I
Synthèse bibliographique
I.1- Aminophénols
I.1.1- Présentation
Les aminophénols (AP), de formule brute C6H7NO, sont des composés organiques
aromatiques qui contiennent deux groupements fonctionnels liés au noyau aromatique, un groupement hydroxyle -OH et un groupement amine -NH2. Ces composés sont également
nommés hydroxyaniline ou amino-hydroxybenzène.
En fonction de la position du groupement amine par rapport au groupement hydroxyle, les aminophénols sont classés sous trois formes d’isomères : en ortho, le 2-aminophénol ou 2AP (1), en méta, le 3-aminophénol ou 3AP (2) et en para, le 4-aminophénol ou 4AP (3) (figure I.1).
Figure I.1 : Différents isomères d’aminophénols selon la position
des groupements –OH et –NH2 sur le cycle aromatique.
I.1.2- Propriétés physiques
A température ambiante, les aminophénols sont des composés solides cristallisés. En milieu aqueux, la solution du 3-aminophénol (3AP) est plus stable que celles du 2-aminophénol (2AP) et du 4-aminophénol (4AP). Ces deux derniers s’oxydent à l’air pour former des sous-produits colorés. A pH acide, les aminophénols sont tous fluorescents (Mitchell et al., 2003).
Le tableau I.1 ci-dessous présente les caractéristiques physiques générales de ces composés citées par Weast (1981).
Tableau I.1: Caractéristiques physiques des aminophénols, selon Weast (1981).
Composé AP N° de CAS Masse molaire (g/mol) Solubilité (g/L) T d’ébullition (°C) T de fusion (°C) 2AP 95-55-6 109,13
Soluble dans l’eau, l’alcool et l’éther. Sa solubilité dans l’eau
est de 17 g/L (20°C)
<153
à 1,47 kPa 174
3AP 591-27-5 109,13
Soluble dans l’eau, l’alcool et l’éther. Sa solubilité dans l’eau
est de 20,1 ± 2,9 g/L (20°C)
164
à 1,47 kPa 123
4AP 123-30-8 109,13
Peu soluble dans l’eau, soluble dans l’alcool. Sa solubilité dans l’eau
est de 6,5 g/L (25°C) 174 à 1,47 kPa 284 à 101,3 kPa 186-7
I.1.3- Propriétés chimiques et réactivité
Le groupement -NH2 sur le cycle aromatique des aminophénols leur confèrent des propriétés
différentes des phénols. Ainsi, ce sont des ampholytes qui se comportent à la fois comme un acide et une base.
Les AP sont des molécules non chargées à pH neutre. Cependant à des pH plus éloignés de la neutralité, les aminophénols cèdent ou gagnent un proton H+ pour devenir des molécules chargées positivement à pH acide (ammonium -NH3+) et négativement à pH basique
Figure I.2 : Ionisation des molécules du 3AP en fonction du pH.
Le tableau I.2 rapporte les différentes valeurs de constantes d’acidité de la littérature relevées par Mitchell et al. (2003). De façon générale, l’acidité de la fonction hydroxyle et le caractère basique de l’amine sont modifiés respectivement par la présence du groupement amine (pour le phénol, pKa = 9,95 à 25°C (Jencks et Regenstein, 2010) ou du groupement hydroxyle (pour l’aniline, pKa = 4,62 à 25°C (Jencks et Regenstein, 2010). Ce phénomène est plus marqué pour le 4AP.
Tableau I.2 : Constantes d’acidité des isomères d’aminophénols dans l’eau citées par
Mitchell et al. (2003).
Composé AP pKa1 pKa2 Température
(°C) 2AP 9,66 15 4,72 21 9,71 22 4,66* 25 3AP 4,17 21 9,87 22 4,31* 25 4AP 5,5 21 10,30 22 5,48* 25 4,86 10,60 30
*Avec 1 % d’alcool éthylique dans l’eau
Les aminophénols sont très réactifs chimiquement et peuvent subir des réactions faisant intervenir le groupement amine, le groupement hydroxyle phénolique, ainsi que des réactions de substitution sur le noyau aromatique. Les groupements amine et hydroxyle sont donneurs d'électrons par effet mésomère, effet qui prédomine sur l’effet attracteur d'électrons par effet inductif. A ce titre, les réactions de substitutions électrophiles sont favorisées et orientées en positions ortho et para des groupements.
Les AP peuvent ainsi intervenir dans des réactions d’alkylation, d’acylation pour former une grande variété de dérivés utilisables dans divers secteurs de l’industrie.
Le groupement amine peut être converti en sel de diazonium par le nitrite de sodium en milieu acide, ces dérivés diazoïques trouvant une utilisation étendue dans l'industrie des colorants (Mitchell et al., 2003).
Le 2AP est particulièrement sensible aux réactions de condensation et de cyclisation en raison de la proximité des groupements -OH et -NH2 sur le cycle aromatique. Un milieu oxydant non
spécifique conduit facilement à la formation de structure de type phénoxazone (telle que la 2-amino-3H-phénoxazine-3-one ou APX) tandis qu’une oxydation plus poussée donne une structure pentacyclique de type triphénodioxazine (Mitchell et al., 2003).
N O O NH2 N O O N 2-amino-3H-phénoxazine-3-one Triphénodioxazine
Une étude spectro-électrochimique réalisée par Salavagione et al. (2004) en milieu très acide (1 M de HClO4) a proposé un mécanisme d’oxydation des aminophénols sur électrode de
platine (figure I.3). L’oxydation par voie électrochimique du 4AP aboutit à la formation de la 1,4-benzoquinone-imine, puis par hydrolyse lente à la 1,4-benzoquinone, NH4+ et CO2 étant
également détectés. Les sous-produits d’oxydation du 3AP sont soit, des polymères réticulés, formés à partir du dimère A, soit des polyéthers linéaires formés à partir du dimère B. Un polymère, de structure constituée d’unités phénoxazine, est quant à lui identifié comme sous-produits de l’oxydation du 2AP.
Figure I.3 : Mécanismes proposés pour l’oxydation électrochimique des aminophénols
à pH très acide selon Salavagione et al. (2004).
Plusieurs études ont été menées sur l’oxydation du 2AP par le dioxygène en solution aqueuse. Le 2AP dans l’eau peut s’oxyder en présence du dioxygène en 2-amino-3H-phénoxazine-3-one (APX) (Prati et al., 1992). Hassanein et al. (2008) ont trouvé que 70 % d’APX se forment après 2 heures d’alimentation du milieu en dioxygène gazeux (pression 1 atm et à 40°C). Cette oxydation atteint son maximum à pH = 9. Deux molécules de 2AP interviennent pour la formation de l’APX (figure I.4).
A
Figure I.4 : Mécanisme de formation d’APX à partir de l’oxydation du 2AP par le dioxygène
selon Hassanein et al. (2008).
L’APX formé absorbe dans le visible à une longueur d’onde maximale de 434 nm (ou 432 nm) avec un coefficient d’extinction molaire ε de 23 200 cm-1.M-1 (Puiu et Oancea, 2004 ; Szigyártό et al., 2006). Gagliardo et Chilton (1992) ont trouvé que les longueurs d’onde maximales dans l’UV-Visible de l’APX sont 432 et 236 nm avec un coefficient d’extinction molaire ε respectivement de 26 000 et 31 000 cm-1.M-1.
L’APX a fait l'objet de nombreuses études en raison de ses propriétés thérapeutiques : il est également connu sous le nom Questiomycin A, antibiotique naturel secrété par Penicillium (Lechevalier, 1975). Les applications de l’APX et autres phénoxazines sont actuellement très étudiées dans le domaine médical car ces composés ont des propriétés antinéoplasiques, inhibant la prolifération des cellules tumorales (Tomoda et al., 1986 ; Szigyártό et al., 2006 ; Miyake et al., 2010).
Hashemi et Beni (1λλλ) ont utilisé une résine échangeuse d’anions Dowex IX8-200, régénérée par une solution d’hypochlorite de sodium pour réaliser l’oxydation d’une série d’amines aromatiques en quinones. Le 2AP et le 4AP ont conduit quantitativement, dans leurs conditions (solvant acétate d’éthyle pour le 2AP, 1,2-diméthoxyéthane pour le 4AP), respectivement aux 1,2-benzoquinone (91 %) et 1,4-benzoquinone (97 %).
I.1.4- Utilisations industrielles et domestiques
Les aminophénols sont des intermédiaires de fabrication de nombreux produits industriels et se retrouvent ainsi dans des produits finis à usage domestique. Les aminophénols sont en effet utilisés dans les industries de teinture (résines, vêtements, fourrures, colorants pour cheveux),
chimiques (synthèse des composés hétérocycliques et des pesticides) et pharmaceutiques (fabrication de médicaments).
Le 2AP et le 4AP sont des agents réducteurs utilisés pour le développement des films photographiques noirs et blancs (Salavagione et al., 2004) sous les noms commerciaux tels que Atomal, Ortol (2AP), Activol, Paranol (4AP) (Mitchell et al., 2003). Ce sont aussi des additifs employés comme agents anti-corrosion et lubrifiants dans des carburants et comme inhibiteur de corrosion dans les peintures (Michalowicz et Duda, 2007).
Les aminophénols sont employés pour synthétiser des résines chélatantes. Kumar et al. (2000) ont ainsi greffé le 2AP sur une résine XAD-2 pour séparer sélectivement les métaux (cuivre, cobalt, cadmium, nickel, zinc) et ainsi permettre leur analyse à l’état de traces dans l’eau. Les aminophénols sont également utilisés en pharmacie (Mitchell et al., 2001 ; Falcone et al., 2011). Le 4AP entre dans la fabrication du paracétamol (acétylaminophénol). L'acide 4-aminosalicylique, produit par carboxylation du 3AP fut l’un des premiers antibiotiques employés pour traiter la tuberculose en association avec la streptomycine (Jain et al., 2008). Le 2AP, par la présence adjacente des groupements amine et hydroxyle, forme facilement des hétérocycles biologiquement actifs, tels que le benzoxazole, dont des dérivés sont utilisés comme antalgiques et anti-inflammatoires.
Les aminophénols sont employés dans la coloration permanente (ou oxydante) des cheveux, soit comme molécule primaire (base d’oxydation), soit couplés à d’autres molécules de synthèse (coupleur) (Nohyneck et al., 2004). Les bases sont des molécules substituées en position ortho ou para, comme par exemple l’ortho-phénylènediamine, la para-phénylènediamine (PPD) ou le 4AP ; ces molécules se retrouvent dans les formulations de colorant pour cheveux dans une gamme comprise entre 0,05 % (tons clairs) à 2,0 % (teinte foncée). Les coupleurs sont eux des molécules substituées en position méta comme la méta-phénylènediamine, le résorcinol ou le 3AP ; ils déterminent la nuance finale, par réaction avec la forme oxydée des molécules primaires, suivie par d'autres réactions de couplage oxydant. Le processus de coloration fait intervenir un oxydant, qui peut être le peroxyde d’hydrogène, en milieu légèrement basique (ammoniaque). Le 3-aminophénol se trouve généralement à une concentration maximale de 2,4 % dans la formulation des colorants de cheveux, et 1,2 % lorsqu’il est mélangé avec du peroxyde d’hydrogène (rapport 1:1).
Figure I.5 : Voies de formation de colorants par oxydation de la PPD,
Nohyneck et al. (2010) d’après Spengler et Bracher (1990).
La figure I.5 présente l’exemple de l’oxydation par H2O2 de la PPD (base) en présence de
résorcinol (coupleur). La réaction va conduire dans un premier temps par oxydation de la para-phénylènediamine à la formation de para-benzoquinone-diimine. La voie de réaction (A) en présence du coupleur (résorcinol) conduit alors à la formation d’un leucocolorant (molécule possédant une forme colorée et une forme incolore) puis aux colorants souhaités dimères et trimères (indoanilines). La voie de réaction hypothétique (B) peut se produire en l'absence de coupleurs, et conduit à une base de Bandrowski.
De façon analogue, l’oxydation du 4AP en présence d’eau oxygénée va aboutir à la formation de la 1,4-benzoquinone-imine. Cette dernière va réagir avec des coupleurs (m-phénylènediamine, 3AP ou résorcinol) pour former des leuco-colorants puis des composés de type indoaniline (Brown et Corbett, 1979b).
Le tableau I.3 présente les couleurs obtenues de l’oxydation du 4AP et ses coupleurs par le peroxyde d’hydrogène.
Tableau I.3 : Couleurs obtenues lors de l’oxydation du 4AP par le peroxyde d’hydrogène en
absence et présence des coupleurs (Tucker, 1967 et Brown et Corbett, 1979b).
Couple Couleur des cheveux
4AP + H2O2 Doré-marron
4AP + m-phénylènediamine + H2O2 Marron-violet
4AP + 2,4-diaminoanisole + H2O2 Rouge-violet
4AP + 3AP + H2O2 Marron
4AP + 5-amino-o-crésol + H2O2 Rouge
4AP + Résorcinol + H2O2 Gris clair
4AP + 4-amino-2-hydroxytoluène + H2O2 Rouge-orange
I.1.5- Présence dans les eaux ou les sols et risques sanitaires
Les aminophénols sont des polluants industriels pouvant être libérés à partir des eaux usées dans les eaux de surface. Ils sont toxiques pour l’homme, les animaux et les plantes. Ce sont des composés absorbés facilement par la peau et les muqueuses en causant des effets mutagènes, génotoxiques, hépatotoxiques et néphrotoxiques (Huang et al., 2009).
La toxicité des phénols chlorés augmente avec le nombre d’atomes de chlore sur le cycle aromatique (Kishino et Kobayashi, 1995). La transformation des AP en dérivés chlorés lors de la chloration de l’eau pourrait de façon similaire augmenter leur toxicité.
En ce qui concerne la toxicité aiguë, le 4AP entraine des irritations de la peau et des yeux, de l’eczéma, de l’asthme, et des anoxies. Cette toxicité est liée d’une part à la formation des quinonimines et d’autre part, aux radicaux superoxydes générés qui endommagent la cellule (Yoshida et al., 1998 ; Michałowicz et Duda, 2007). Ainsi, par l’intermédiaire des quinonimines formées, le 4-AP altère les membranes cellulaires et présente une néphrotoxicité pour des doses de 200 mg/kg de poids du corps (Kanbak et al., 1996). Les doses létales du 4AP pour l’homme sont estimées autour de 50 à 500 mg/kg de poids du corps (Michałowicz et Duda, 2007). Le 4AP peut aboutir à une inflammation chronique du colon (Pedersen et al., 2002).
Les colorations pour cheveux, contenant des amines aromatiques, sont susceptibles de provoquer des allergies mais sont également suspectées d’induire des cancers de la vessie (Irigaray et al., 2007 ; Kane et al., 1984). Cependant, la relation entre l'utilisation de colorations pour cheveux et cancer de la vessie n’est pas clairement établie. Ainsi, une méta-analyse réalisée par Huncharek et Kupelnick (2005) portant sur la période 1966 à 2003 indique que l'utilisation personnelle des produits de colorations capillaires augmente le risque de cancer de la vessie de 22 % à 50 % par rapport à la non-utilisation. D’autres membres de la communauté scientifique ne confirment néanmoins pas ces résultats (Nohynek et al., 2004 ; Bolt et Golka, 2007). Très récemment, Lewis et al. (2013) se sont interrogés sur la possibilité de former des nitrosamines à partir des dérivés trimères indoanilines possédant un groupement amine secondaire (figure I.5).
2AP
Puiu et ses collaborateurs (Puiu et Oancea, 2004 ; Puiu et al., 2007 ; Puiu et al., 2008) se sont intéressés à l’oxydation du 2AP comme modèle de polluant phénolique souvent libéré dans les eaux usées industrielles. L’oxydation du 2AP en présence de l’oxygène dissous aboutit à la formation de l’APX qui est un sous-produit non toxique formé naturellement. Cette réaction peut être catalysée par les métaux comme le cuivre (II) (Puiu et Oancea, 2004 ; Puiu et al., 2007). D’autres études faites sur l’oxydation du 2AP par le dioxygène catalysée par la peroxydase ont montré que l’APX formé est stable. Cette oxydation peut être développée sur des eaux usées pour transformer le 2AP en APX (Puiu et al., 2008).
L’incorporation des substances allélopathiques dans la gestion de l’agriculture joue un rôle d’interaction biochimique d’une plante sur une autre (ou sur un microorganisme) et peut réduire l’utilisation des pesticides. Ainsi, les benzoxazolinones et les benzoxazinones, utilisés dans la culture du blé, du seigle et du maïs peuvent se transformer dans le sol en APX en passant par une oxydation du 2AP à l’air en présence de microorganismes (Gents et al., 2005 ; Fomsgaard et al., 2004 ; Zikmundová et al., 2002 ; Friebe et al., 1998 ; Friebe et al., 1996 ; Kumar et al., 1993 ; Gagliardo et Chilton, 1992).
3AP
Dans l’environnement, le 3AP a été détecté comme sous-produits de dégradation du phenmédiphame et du desmédiphame, des herbicides de la famille des phényl-carbamates épandus sur les sols pour léser les plantules à la levée. Ces pesticides ont été utilisés souvent dans la culture de la betterave à sucre et des fraises (Schettgen et al., 2001 ; U.S. EPA, 1996 ; Sonowane et al., 1971).
Le phenmédiphame est décomposé dans les sols légèrement acides, de faible teneur en humus, avec un temps de demi-vie de 28 à 55 jours en fonction des conditions expérimentales et du type de sol. Le phenmédiphame est hydrolysé en 3AP via le méthyl-N-(3-hydroxyphényl) carbamate (MHPC). Les auteurs suggèrent la possibilité de former un complexe humus-3AP (Sonawane et Knowles, 1971).
Dans un avis de l’Agence Française de Sécurité Sanitaire des Aliments (AFSSA, 2009) relatif à une demande d'autorisation de mise sur le marché d’une préparation commerciale à base de phenmédiphame, la concentration prévisible maximale dans les sols de la molécule mère et de ses principaux métabolites a pu être estimée en considérant notamment les paramètres suivants : temps de demi-vie de 43 jours pour le phenmédiphame, pourcentage maximal de métabolite formé par dégradation du phenmédiphame de 54 % pour le méthyl-(3-hydroxy-phényl)carbamate (MHPC) et de 17,8 % pour le 3-aminophénol. Ainsi, pour une application de 960 g substance active/ha/an, les concentrations prévisibles dans les sols seraient de 0,31 et 0,066 mg/kg sol, respectivement pour le MHCP et le 3AP.
Le 3AP a également été détecté comme métabolite majeur du pesticide phenmédiphame dans l’urine des rats (Schettgen et al., 2001) (figure I.6).
Figure I.6 : Métabolisation chez le rat du pesticide phenmédiphame.
4AP
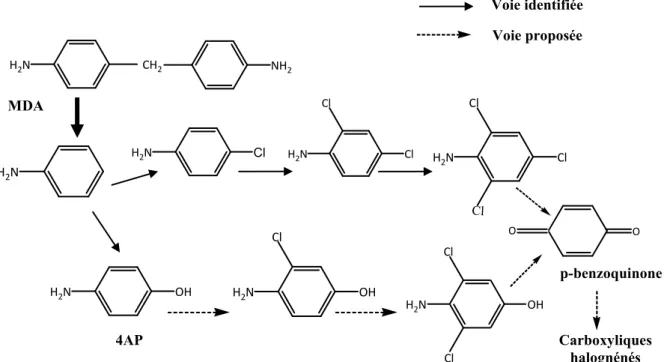
Tomiyama et al. (2008) se sont intéressés à la réaction du chlore sur la 4,4’-méthylènedianiline (MDA). Ce composé a été utilisé jusqu’en 1λ86 comme durcisseur pour résine époxy (revêtement intérieur de canalisations de distribution d’eau potable), avant d’être classé cancérigène et mutagène. Tomiyama et al. (2008) ont montré que dans les conditions de chloration rencontrées en réseau d’eau potable, la réaction du chlore est rapide et conduit à la formation de p-(chlorométhyl)aniline, p-aminobenzaldéhyde, p-méthoxyaniline, chloroanilines (4-chloroaniline, 2,4-dichloroaniline et 2,4,6- trichloroaniline), et 4AP. Tomiyama et al. (2008) suggèrent la formation du p-benzoquinone à partir de la MDA en passant par l’intermédiaire du 4AP (figure I.7).
Figure I.7 : Une voie d’oxydation de la MDA par le chlore en solution aqueuse tamponnée
proposée par Tomiyama et al. (2008).
Le paracétamol, (4-acétylaminophénol ou N-acétyl-para-aminophénol), est un médicament antalgique qui peut exister à faible teneur dans les eaux usées et les eaux naturelles. Kasprzyk-Hordern et al. ont ainsi mis en évidence des concentrations en paracétamol jusqu’à 1 µg/L dans des eaux de surface britanniques (Kasprzyk-Hordern et al., 2008). Sa chloration en solvant organique aboutit à la formation du N-acétyl-2,3,6-trichloro-4-aminophénol et de la N-acétyl-2,3,5,6-tétrachloro-1,4-benzoquinone imine (Avdeenko et Marchenko, 2001). Le 4AP est un métabolite actif du paracétamol retenu dans la « liste des médicaments humains et de leurs métabolites actifs prioritaires à rechercher dans les eaux » préconisée par l’Agence Française de Sécurité de Aliments (AFSSA, 2008).
I.1.6- Analyse
La spéciation et l’analyse des isomères d’aminophénols peuvent se faire par des méthodes spectrophotométriques, chromatographiques, électrochimiques et par électrophorèse capillaire. La limite de détection varie selon chaque méthode de détection et d’analyse.
Les aminophénols sont analysables par spectrophotométrie à des longueurs d’onde de 220, 254 et 275 nm (Mitchell et al., 2003).
Su et al., (2011) ont développé une méthode électrochimique à l’aide d'électrodes de carbone de type sérigraphié pour la détermination des aminophénols dans les eaux naturelles. Par dopage d’eaux de rivières (entre 30 et 70 µM en AP), ils ont pu analyser simultanément les trois isomères avec de bons rendements de recouvrement (proches de 100 %). Ces auteurs suggèrent l’application possible de leur technique pour la surveillance des polluants
H2N OH Cl Cl MDA Carboxyliques halognénés H2N H2N Cl H2N Cl Cl H2N Cl Cl Cl H2N OH H2N OH Cl O O 4AP H2N CH2 NH2 p-benzoquinone Voie proposée Voie identifiée
Su et al., (2011) ont comparé les différentes méthodes d’analyse des aminophénols et leurs limites de détection citées dans la littérature (tableau I.4).
Tableau I.4 : Comparaison des différentes méthodes d’analyse des aminophénols et leurs
limites de détection (Su et al., 2011). Méthode d’analyse Composés Limite de détection
(µM) Références
Chromatographie gazeuse
2AP 23,45 Tanada et al. (1994)
Kumar et Panwar (1993) 3AP 29,95 4AP 11,82 Chromatographie liquide 2AP 4,2 Wang et Huang (2005) 3AP 2,7 4AP 1,6 Electrophorèse capillaire 2AP 0,75 Yang et al. (2001) 3AP 2,64 4AP 1,16 Spectrophotométrie
2AP - Lopez-Cueto et al.
(1996) Filik et Taveman (2006) 3AP - 4AP 1,83 Electrochimie
2AP 0,04-0,05 Wang et al. (2009)
Su et al. (2011) Duan et al. (2013)
3AP 0,03-0,05
4AP 0,06-0,02
I.2- Chlore
I.2.1- Propriétés physiques du chlore
Le désinfectant chlore peut exister sous forme gazeuse (Cl2), liquide (hypochlorite de sodium
NaOCl) ou solide (hypochlorite de calcium, Ca(OCl)2) (tableau I.5).
Le chlore gazeux et l’hypochlorite de sodium sont fréquemment appliqués en désinfection des eaux potables et des eaux de piscines.
L’hypochlorite de calcium solide est aussi destiné à désinfecter des eaux de piscines et est utilisé comme un agent décolorant dans les industries.
Tableau I.5 : Propriétés physiques du chlore. Chlore Etat physique Formule chimique Couleur Température de fusion (°C) Température d’ébullition (°C) Dichlore Gazeux Cl2 Jaune verdâtre -101 -34,6 Hypochlorite
de sodium Liquide NaOCl Jaune clair 18 111
Hypochlorite
de calcium Solide Ca(OCl)2 Blanc-gris 100 175
I.2.2-
Chimie du chlore dans l’eau
Lors du traitement des eaux, le chlore gazeux (Cl2) est souvent utilisé pour la désinfection.
Dans l’eau, le chlore gazeux s’hydrolyse très rapidement en acide hypochloreux (HOCl) et en ions chlorure (Cl-) (Connick et Chia, 1959) :
Cl2 + H2O → HOCl + Cl- + H+ k1 = 22,3 s-1
HOCl + Cl- + H+→ Cl2 + H2O k-1 = 4,3.104 M2.s-1
où k1 et k-1 sont calculées selon Wang et Margerum (1994) à = 0 M et à 25°C.
Ainsi, la constante d’équilibre KCl2 de la réaction de dismutation du chloreest comprise entre
1,3.10-4 M2 à 0°C et 5,1.10-4 M2 à 25oC (Wang et Margerum, 1994). Réaction de dismutation du chlore :
Cl2 + H2O ⇌ HOCl + Cl- + H+ KCl2= k1/k-1 = 1,3.10
-4
à 5,1.10-4 M2 (de 0 à 25oC) L’acide hypochloreux est un acide faible qui se dissocie dans l’eau en ion hypochlorite. HOCl ⇌ ClO- + H+ KHOCl = 1,5.10-8 à 2,9.10-8 (de 0 à 25oC) (Morris, 1966)
Le pKa (HOCl/ClO-) est ainsi de 7,82 et 7,54 respectivement à 0°C et 25°C (Morris, 1966). Dans les conditions habituelles de chloration de l’eau potable (6,5 ≤ pH ≤ 8,5), les espèces HOCl et ClO- sont majoritaires dans le milieu. La figure I.8 représente la distribution des différentes espèces du chlore en fonction du pH pour une concentration en ions chlorure de 5.10-3 M et à 25°C (Deborde et von Gunten, 2008).
Figure I.8: Distribution des espèces de chlore en solution aqueuse
en fonction du pH (25°C ; [Cl-] = 5.10-3 M).
Dans certaines conditions, le chlore peut former l’ion trichlorure (Cl3-) (Zimmerman et
Strong, 1957), l’hémioxyde de chlore (Cl2O) et l’ion H2OCl+ (Arotsky et Symons, 1962 ;
Reinhard et Stumm, 1980, Cherney et al., 2006).
L’ion H2OCl+ pourrait être présent à des pH inférieurs à 4 et responsable des réactions du
chlore catalysées en milieu acide. Il est reconnu plus réactif que l’acide hypochloreux mais sa forme constitue une fraction négligeable du chlore total dans les conditions habituelles de chloration de l’eau.
Cl2 + Cl-⇌ Cl3- KCl3− = 1,91.10
-1
(25oC) Zimmermann et Strong, 1957 2HOCl ⇌ Cl2O+ H2O KCl2O = 8,7.10-3 (25°C) Reinhard et Stumm, 1980
H2OCl+ + H2O ⇌ HOCl + H3O+ KH2OCl+= 10
-3
- 10-4 (25oC) Arotsky et Symons, 1962
Le chlore total dans l’eau englobe la totalité du chlore libre (Cl2, HOCl et ClO-, H2OCl+, Cl3-,
Cl2O) et du chlore combiné (chloramines minérales et organiques).
I.2.3- Réactivité du chlore sur les composés minéraux
Le chlore réagit sur les composés inorganiques dissous dans l’eau comme l’azote ammoniacal, les ions bromure et iodure, les ions métalliques et d’autres. La consommation rapide du chlore par ces derniers peut réduire le chlore résiduel nécessaire à maintenir pour la désinfection dans les réseaux de distribution d’eau potable.
I.2.3.1- Oxydation de l’azote ammoniacal
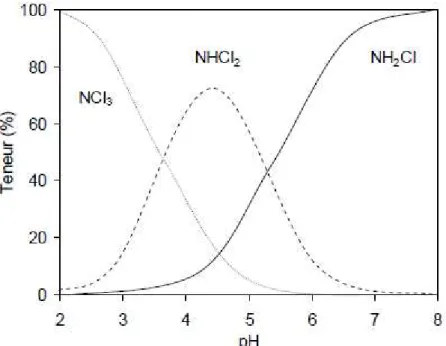
Dans l’eau, le chlore oxyde l’azote ammoniacal en diazote et ions nitrate par une suite de réactions qui mènent en premier temps à la formation de chloramines inorganiques. Les chloramines inorganiques sont la monochloramine (NH2Cl), la dichloramine (NHCl2) et la
trichloramine (NCl3).
Les quantités relatives de chacune de ces chloramines inorganiques sont déterminées par le pH de l’eau, les concentrations relatives du chlore et de l’azote ammoniacal, le temps de réaction et la température.
NH3+ HOCl → NH2Cl + H2O k = 1,5.1010 M-1.h-1 (25oC) Morris et Isaac (1983)
NH2Cl + H2O → NH3 + HOCl k = 7,6.10-2 h-1 (25oC) Morris et Isaac (1983)
NH2Cl + HOCl → NHCl2 + H2O k = 1,0.106 M-1.h-1 (25oC) Margerum et al. (1978)
NHCl2 + H2O → NH2Cl + HOCl k = 2,7.10-3 h-1 (25oC) Margerum et al. (1978)
NHCl2+ HOCl → NCl3 + H2O k = 7,6.103 M-1.h-1 (25oC) Morris et Isaac (1983)
NCl3 + H2O → NHCl2 + HOCl k = 0,16 h-1 (25oC) Morris et Isaac (1983)
Figure I.9 : Courbe de break point d’une solution avec [NH4+] = 1 mg N.L-1.
Lorsque le chlore est ajouté à une eau contenant de l’azote ammoniacal, il est consommé instantanément pour former le chlore combiné (figure I.9).
Ainsi, au pH des eaux naturelles, et pour des rapports molaires N/Cl supérieurs à 1, la monochloramine est formée.
Pour des rapports molaires N/Cl compris entre 1 et 0,7, la dichloramine est formée dans le milieu. Le chlore résiduel diminue avec la décomposition de ces chloramines jusqu’à atteindre le point de rupture. L’ajout de chlore au-dessus du point de rupture (ou break point) fait réaugmenter le chlore résiduel sous forme libre (figure I.9).
La trichloramine est la seule chloramine stable qui peut se former à des pH <3 et pour des rapports molaires N/Cl plus faibles.
I.2.3.2- Oxydation des halogénures
L’acide hypochloreux HOCl peut oxyder les ions bromure et les ions iodure présents dans l’eau. La réactivité de HOCl sur les halogénures est supérieure à celle de ClO
avec une constante de vitesse k(HOCl)≥ 106 k(ClO−) (Deborde et von Gunten, 2008).
Le mécanisme proposé pour la chloration de ces halogénures (X-) est le suivant: (Kumar et al., 1986 ; Kumar et Margerum, 1987 ; Johnson et Margerum, 1991) HOCl + X-→ XCl + OH
-XCl + 2 OH- → OX- + Cl- + H2O
Lorsqu’une eau contenant des ions Br
et I- est chlorée, il y a une formation rapide de HOBr et HOI qui peuvent être plus réactifs que HOCl sur les composés présents dans l’eau. Ces halogénures sont considérés comme des catalyseurs pour les réactions d’oxydation par le chlore en solution aqueuse.
Notamment, les bromures peuvent catalyser l’oxydation de la matière organique dissoute par le chlore et renforcer la formation de sous-produits de désinfection comme les trihalométhanes (THM) et les acides haloacétiques (HAA) (Yang et Shang, 2004).
Les valeurs des constantes de vitesse apparente d’ordre 2 de la chloration de ces halogénures sont représentées dans le tableau I.6.
I.2.3.3- Oxydation des nitrites et des sulfites
La chloration des nitrites dépend du pH. La réaction est stœchiométrique et rapide à pH neutre, par contre elle devient lente à pH 9 (Morris, 1978).
NO2-+ HOCl → NO3- + H+ + Cl
-Johnson et Margerum (1991) ont proposé deux voies de mécanisme de l’oxydation des nitrites en passant par la décomposition du chlorure de nitrile (NO2Cl) en nitrate (NO3-).
NO2Cl provient d’une attaque électrophile de Cl+ sur les nitrites (Fogelman et al., 1989 ;
NO2Cl ⇌ NO2+ + Cl-
NO2+ + OH-→ NO3- + H+
Et/ou
NO2Cl + NO2-⇌ N2O4 + Cl
-N2O4 + OH-→ NO3- + NO2- + H+
De même l’oxydation des sulfites par HOCl aboutit à la formation de ClSO3- qui va se
décomposer en SO42-. La constante de vitesse de cette réaction est citée dans le tableau I.6.
Le tableau I.6 ci-dessous présente les constantes de vitesse d’oxydation des composés minéraux par le chlore citées par Deborde et von Gunten (2008).
Tableau I.6 : Constantes de vitesse de chloration des composés minéraux
d’après Deborde et von Gunten (2008). Composés inorganiques pKa kHOCl (M-1.s-1) (25°C) kapp2 (M-1.s-1) (25°C)d Références Azote ammoniacal (NH3) 9,25 3,07.106 4,2.106 2,9.106 1,3.104a 1,8.104a 1,3.104a Qiang et Adams (2004) Morris et Isaac (1983) Margerum et al. (1978) Monochloramine (NH2Cl) 1,5.102 3,5.102 1,2.102a 2,7.102a Margerum et al. (1978) Morris et Isaac (1983)
Chlorure (Cl-) ≤ 0,16 ≤ 0,13a Gerritsen et Margerum
(1990) Bromure (Br-) 6,84.103 2,95.103 1,55.103 5,3.103a 3,8.103a 2,3.103a 1,2.103a Bousher et al. (1986) Rebhun et al. (1987) Farkas et al. (1949) Kumar et Margerum (1987)
Iodure (I-) 1,4.108 1,1.108a Nagy et al. (1988)
Sulfite (SO32-) 7,2 7,6 (±0,4).108 2,3.108a Fogelman et al. (1989)
Cyanure (CN-) 9,2 1,22 (±0,03).109 6.106a Gerritsen et Margerum (1990)
Acide arsénique
(As(III)) 2,9.10
5a
As(OH)3 9,2 4,3 (±0,8).103 Dodd et al. (2006)
As(OH)2O- 12,1 5,8 (±0,1).107 Dodd et al. (2006)
As(OH)22- 12,7 1,4 (±0,1).109 Dodd et al. (2006)
Fer (Fe(II)) 1,7 (± 0,1) .104b Folkes et al. (1995)
Manganèse
(Mn(II)) ≈ 6,4.10
-4c
Hao et al. (1991)
a
à pH 7 et en considérant pKa (HOCl) = 7,54
b
à pH 4
c
à pH 8 et à 22°C
d
I.2.4- Réactivité du chlore sur les composés organiques
Le chlore a une forte réactivité sur la matière organique dissoute de l’eau qui constitue les précurseurs de nombreux sous-produits de désinfection (SPD) produits sur une filière de traitement d’eau potable et pouvant affecter la santé humaine. Les trihalométhanes (THM) et les acides haloacétiques (HAA) sont très étudiés parmi ces SPD. Ils sont limités dans l’eau par l’U.S. EPA (2001) à des concentrations respectives de 80 et 60 µg/L.
L'acide hypochloreux est l'espèce réactive impliquée dans la plupart des réactions avec les composés organiques. Grâce à son pouvoir oxydant et sa structure, caractérisée par la polarisation de la liaison Cl-O (+ClOH-), trois types de réactions peuvent exister : les réactions d'oxydation, les réactions d'addition sur des liaisons insaturées, les réactions de substitution électrophile sur des sites nucléophiles (Doré, 1989).
I.2.4.1- Oxydation des composés organiques azotés
I.2.4.1.1- Amines aliphatiques
L’acide hypochloreux est très réactif sur les amines aliphatiques primaires (RNH2),
secondaires (R2NH) et dans une moindre mesure, tertiaires (R3N).
Abia et ses collaborateurs (1998) ont identifié quatre réactions possibles, mettant en jeu les quatre espèces réactives (HOCl, ClO-, l’amine neutre R2NH, l’amine protonée R2NH2+) :
R2NH2+ + ClO-→ R2NCl + H2O (1)
R2NH + ClO-→ R2NCl + OH- (2)
R2NH2++ HOCl → R2NCl + H3O+ (3)
R2NH + HOCl → R2NCl + H2O (4)
Selon ces auteurs, les espèces chargées de chlore et d’amine ne sont pas réactives dans le milieu et les réactions (2), (3) et (4) sont négligeables (Abia et al., 1998). Ainsi, la réaction de chloration des amines est déterminée par l’équation (4) entre les deux formes neutres du chlore HOCl et de l’amine R2NH (avec une constante cinétique élémentaire kHOCl). Selon
Abia et al. (1998), la réaction procède par un transfert de Cl sur l’azote de l’amine pour former une liaison N-Cl.
La vitesse apparente de réaction entre le chlore et les amines suit un ordre global 2, ordre 1 par rapport à l’oxydant, ordre 1 par rapport à l’amine, selon l’équation suivante :
V = kapp2.[chlore].[amine]
Avec
V : vitesse de réaction.
kapp2 : constante de vitesse apparente d’ordre 2.
[chlore] et [amine] : concentrations en chlore total (HOCl et ClO-) et en amine totale (forme neutre et protonée).
La réactivité de HOCl sur les amines augmente avec la basicité de l’amine.
La vitesse de réaction atteint une valeur maximale pour un pH max = (pKa + pKc)/2 (pKa et
pKcsont respectivement les valeurs des pKa de l’amine et du couple HOCl/ClO-).
Le tableau I.7 donne les constantes cinétiques de la chloration des amines citées par Deborde et von Gunten (2008).
Tableau I.7 : Constantes cinétiques de chloration des amines aliphatiques (à 25°C). Composés amines pKa kHOCl (M-1.s-1) kapp2 (M-1.s-1) à pH 7 Références Amines primaires MeNH2 10,66 1,9.108 3,6.108 3,2.104a 6,1.104a 4,23.104c
Margerum et al.(1978) cité par Abia et al. (1998) et par Yang et Shang (2004)
Morris (1967) calculé par Weil et Morris (1949) Yoon et Jensen (1993) calculé par Gray et al.
(1978)
EtNH2 10,81 1,98.10
8
2,4.104a Abia et al. (1998) calculé par Antelo et al.(1995)
PrNH2 10,56 1,83.10
8
3,9.104a Abia et al. (1998) calculé par Antelo et al.(1995)
BuNH2 10,49
1,63.108 1,03.108
4,1.104a 2,6.104a
Abia et al. (1998) calculé par Antelo et al.(1995), Antelo et al. (1992)
iPrNH2 10,67 1,88.10
8
3,1.104a Abia et al. (1998) calculé par Antelo et al.(1995)
iBuNH2 10,49
1,57.108 8,68.107
3,9.104a 2,2.104a
Abia et al. (1998) calculé par Antelo et al.(1995), Antelo et al.(1992) sBuNH2 10,56 8,9.107 5,16.107 1,9.104a 1,1.104a
Abia et al. (1998) calculé par Antelo et al.(1995), Antelo et al.(1992) tBuNH2 10,69 5,44.107 3,2.107 8,6.103a 5,1.103a 2,5.103d
Abia et al. (1998) calculé par Antelo et al.(1995), Antelo et al.(1992) Pattison et Davis (2001) Amines secondaires Me2NH 10,72 6,05.107 3,3.108 5.107 8,9.103a 4,9.104a 7,4.103a Abia et al. (1998)
Morris (1967) calculé par Weil et Morris (1949) Morris (1967) calculé par Edmond et Soper (1949)
MeEtNH 10,92 6,45.107 6.103a Abia et al. (1998)
Morris (1967) calculé par Edmond et Soper (1949)
Et2NH 11,02 4,14.107 1,4.107 1,4.108 3,1.103a 1.103a 1.104a Abia et al. (1998)
Morris (1967) calculé par Edmond et Soper (1949) Morris (1967) calculé par Friend (1954)
Pr2NH 10,94 3,81.107 4,3.107 3,4.103a 3,8103a Abia et al. (1998)
iPr2NH 11,48 1,8.107 4 ,6.102a Abia et al. (1998)
iBu2NH 10,41
b
2,2.107 6,6.103a Abia et al. (1998)
Amines tertiaires
Triméthylamine 9,75 5.104 6,9.101a Abia et al. (1998) calculé de Antelo et al.(1985)
(N-Me)-pipéridine 10,08 8.10
4
5,2.101a Canle (1994) cité par Abia et al.(1998)
Diéthyléthanol-amine 9,82 1,4.10
5
1,6.102a Abia et al.(1998) calculé par Antelo et al. (1985)
Diméthyléthanol-amine 9,26 3.10
4
1,3.102a Abia et al.(1998) calculé par Antelo et al. (1985)
Méthyldiéthanol-amine 8,52 6,4.10
3
1,5.102a Abia et al.(1998) calculé par Antelo et al. (1985)
Ethyldiéthanol-amine 8,92 1,6.10
4
1,5.102a Abia et al.(1998) calculé par Antelo et al. (1985)
I.2.4.1.2- Aniline
L’aniline est une amine aromatique primaire très soluble dans l’eau (solubilité = 35 g/L à 20°C). C’est un polluant industriel toxique pour l’homme et l’environnement (Canadian Council of Ministers of the Environment, 1999 ; Fiche toxicologique FT 19 INRS, 2010). Les concentrations en aniline dans les eaux de rivière varient entre 4,2 et 31 ng/L (Okumura et al., 1996).
L’aniline est très réactive dans l’eau en présence du chlore. De Laat et al. (1982) ont déterminé une demande en chlore pour l’aniline de 8,3 mol/mol après 15 h de réaction à 20°C et à pH 7 ([chlore]0 = 2 mM ; [composé]0 = 0,1 mM ; tampon phosphate 5 mM).
Guo et Chen (200λ) ont trouvé que l’aniline consomme plus de chlore que le phénol, le nitrobenzène, l’acide benzoïque et le chlorobenzène. Le groupement amine de l’aniline, donneur d’électron, augmente en effet sa réactivité vis-à-vis du chlore, contrairement aux composés aromatiques qui ont un groupement accepteur d’électron, moins consommateurs de chlore. La demande en chlore de l’aniline obtenue par ces auteurs, plus faible que celle déterminée par De Laat et al. (1λ82), est d’environ 4,3 mol/mol après 24 h de réaction à 25°C et à pH 7,5.
L’aniline est également considérée comme un précurseur des SPD générés par la chloration des eaux. Ainsi, elle forme 46,25 mmol/mol d’acides haloacétiques (HAA) répartis en 0,56 mmol/mol d’acide monochloroacétique, λ,62 mmol/mol d’acide dichloroacétique et 36,07 mmol/mol d’acide trichloroacétique (24 h de réaction à 25°C et à pH 7,5 ; Guo et Chen, 2009).
(Neale et al. (1λ64) ont montré que la chloration de l’aniline et de N-alkylanilines conduit à la formation de dérivés o- et p-chloroanilines par réarrangement d’intermédiaires N-chlorés formés dans une première étape. Les dérivés o-chloroanilines correspondant sont alors formés très majoritairement.
En étudiant le réarrangement de N-alkyl-N-chloroanilines parasubstituées, Gassman et Campbell (1972) ont établi que la rupture de la liaison N-Cl se produit par l'intermédiaire d'un état de transition qui implique une perte d'anion chlorure et la génération d’une charge positive sur l'azote (figure I.10).
N R + + Cl -N R N R N R Cl NH R HOCl NH R NH R Cl Cl N R
Figure I.10 : Réarrangement de N-alkyl-N-chloroanilines selon Gassman et Campbell
(1972).
Ainsi, Hwang et ces collaborateurs (1990) ont mis en évidence la formation comme premiers sous-produits d’oxydation de chloroaniline (majoritaire) et de dichloroaniline par chloration de l’aniline (pH = 8 ; temps de réaction : 10 minutes ; [aniline]0 = 2.10-4 M ; Rmolaire
chlore/aniline = 1,5).
I.2.4.2- Oxydation des hydroxybenzènes
I.2.4.2.1- Phénol
Les phénols sont des micropolluants organiques toxiques qui peuvent contaminer l’eau suite à des applications industrielles notamment la fabrication de pesticides, de colorants, de composés pharmaceutiques, de plastiques, et d’autres. Ces composés sont caractérisés par leur forte solubilité dans l’eau, leur forte réactivité et leur faible biodégradabilité et peuvent avoir des impacts négatifs sur l’homme et l’environnement. Ils constituent également la grande partie des substances humiques qui composent majoritairement la matière organique naturelle dissoute.
Les phénols sont détectés dans les eaux à des concentrations maximales moyennes de 10 µg/L et sont responsables des goûts indésirables et de la formation des dérivés plus toxiques en contact avec les oxydants lors du traitement des eaux potables (Acero et al., 2005).
En plus des réactions spécifiques à certains groupements liés au cycle aromatique, le chlore réagit avec les composés aromatiques essentiellement par des substitutions électrophiles. En
raison de l'activation du noyau aromatique en position ortho et para du groupement hydroxyle, la chloration du phénol conduit à des phénols substitués en positions 2, 4 et 6.
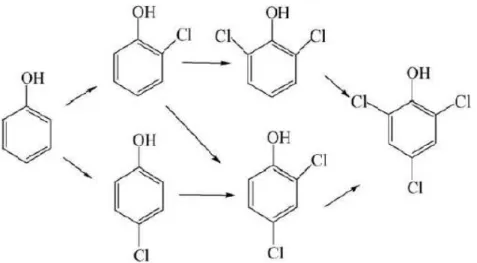
Les chlorophénols, incluant les monochlorophénols (2-chlorophénol et 4-chlorophénol), les dichlorophénols (2,6-dichlorophénol et le 2,4-dichlorophénol) et le 2,4,6-trichlorophénol sont formés à partir des substitutions électrophiles successives du chlore sur le cycle phénolique (figure I.11).
Figure I.11 : Formation successives des chlorophénols
lors de la chloration du phénol (Ge et al., 2006).
Lors de la chloration, la consommation du phénol et la répartition des chlorophénols dans le milieu dépend du pH (Ge et al., 2006). La réaction de HOCl avec l’ion phénolate contrôle la réaction globale lorsque pH ≥ 5. De plus, la distribution de la densité de charge sur le cycle phénolique dépend du pH. En effet, la position para est plus activée à pH acide tandis que la position ortho l’est plutôt à pH neutre et basique (Nunez-Gaytan et al., 2010).
Ainsi, le phénol est lentement transformé en monochlorophénols à pH 4,8. Cette réaction devient plus rapide en augmentant le pH et les monochlorophénols sont majoritaires à pH légèrement acide (pH 6 - 6,5). A pH basique ou légèrement basique, les dichlorophénols et le trichlorophénol sont les formes majoritaires dans le milieu (Ge et al., 2006).
Selon Nunez-Gaytan et al. (2010), deux voies réactionnelles compétitives coexistent à pH neutre ou basique et selon la teneur en chlore. Ainsi, une faible teneur en chlore de 1 ppm favorise la transformation rapide des monochlorophénols en trichlorophénol via des substitutions successives, suivie d’un clivage du cycle aromatique. Cette voie est favorisée à pH basique (pH 9) avec une faible ou moyenne force ionique. Par contre, pour une teneur en chlore plus élevée de 5 ppm, les monochlorophénols sont oxydés par voie directe en composés aromatiques oxygénés (dihydroxybenzènes, benzoquinones) rapidement dégradés en composés aliphatiques.
La formation des THM peut provenir de réactions rapides ou lentes du chlore sur diverses fractions de la matière organique (Gallard et von Gunten, 2002a). Ainsi, les structures phénoliques, notamment celle du type résorcinol (1,3-dihydroxybenzène), peuvent être les
Guo et Chen (2009) ont trouvé que la chloration du phénol génère 49,07 mmol/mol de HAA dont la fraction la plus grande est représentée par l’acide trichloroacétique (31,78 mmol/mol). Le reste est réparti en 16,45 mmol/mol d’acide dichloroacétique et 0,84 mmol/mol d’acide monochloroacétique (après 24 h à 25°C et à pH 7,5). Cette production de HAA diminue avec l’augmentation du pH de 6 à λ.
La consommation du chlore par le phénol et quelques dérivés est présentée dans le tableau I.8.
Tableau I.8 : Demandes en chlore du phénol, de ses dérivés chlorés et des aminophénols à
pH 7 et à 20°C. Composé phénolique Temps de contact (h) Chlore consommé (mol/mol) Références Phénol 15 9,8a De Laat, 1981 72 9,76b Reckhow et Singer, 1985 4-24 8,7c Rook, 1980 2-chlorophénol 15 8,9a De Laat, 1981 3-chlorophénol 15 8,8a De Laat, 1981 4-chlorophénol 15 8,7a De Laat, 1981 2-aminophénol 15 3,9a De Laat, 1981 3-aminophénol 15 7,7a De Laat, 1981 4-aminophénol 15 5,4a De Laat, 1981 a
[chlore]0 = 2 mM ; [composé]0 = 0,1 mM ; pH = 7 ; tampon phosphate 5 mM ; T = 20°C.
b
[chlore]0 = 0,3 mM ; [composé]0 = 7 à 22 µM ; pH = 7.
c
pH 7,5 ; T = 22°C.
La demande en chlore des phénols diminue avec l’augmentation du nombre de chlore substitué sur le cycle aromatique (Gallard et von Gunten, 2002a).
Le phénol dans l’eau est en équilibre avec l’ion phénolate chargé négativement. Lors de la chloration du phénol, l’acide hypochloreux peut réagir avec ces deux espèces pour former des sous-produits de chloration. La réactivité de l’ion hypochlorite ClO- sur le phénol est considérée comme négligeable. Des réactions élémentaires ont été proposées dans la littérature afin d'expliquer et de modéliser la réaction de chloration du phénol pour un pH donné ; elles comprennent :
- les réactions de HOCl avec la forme ionisée et la forme neutre,
- la réaction catalysée en milieu acide de HOCl avec la forme neutre (Gallard et von Gunten, 2002b).
Phénol ⇌ Phénolate + H+
Phénol + HOCl ⇌ Produits k1
Phénolate + HOCl ⇌ Produits k2
La vitesse apparente de chloration du phénol suit un ordre global 2. Ainsi l’équation de vitesse peut être exprimée :
V = - d[Phénol ] dt
= kapp2 [Phénol] [HOCl]
Cette vitesse augmente avec le pH. La constante de vitesse apparente 2 (kapp2) de réaction
atteint une valeur maximale à des pH compris entre 7 et 8 (tableau I.9).
Tableau I.9 : Constantes de vitesse apparentes (kapp2) obtenues à pH 6, 7 et 8 (25°C) pour le
phénol et ses dérivés chlorés (Acero et al., 2005). Composés phénoliques kapp2 (M-1.s-1) à 25°C pKa pH 6 pH 7 pH 8 Phénol 9,99 3,26 23,5 76,5 2-chlorophénol 8,56 6,7 50,2 133,5 4-chlorophénol 9 ,43 0,96 7,5 24 2,4-dichlorophénol 7,85 4,1 28,8 45,2 2,6-dichlorophénol 6,97 18,3 77,9 45,7 2,4,6-trichlorophénol 6,15 5,2 8,7 3,2
D’après les modélisations cinétiques de chloration du phénol, l’espèce phénolate est connue comme la forme la plus réactive avec l’acide hypochloreux (Soper et Smith, 1λ26). La constante de vitesse élémentaire k2 du phénolate avec l’acide hypochloreux est supérieure à
celle du phénol k1d’un facteur de 103-105.
Les substituants sur le noyau aromatique ont une influence sur la vitesse de la réaction de substitution : des groupements donneurs d'électrons vont augmenter la densité de charge du noyau aromatique et vont ainsi conduire à une réaction de substitution plus rapide.
Les constantes de vitesse élémentaires de la chloration du phénol et de ses dérivés chlorés sont représentées dans le tableau I.10.
Tableau I.10 : Constantes cinétiques élémentaires de chloration du phénol et de ses dérivés
chlorés en solution aqueuse (25°C). Composés
phénoliques k1 (M
-1
.s-1) k2 (M-1.s-1) k3 (M-2.s-1) Références
Phénol
3,05.104 Cité par Rebenne et al. (1996) à partir de Lee (1967) 3,3.105
Cité par Rebenne et al. (1996) à partir de Soper et Smith
(1926) 3,52 (±0,19).104 Lee et Morris (1962) 0,36 (±0,28)a 2,19 (±0,08).104a 2,49 (±0,98).102a
Gallard et von Gunten (2002b)
2-chlorophénol
2150 Cité par Rebenne et al. (1996)
à partir de Lee (1967) 5.9.104
Cité par Rebenne et al. (1996) à partir de Soper et Smith
(1926) 2,42
(±0,08).103 Lee et Morris (1962)
4-chlorophénol
2680 Cité par Rebenne et al. (1996)
à partir de Lee (1967) 4.9.104 Cité par Rebenne et al. (1996)
à partir de Soper et Smith (1926) 3,16 (±0,22) .103 Lee et Morris (1962) 0,02 (±0,005)a 2,17 (±0,33) .103a 1,6 (±0,4).101a
Gallard et von Gunten (2002b) 2,4-dichlorophénol 3,03 (±0,09) .102 Lee et Morris (1962) 2,6-dichlorophénol 1,94 (±0,11) .102 Lee et Morris (1962) 2,4,6-trichlorophénol 12,84 (±0,69) Lee et Morris (1962) a à 22°C I.2.4.2.2- Résorcinol
Le résorcinol (1,3-dihydroxybenzène) et ses dérivés sont des précurseurs importants de la formation des THM (Boyce et Hornig, 1983).
Heasley et al. (1989) ont identifié le 2-chloro-, 4-chloro-, 2,4-dichloro-, 4,6-dichloro- et 2,4,6-trichlororésorcinol comme dérivés chlorés du résorcinol suite à des substitutions électrophiles successives du chlore sur le cycle aromatique. Ensuite, le trichlororésorcinol se transforme en un intermédiaire pentachloré, le 2,2,4,4,6-pentachloro-5-cyclohexen-1,3-dione, qui va