HAL Id: hal-02055800
https://hal.archives-ouvertes.fr/hal-02055800
Submitted on 4 Mar 2019
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Mesoporous silica nanoparticles functionalised with a
photoactive ruthenium( ii ) complex: exploring the
formulation of a metal-based photodynamic therapy
photosensitiser
Younes Ellahioui, Malay Patra, Cristina Mari, Rim Kaabi, Johannes Karges,
Gilles Gasser, Santiago Gómez-Ruiz
To cite this version:
Younes Ellahioui, Malay Patra, Cristina Mari, Rim Kaabi, Johannes Karges, et al.. Mesoporous silica
nanoparticles functionalised with a photoactive ruthenium( ii ) complex: exploring the formulation of a
metal-based photodynamic therapy photosensitiser. Dalton Transactions, Royal Society of Chemistry,
2018, 48 (18), pp.5940-5951. �10.1039/C8DT02392A�. �hal-02055800�
Mesoporous silica nanoparticles functionalised
with
a
photoactive
ruthenium(II)
complex:
Exploring the formulation of a metal-based
photodynamic therapy photosensitiser
Younes Ellahioui 1, Malay Patra 2, Cristina Mari 2, Rim Kaabi 1, Johannes Karges 3, Gilles Gasser 3,* and Santiago Gómez-Ruiz 1,*
1
Departamento de Biología y Geología, Física y Química Inorgánica, ESCET, Universidad Rey Juan Carlos, Calle Tulipán s/n, E-28933, Móstoles (Madrid), Spain; santiago.gomez@urjc.es
2
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland 3
Chimie ParisTech, PSL University, Laboratory for Inorganic Chemical Biology, F-75005 Paris, France;
gilles.gasser@chimieparistech.psl.eu
* Correspondence: santiago.gomez@urjc.es ( https://santiago-gomez-ruiz.webnode.es/comet-nano-group/); gilles.gasser@chimieparistech.psl.eu (www.gassergroup.com).
Abstract: A series of nanomaterials based on mesoporous silica have been synthesised and
functionalised with a photoactive polypyridyl ruthenium(II) complex, namely [Ru(bipy)2
-dppz-7-hydroxymethyl][PF6]2 (bipy = 2,2’-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine) by various
methods. The functionalisation reactions were based on the covalent binding to different ligands attached to the pores of the mesoporous nanoparticles and a simple physisorption using polyamino-functionalised mesoporous silica nanoparticles. The resulting nanostructured systems have been characterised by XRD, XRF, BET, SEM and TEM, observing the incorporation of the metallodrug onto the nanostructured silica in a different way depending on the synthetic method used in the loading reactions. In our studies, we have also observed that functionalisation with the metallodrug causes changes in the structural and textural features of the materials. The phototherapeutic activity of the ruthenium-functionalised materials in HeLa cervical cancer cells have been tested and the preliminary results are presented herein.
Keywords: cytotoxicity; mesoporous silica nanoparticles; photodynamic therapy; ruthenium.
Abbreviations: BET = Brunauer, Emmett and Teller; bipy = 2,2’-bipyridine; BJH = Barrett,
Joyner and Halenda; CP = cross polarization; CTAB = cetyltrimethylammonium bromide; DMEM = Dulbecco's Modified Eagle's medium; dppz = dipyrido[3,2-a:2′,3′-c]phenazine; DR-UV = diffuse reflectance Ultraviolet spectroscopy; EPR = enhanced permeability and retention effect; FBS = Foetal Bovine Serum; FT-IR = Fourier Transformed Infrared spectroscopy; ICP-MS = Inductively coupled plasma mass spectrometry; MAS = magic angle spinning; MDR = multidrug resistance; MSN = mesoporous silica nanoparticles; PDT = photodynamic therapy; SEM = Scanning Electron Microscopy; TEM = Transmission Electron Microscopy; TEOS = tetraethylorthosilicate; XRD = X-ray diffraction; XRF = X-ray fluorescence.
1. Introduction
The use of metallodrugs to treat cancer was greatly developed after the serendipitous discovery of cisplatin (cis-diamminodichloroplatinum(II)) by the research team of Rosenberg in the late 1960’s.1 Since then, metal-based drugs have been investigated for different medicinal or diagnostic applications against various cancer-related issues.2,3 However, the full exploitation of the specific physicochemical properties of metallodrugs has been, and is still, hampered by the fact that some of the most interesting synthesised agents present severe side effects,4 are becoming less active or even inactive after several treatments due to the development of resistance5 and, usually, possess low stability in aqueous solutions,6 precluding their administration to humans. Nevertheless, the associated problems to the use of metal-based drugs in cancer treatment or for diagnosis, have also boosted the search for novel alternatives using different metals and ligands with improved properties against cancer, especially when speaking about the development of multidrug resistance (MDR).7,8
When one thinks about the use and application of metallodrugs in humans, many questions arise regarding their stability, transport to cell, membrane crossing, targeting and some other processes which usually lead to the transformation of the metallodrug to give the final active species which interacts with the biological target(s).9 Therefore, the administration of metallodrugs is a very important issue that needs to be addressed to minimise the speciation reactions.
In this context, in recent years, several approaches based on different nanostructured materials as vectors for metallodrugs have been used for protecting and subsequently delivering the metal-containing active species to the biological target(s).10,11 Thus, many different nanosystems have been tested in vitro and in vivo10 with a very extensive study of nanostructured mesoporous silica loaded with titanocene derivatives.11-20 organotin compounds20-22 or other metallodrugs.10,23. This approach has been shown to be highly effective against cancer cell lines.10,11
At our end, in most of our studies, we have observed that metallodrug-functionalised silica-based nanomaterials usually work as “non-classical” drug-delivery nanosystems because they do not release the metal-based drug, but, in most of the cases, they act as an entire nanoparticulated therapeutic system, presumably by the action of the metallodrug supported onto the silica particles. These systems base their improved cytotoxic action on a higher cell uptake of metal-containing active species of metallodrug-functionalised silica-based systems, compared to “free” metallodrugs, because of a high uptake of silica particles by the cancer cells due to the enhanced permeability and retention (EPR) effect of malignant cells. In addition, the support of the metallodrugs onto different nanostructured systems usually leads to a different mechanism of cytotoxic action compared with that of non-encapsulated “free” metallodrugs 10,11. In brief, the dynamics of apoptotic morphological and functional changes is modified when the metallodrug are incorporated into nanostructured silica-based systems.16,18
All our studies have been carried out using cytotoxic titanium- or tin-based metallodrugs and have been focused on the potential use of these nanostructured systems in chemotherapy, however, we decided to expand to other metal complexes with improved therapeutic properties. We therefore became interested in ruthenium complexes as their photoactivation is of great interest nowadays because of their potential therapeutic mild processes against cancer.24-29 Thus, a wide variety of dual photoreleasing and photoactive systems have been studied and have been considered as interesting alternatives to current treatments.30-33 Lately, several photoactivatable Ru(II) complexes with interesting biological properties have been published.25,34-40 As a highlight, one of such compounds, namely TLD-1433 from the McFarland group, has now entered clinical trial as a PDT photosensitiser to treat bladder cancer 41. However, to the best of our knowledge, there are only a few reports on the use of ruthenium compounds with therapeutic properties supported onto mesoporous silica,42,43 which have been synthesised using a very similar approach to the one that we published in 200910,12 and have been tested in terms of regular cytotoxicity. Additionally, some other more advanced cytotoxic systems based on silica have also been reported.44-46 Only very recently, a Ru(II) complex was
covalently bound to mesoporous silica nanoparticles (MSN) for a subsequent cellular uptake of the particles and release of the ruthenium complexes to be transformed upon light irradiation into a cytotoxic complex47 and a similar approach has been studied using other ruthenium complexes48 or silicon nanoparticles.49 or other non-polymeric nanoparticles.50
In this context and in view of the low number of examples of ruthenium-functionalised silica-based materials with phototherapeutic properties, we decided to combine the experience of our groups on the synthesis, characterization and phototherapeutic studies of ruthenium complexes51-53 with our knowledge on the field of metallodrug-functionalised nanostructured materials,11-22 preparing a series of MSN functionalised with a photoactive compound using different synthetic protocols. All the materials have been characterised by different methods and a preliminary study of the potential biological application of the materials in photodynamic therapy is described.
2. Materials and Methods
2.1. General conditions
All the functionalisation reactions were performed using standard Schlenk tube techniques in an atmosphere of dry nitrogen. Solvents were distilled from the appropriate drying agents and degassed before use. The reagents used in the preparation of the ruthenium derivative were purchased from Sigma Aldrich and used directly without further purification. 3-chloropropyltriethoxysilane, 3-isocyanatopropyltriethoxysilane and tris(2-aminoethyl)amine, TEOS, NaOH and CTAB were purchased from Sigma Aldrich and used directly without further purification. The ruthenium complex, [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (bipy =
2,2’-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine, was prepared according to the literature procedure reported by us.35
2.2. General conditions on the characterization of the materials 1
H, 13C-CP MAS and 29Si MAS NMR spectra and 31P NMR were recorded on a Varian-Infinity Plus Spectrometer at 400 MHz operating at 100.52 MHz proton frequency (4 µs 90° pulse, 4000 transients, spinning speed of 6 MHz, contact time 3 ms, pulse delay 1.5 s). X-ray diffraction (XRD) pattern of the silicas were obtained on a Philips Diffractometer model PW3040/00 X'Pert MPD/MRD at 45 KV and 40 mA, using a wavelength Cu Kα (λ = 1.5418 Å). Ru wt % determination by X-ray fluorescence was carried out with an X-ray fluorescence spectrophotometer Philips MagiX with a X-ray source of 1 kW and a Rh anode using a helium atmosphere. The quantification method is able to analyse from 0.0001% to 100% ruthenium. N2
gas adsorption-desorption isotherms were performed using a Micromeritics ASAP 2020 analyser. Scanning electron micrographs and morphological analysis were carried out on a XL30 ESEM Philips with an energy dispersive spectrometry system (EDS). The samples were treated with a sputtering method with the following parameters: Sputter time 100 s, Sputter current 30 mA, film thickness 20 nm using a Sputter coater BAL-TEC SCD 005. Conventional transmission electron microscopy (TEM) was carried out on a TECNAI 20 Philips, operating at 200 kV.
2.3. Synthesis of mesoporous silica nanoparticle (MSN)
The mesoporous silica nanoparticles (MSNs) were synthesised with slight modifications of the method described by Zhao et al..54 In brief, hexadecyltrimethylammonium bromide (CTAB, 2.00 g, 5.48 mmol) was added to 960 mL of nanopure water, and subsequently 7 mL of 2.00 M sodium hydroxide aqueous solution were added and the mixture stirred at 80 ºC. Subsequently, tetraethylorthosilicate (TEOS, 10 mL, 45 mmol) was added dropwise to the surfactant solution under vigorous stirring. The mixture was then stirred for 2 h to give a white precipitate. The resulting product was isolated by filtration, washed with nanopure water and methanol and dried
under vacuum overnight. The surfactant template (CTAB) was removed after calcination at 550 ºC for 12 h.
2.4. Synthesis Cl-functionalized MSN (MSN-CL)
A solution of 3-chloropropyltriethoxysilane (5.80 mL, 22.6 mmol) in toluene (100 mL) was added to dehydrated MSN (5.00 g) and the mixture was stirred 48 h at 110 ºC. The slurry was filtered through fritted discs and the solid residue washed with toluene (5 × 200 mL) and diethylether (5 × 200 mL). The resultant solid was dried under vacuum at room temperature for 24 h to give a white free flowing powder.
2.5. Synthesis of CNO-functionalized MSN (MSN-CNO)
A solution of 3-isocyanatopropyltriethoxysilane (1.00 g) in toluene (30 mL) was added to dehydrated MSN (1.00 g) and the mixture was stirred 48 h at 110 ºC. The slurry was filtered through fritted discs and the solid residue washed with toluene (5 × 50 mL) and diethylether (5 × 50 mL). The resultant solid was dried under vacuum at room temperature for 24 h to give a white free flowing powder (MSN-CNO).
2.6. Synthesis of triamine-functionalized MSN (MSN-TRI)
A solution of tris(2-aminoethyl)amine (TRI 2.40 mL, 16.0 mmol) in toluene (50 mL) was added to a suspension of MSN-CL (1.00 g) in toluene (50 mL) and the mixture was stirred for 48 h at 110 ºC. The slurry was filtered through fritted discs and the solid residue was washed with toluene (5 × 200 mL), ethanol (5 × 200 mL), methanol (5 × 200 mL) and diethylether (5 × 200 mL). The resultant solid was dried under vacuum at room temperature for 24 h giving a white free flowing powder (MSN-TRI)
2.7. Synthesis of MSN-CL-Ru
A suspension of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (10 mg) (to obtain a theoretical
level of 5% ruthenium/MSN) in ethanol (100 mL) was added to MSN (190 mg) and the mixture was stirred at room temperature for 10 minutes, subsequently an excess of the stoichiometric amount of trimethylamine (1.5:1 with [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2) was added to the
mixture which was then stirred under reflux during 48 h. The slurry was filtered through fritted discs and the solid residue was washed several times with ethanol (5 × 200 mL) water (5 × 200 mL) and diethylether (5 × 200 mL) and then dried under vacuum at room temperature for 24 h to give a pale orange free flowing powder (MSN-CL-Ru)
2.8. Synthesis of MSN-CNO-Ru
A suspension of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (10 mg) (to obtain a theoretical
level of 5% Ru/MSN-CNO) in ethanol (100 mL) was added to MSN-CNO (190 mg) and the mixture was stirred at room temperature for 10 minutes, subsequently an excess of the stoichiometric amount of pyridine (1.5:1 molar ratio with [Ru(bipy)2
-dppz-7-hydroxymethyl][PF6]2) was added to the mixture which was then stirred under reflux during 48 h.
The slurry was filtered through fritted discs and the solid residue was washed several times with ethanol (5 × 100 mL) water (5 × 100 mL) and diethylether (5 × 100 mL) and then dried under vacuum at room temperature for 24 h to give a pale orange free flowing powder.
2.9. Synthesis of MSN-TRI-Ru
A suspension of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (48 mg) (to obtain a theoretical
level of 20% Ru/MSN-TRI-NH) in ethanol (100 mL) was added to MSN-TRI-NH (190 mg, respectively) and the mixture was stirred at room temperature for 10 minutes and then stirred under reflux during 48 h. The slurry was filtered through fritted discs and the solid dried under vacuum at room temperature for 24 h to give an orange free flowing powder.
2.10. Biological studies: cell culture conditions, Cytotoxicity and phototoxicity assays.
HeLa (human cervical cancer) cells were grown in DMEM (Gibco) medium supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin. Cells were cultured at 37
⁰C in a humidified atmosphere containing 5% CO2.
The cytotoxicity of the ruthenium complex and its three MSN conjugates against HeLa cells in the presence and absence of UV irradiation was measured using a resazurin (Promocell GmbH) based fluorometric cell viability assay. In 96-well plates, cells (at a density of 4 x 103 cells per well) were seeded in triplicate in 100 µL medium 24 h prior to treatment. The following day, cells were treated with increasing concentrations of compounds. For this purpose, freshly prepared stocks (note: due to their insolubility in aqueous media, MSN and ruthenium-MSN conjugates are added as suspension) were diluted using the respective cell culture media and 100 µL was added to each well (total volume 200 µL/well). For evaluation of cytotoxicity, cells were incubated with compounds for 48 h before the medium was replaced by 100 µL complete medium containing resazurin (0.2 mg/mL). After 4 h of incubation, fluorescence of the highly fluorescent resorufin metabolite at 580 nm emission was quantified using a SpectraMax M5 microplate reader with an excitation wavelength of 540 nm.
To assess phototoxicity, cells were treated with increasing concentrations of compounds for 4 h only. After that, the medium was removed and replaced by fresh complete medium and either kept in the dark or irradiated for 10 min with 350 nm UVA (2.58 J.cm-2). Cells were then returned to the incubator for an additional 44 h. After incubation, the viability was quantified using the resazurin assay as described above. The resorufin fluorescence intensities were normalized to untreated control wells and plotted as concentration of test compound versus % cell viability. IC50 values were interpolated from the resulting dose-dependence curves. The
reported IC50 values are the average from at least three independent experiments. 2.11. Photophysical studies: emission, luminescence quantum yield, lifetimes
For the determination of the emission and luminescence quantum yields (Φem), the samples
were prepared in an CH3CN solution with an absorbance of 0.1 at 355 nm. This solution was
irradiated in fluorescence quartz cuvettes (width 1 cm) using a NT342B OPO pulse laser Nd-YAG pumped optical parametric oscillator (Ekspla) at 355 nm. The emission signal was focused and collected at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator equipped with 1200 g/mm grating blazed at 500 nm. As a detector a XPI-Max 4 CCD camera (Princeton Instruments) has been used. The luminescence quantum yields were determined by comparison with the reference [Ru(bipy)3]Cl2 in CH3CN
(Φem=5.9%) 55
applying the following formula:
Φ , = Φ , ∗ F ∗II ∗ nn
F = 1 − 10
Φem = luminescence quantum yield, F = fraction of light absorbed, I = integrated emission
intensities, n = refractive index, A = absorbance of the sample at irradiation wavelength
For the determination of the lifetimes, the samples were prepared in an air saturated and in a degassed CH3CN solution with an absorbance of 0.1 at 355 nm. This solution was irradiated in
fluorescence quartz cuvettes (width 1 cm) using a NT342B Nd-YAG pumped optical parametric oscillator (Ekspla) at 355 nm. The emission signal was focused and collected at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator equipped with 1200 g/mm grating blazed at 500 nm. As a detector a R928 photomultiplier tube (Hamamatsu) has been used.
2.11. Photophysical studies: Singlet oxygen measurement
The singlet oxygen production of a sample has been determined by two methods (direct and indirect) as already descibed in previous publications of our group.35,51,53,56
The samples were prepared in an air saturated CH3CN or D2O solution with an absorbance of
0.2 at 450 nm. This solution was irradiated in fluorescence quartz cuvettes (width 1 cm) using a mounted M450LP1 LED (Thorlabs) whose irradiation, centred at 450 nm, has been focused with aspheric condenser lenses. The intensity of the irradiation has been varied using a T-Cube LED Driver (Thorlabs) and measured with an optical power and energy meter. The emission signal was focused and collected at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator equipped with 600 g/mm grating blazed at 1200 nm. A longpass glass filter was placed in front of the monochromator entrance slit to cut off light at wavelengths shorter than 850 nm. The slits for detection were fully open. As a detector an EO-817L IR-sensitive liquid nitrogen cooled germanium diode detector (North Coast Scientific Corp.) has been used. The singlet oxygen luminescence at 1270 nm was measured by recording spectra from 1100 to 1400 nm. For the data analysis, the singlet oxygen luminescence peaks at different irradiation intensities were integrated. The resulting areas were plotted against the percentage of the irradiation intensity and the slope of the linear regression calculated. The absorbance of the sample was corrected with an absorbance correction factor. As reference for the measurement in an CH3CN solution phenalenone (Φphenaleone=95%)
57
and for the measurement in a D2O solution [Ru(bipy)3]Cl2 (ΦRu(bipy)3Cl2=22%) 58
was used and the singlet oxygen quantum yields were calculated using the following formula:
Φ = Φ ∗ SS ∗II
I = I ∗ (1 − 10 )
Φ = singlet oxygen quantum yield, S = slope of the linear regression of the plot of the areas of the singlet oxygen luminescence peaks against the irradiation intensity, I = absorbance correction factor, I0 = light intensity of the irradiation source, A = absorbance of the sample at
irradiation wavelength. - Indirect evaluation
For the measurement in CH3CN: The samples were prepared in an air-saturated CH3CN
solution containing the complex with an absorbance of 0.1 at the irradiation wavelength, N,N-dimethyl-4-nitrosoaniline aniline (RNO, 24 µM) and imidazole (12 mM). For the measurement in PBS buffer: The samples were prepared in an air-saturated PBS solution containing the complex with an absorbance of 0.1 at the irradiation wavelength, N,N-dimethyl-4-nitrosoaniline aniline (RNO, 20 µM) and histidine (10 mM). The samples were irradiated on 96 well plates with an Atlas Photonics LUMOS BIO irradiator for different times. The absorbance of the samples was measured during these time intervals with an SpectraMax M2 Microplate Reader (Molecular Devices). The difference in absorbance (A0-A) at 420 nm for the CH3CN solution or
at 440 nm a PBS buffer solution was calculated and plotted against the irradiation times. From the plot the slope of the linear regression was calculated as well as the absorbance correction factor determined. The singlet oxygen quantum yields were calculated using the same formulae as those used for the direct evaluation.
3. Results and Discussion
3.1. Synthesis and characterisation of ruthenium-functionalized nanostructured materials
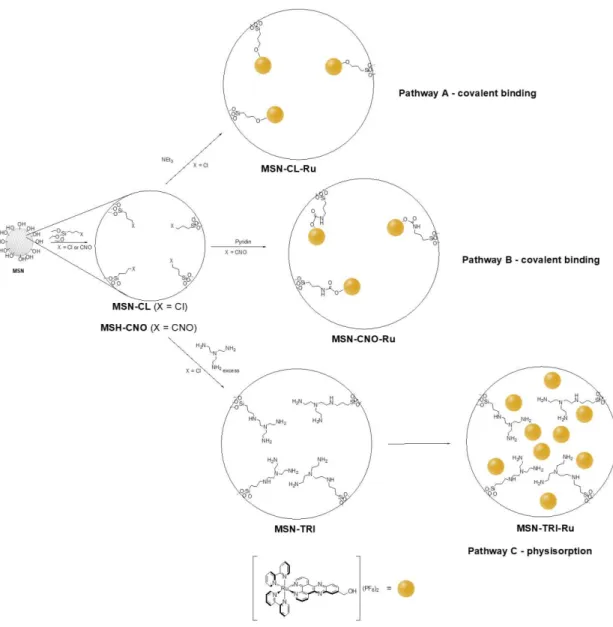
The preparation of the ruthenium-functionalised materials MSN-CL-Ru, MSN-CNO-Ru and
MSN-TRI-Ru was carried out using three different synthetic methods. For the preparation of MSN-CL-Ru the pathway A (Scheme 1) was followed. This consists of a simple grafting reaction
of 3-chloropropyltriethoxysilane on dehydrated (treated for 24 hours at 150 ºC under vacuum) MSN to give MSN-CL following reported procedures.59,60 Subsequently, a functionalisation reaction with the ruthenium polypyridyl complex, [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2, was
carried out using a theoretical 5% of Ru wt.% (Ru/MSN). The reaction was performed in the presence of triethylamine (1.5 equivalent per equivalent of ruthenium) to promote the deprotonation of the alcohol group of the polypyridyl ruthenium compound and elimination of triethylammonium chloride to give the covalently supported ruthenium complex.
Scheme 1. % of Ru grafted on the mesoporous silica nanoparticles.
The theoretical amount of ruthenium in the reaction was adjusted according to a previous loading study that was carried out using different starting quantities of ruthenium (2.5, 5, 10 and 15% wt. Ru/MSN) which showed the best efficacy in functionalisation when using 5% wt. ruthenium (Table 1).
Table 1. % of Ru grafted on the mesoporous silica nanoparticles.
Material Theoretical %Ru wt. Experimental %Ru wt.1 Experimental mmol of Ru/g material MSN-CL-Ru 5 2.1 0.21 MSN-CNO-Ru 5 3.1 0.31 MSN-TRI-Ru 20 7.5 0.74 1
Determined by X-ray fluorescence.
Analogously, for the preparation of MSN-CNO-Ru the pathway B (Scheme 1) was performed. This consists of the functionalisation of MSN with 3-isocyanatopropyltriethoxysilane
to give MSN-CNO and the subsequent reaction with the ruthenium polypyridyl complex (using a theoretical 5% of Ru wt.% Ru/MSN) in the presence of pyridine (1.5 equivalent per equivalent of ruthenium). This synthetic procedure led to the covalent functionalisation of the material with the ruthenium complex via the formation of the corresponding urethane derivative (Scheme 1, pathway B).
Finally, material MSN-CL was treated with tris(2-aminoethyl)amine giving rise to a polyamine-functionalized MSN (MSN-TRI) which was treated with an ethanolic suspension of the ruthenium polypyridyl complex (with a theoretical 20 wt.% Ru/MSN) under reflux conditions for 48 h to obtain the ruthenium-physiosorbed material MSN-TRI-Ru (Scheme 1, Pathway C). The material was not washed to avoid the leaching of the adsorbed complex. In this case, an adsorption of the ruthenium derivative is presumably achieved via intermolecular interactions between the polar groups of ruthenium complex and the amino groups attached to the material, although electrostatic interactions between protonated amines of the material, the anionic PF6
-and the cationic ruthenium moiety cannot be ruled out. 3.1.1. X-ray fluorescence
The ruthenium-loaded materials MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru were characterised by XRF observing a very different quantity of Ru wt. %. For the covalent loaded ruthenium complexes MSN-CL-Ru and MSN-CNO-Ru only 2.1% and 3.1% of Ru was found, while in the case of the material with the physiosorbed Ru complex a much higher quantity of 7.5% wt. Ru was observed (Table 1). It is important to note that we tried some other synthetic approaches for increasing the quantity of loaded Ru by covalent modification or physical adsorption. However, we did not succeed in achieving this using other methods and different quantities of the ruthenium compound (See supplementary material). The observed low functionalisation using pathways A and B was attributed to the weak acidity of the alcoholic proton of the ruthenium complex and also to the partial saturation of the pores of the mesoporous silica nanoparticles with the different ligands (chloroproyltriethoxysilane or isocianatopropyltriethoxysilane in MSN-CL or MSN-CNO, respectively) which reduced the degree of Ru-functionalisation.
3.1.2. Powder X-ray diffraction
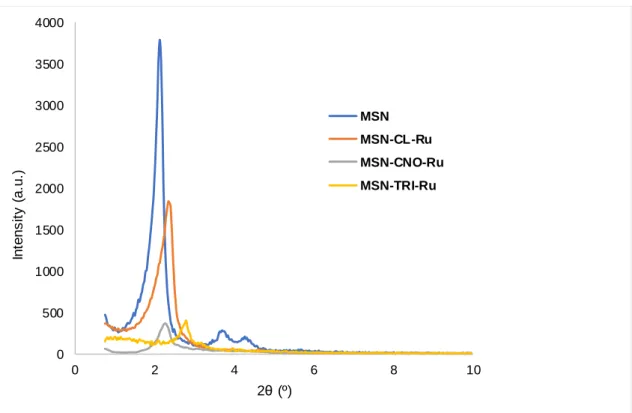
All the materials have been characterised by XRD. In all cases, the diffractograms show typical reflections for hexagonally ordered mesoporous materials, with relatively broad diffraction peaks. Non-modified MSN material gave a sharp peak at a 2θ value of ca. 2.1º corresponding to the Miller plane (100), another two peaks of very low intensity were observed at ca. 3.8° and 4.2º which correspond with the (110) and (200) Miller planes, respectively. (Figure 1).
Interestingly, after the consecutive functionalization reactions with the different organic fragments and finally with the ruthenium photosensitiser, the position of the peaks slightly shifted to higher 2θ values and the intensity of the peaks notably decreased. The decrease in the intensity of the peaks is presumably due to the partial blocking of the dispersion points of the material while the slight shift towards higher diffraction angles confirm that the structural mesoporous order is retained after functionalisation reactions and that, apparently, the pore size notably decreases.
Figure 1. XRD Diffractograms of MSN, MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru.
3.1.3. N2 adsorption-desorption isotherms
The unmodified MSN and the ruthenium-functionalised materials MSN-CL-Ru,
MSN-CNO-Ru and MSN-TRI-MSN-CNO-Ru have been characterised by nitrogen adsorption / desorption isotherms. In
the cases of MSN, MSN-CL-Ru and MSN-CNO-Ru, the observed isotherms are between type IV and type VI (Figure 2) according to IUPAC classification.61 These isotherms are associated with mesoporous silica nanoparticles and show three different parts, namely, below P/P0 ca.
0.4, between P/P0 0.4 and 0.9 and above P/P0 0.9. A hysteresis loop was observed in these
cases between P/P0 1.0 and 0.85 which is correlated with the existence of capillary
condensation into the mesoporous structure of the porous system. Furthermore, an additional hysteresis loop was observed between P/P0 0.4 and 0.15.
On the other hand, the isotherm found for material MSN-TRI-Ru is a type III isotherm (Figure 2) with a hysteresis loop between P/P0 1.0 and 0.85 which is indicative of a non-porous
system as it corresponds after the physical adsorption of the ruthenium complex inside the pores.
Table 2. Physical parameters of MSN, MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru
measured by N2 adsorption-desorption isotherms
Material SBET (m 2
/g) Total pore volume (cm3/g) DP (BJH) 1 (nm) MSN 853 0.728 2.54 MSN-CL-Ru 717 0.563 <2.0 MSN-CNO-Ru 295 0.299 <2.0 MSN-TRI-Ru 7 0.022 <2.0 1
Barrett, Joyner and Halenda. 0 500 1000 1500 2000 2500 3000 3500 4000 0 2 4 6 8 10 In te n s it y ( a .u .) 2θ(º) MSN MSN-CL-Ru MSN-CNO-Ru MSN-TRI-Ru
Table 2 gives all the physical parameters that show that starting MSN material has a very high SBET of 853 m
2
g-1 and a BJH pore diameter of 2.5 Å. In addition, a very narrow distribution of the pore diameter was observed (see supplementary material). After functionalisation with the different ligands and ruthenium complex, the SBET significantly decreased to 717 and 295 m
2
g-1 for MSN-CL-Ru and MSN-CNO-Ru, respectively. This decrease indicates that the functionalisation takes place inside the pores of the mesoporous system. In the case of
MSN-TRI-Ru, the decrease in the BET surface is dramatic going down to 7 m2g-1 owing to the complete blocking of the pores by the incorporation of both the tris-amino ligand and the ruthenium complex. A very high congestion of the pores is already observed after functionalization with the tris amine in MSN-TRI as the BET surface is already as low as 47 m2g
-1
(Figure S8 and Table S1 of supplementary material) which is the main reason of the relatively low physisorption of the ruthenium compound. A similar behaviour has already been found for analogous systems in previous studies.59 Nevertheless, one cannot rule out that a plausible explanation of the very low surface area may also be an inhomogeneous functionalization.
It is important to note that, after the functionalisation with the different ligands and the ruthenium complex, the resulting materials gave pore size distributions with a maximum below 2.0 nm, which is a significant decrease of more than 0.5 nm compared with the unmodified
MSN, indicating, again, that the functionalisation may have been taken place inside the pores of
the nanostructured system.
Figure 2. N2 adsorption / desorption isotherms of MSN, MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru. 3.1.4. Solid-state NMR spectroscopy
The 13C CP MAS spectra of materials MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru. (Figure 3) have been recorded. The spectra show the signals associated with the different carbon atoms of the different ligands and the ruthenium complex that are attached to the mesoporous silica nanoparticles. In general, two different set of signals are found in each of the
spectra of the different studied materials. One set of signals was recorded between ca. 0 and 60 ppm and was assigned to the aliphatic carbon atoms of the fragment coming from the 3-chloropropyl moiety in MSN-CL-Ru, from the 3-isocyanatopropyl moiety in MSN-CNO-Ru or from the tris-amine in MSN-TRI-Ru. In addition, the aliphatic carbon atoms of the alcoholic fragment of the ruthenium complex are also part of that set of signals. The second set of signals appear between 110 and 170 ppm and is assigned to the aromatic carbon atoms of the covalently bound (for MSN-CL-Ru, MSN-CNO-Ru) or physically adsorbed (in MSN-TRI-Ru) polypyridyl ruthenium complex in the different synthesised materials. It is important to note that the intensity of the signals of the carbon atoms of the ruthenium complex are not very high in the case of the materials MSN-CL-Ru and MSN-CNO-Ru (covalently bound) as it corresponds with a low ruthenium functionalisation rate. However, in the case of the material MSN-TRI-Ru, the relative intensity of the signals of the carbon atoms of the polypyridyl ligand of the ruthenium complex are much higher, as the quantity of immobilised complex is also much higher.
Figure 3. 13C CP MAS NMR spectra of MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru.
3.1.6. IR and UV spectroscopy
DR-UV-vis spectra of the studied ruthenium containing materials MSN-CL-Ru,
MSN-CNO-Ru and MSN-TRI-MSN-CNO-Ru were recorded. The spectra show a very intense absorption peak at 286
nm and a small shoulder of absorption at 443 nm which correspond with the incorporation of the ruthenium complex (see supplementary material). In addition, FT-IR spectra of the materials show the appearance of typical C-H and N-H small intensity bands due to the immobilisation of the ruthenium complex together with the typical characteristic bands of the silica intense broad bands between 3450 and 3200 cm–1, (O–H stretching of the silanol groups of the material and the adsorbed water molecules), a broad band at around 1100 cm–1 (assigned to the siloxane (Si–O–Si) groups) and the stretching band of the Si–O bonds at ca. 900 cm–1. Finally, a band at
1630 cm–1 was observed and corresponds with the deformation vibrations of adsorbed water molecules (see supplementary material).
3.1.7. SEM and TEM
The ruthenium-functionalised materials MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru were characterised by Scanning Electronic Microscopy (SEM) and Transmission Electronic Microscopy (TEM) to observe their morphology, particle size and pore size.
In the SEM micrographs, one can see the agglomeration of the spherical nanoparticles (Figure 4). It seems that the functionalisation with the ruthenium complex, either by covalent binding or physical adsorption, do not lead to changes in the spherical morphology of the nanoparticles.
Figure 4. SEM images of MSN, MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru.
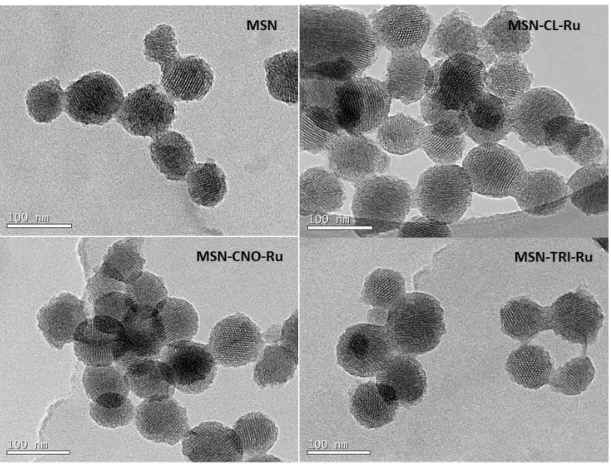
TEM images of the ruthenium-functionalised nanomaterials MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru (Figure 5) show the porous spheres with hexagonal ordered porous parallel channels. The particle size distribution calculated by using ImageJ® program is very narrow and is of 77±13 nm for MSN-CL-Ru, 72±8 nm for MSN-CNO-Ru and 77±10 nm for MSN-TRI-Ru. The differences in size with the different functionalization are only probably due to a statistic factor. It seems, therefore, that the functionalisation with the ruthenium complex does not have any significant effect on the morphology or particle size of the materials.
Figure 5. TEM images of MSN, MSN-CL-Ru, MSN-CNO-Ru and MSN-TRI-Ru.
3.2. Biological studies
The (photo-)toxic potential of the new MSNs conjugated to the ruthenium photosensitizer were evaluated on human cervical HeLa cancer cells. In this assay, we chose to investigate
MSN-CNO-Ru (3.1 % Ru loading) and MSN-TRI-Ru (7.5 % Ru loading), which have the highest Ru
loading. The parent Ru(II) complex was also included as control in our experiments. Of note, for a fair comparison purpose, the concentrations are given as µM of the ruthenium complex in the materials. The results are summarised in Table 3 and representative dose-response plots are shown in Figure 6. The cells were treated with increasing concentrations of materials and incubated for 4 h and 48 h to test cytotoxicity in the dark. As shown in Table 3, very low toxicity (IC50 values 183-278 µM, Table 3) was noticed for both MSNs and the parent Ru(II) complex. In
the 4 h incubation assay in the dark, only MSN-TRI-Ru showed some toxicity with an IC50 value
of 252 µM. The parent Ru(II) complex and MSN-CNO-Ru turned out to be non-toxic up to a 400 µM concentration. These results are in line with our expectations considering that MSN-TRI-Ru has the highest Ru loading of the material series. Unfortunately, as shown in Table 3, no significant differences in the IC50 values were observed when cells treated with our compounds
were irradiated with 350 nm light for 10 min (2.58 J.cm-2). We note that this light dose is not toxic for cells.
In order to rationalise the modest results obtained when using 350 nm light, two hypothesises can be envisioned, namely 1) a quenching of the Ru(II) complex due to a possible surface quenching or 2) a too low Ru loading. Regarding 2), our efforts to quantify the uptake of these compounds using either ICP-MS or fluorescence microscopy was fruitless due to precipitation of the ruthenium-containing MSNs in cell culture media. For 1), we have carried out emission, luminescence, lifetimes and 1O2 quantum yields measurements on the most active material,
Table 3. (Photo-)toxicity of MSNs and parent Ru(II) complex in HeLa cells given in µM of
ruthenium complex for Ru-complex or µM of loaded ruthenium complex for materials
MSN-CNO-Ru and MSN-TRI-Ru. Data in brackets correspond with the quantity of functionalised
materials MSN-CNO-Ru and MSN-TRI-Ru in µg/mL.
Compounds 48 h / dark 4 h / dark 4 h / 350 nm light, 10 min (2.58 J.cm-2) MSN-CNO-Ru 278±14 [903±40] >400 [>1300] >400 [>1300] MSN-TRI-Ru 219±21 [295±26] 252±16 [340±19] 216±18 [291±22] Ru-complex 183±19 >400 329±21
Figure 6. Representative dose-response curves of the phototoxicity assays with MSNs and
parent Ru complex in Hela cells. Note: Concentrations are given as µM of the ruthenium complex in the materials.
3.3. Photophysical Studies
3.3.1 Emission, Luminescence and Lifetimes
The low biological activity observed in section 3.2 could be potentially explained by a quenching of the excited state of the Ru(II) complex in the nanoparticle. In order to exclude this possibility, emission, luminescence and lifetimes measurements were undertaken on the most active nanoparticles (TRI-Ru) and compared to the Ru(II) complex itself. The values of
MSN-TRI-Ru and the Ru(II) complex presented in Table 4 are very similar and seem to rule out any
quenching of the excited state of the Ru(II) complex.
Table 4. Spectroscopic properties of MSN-TRI-Ru in CH3CN at room temperature.
Compound Emission λem / nm Φem / % Lifetime / ns
air degassed
MSN-TRI-Ru 622 1.9 205 952
3.3.2 Singlet oxygen production
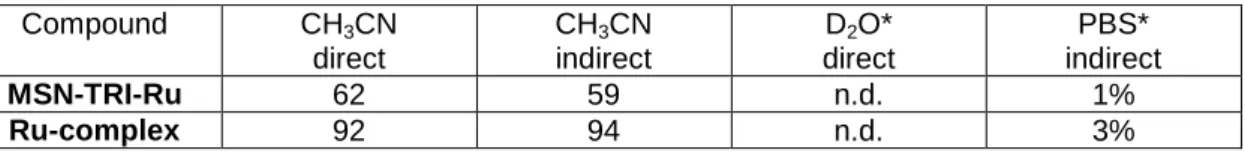
To further explore a potential quenching of the excited state of the Ru(II) complex, the singlet oxygen quantum yields of MSN-TRI-Ru were determined by direct and indirect methods, as previously described by some of us.35,51,53,56 The values presented in Table 5 are in the same order of magnitude and seem to exclude a quenching of the excited state. Overall, the results presented in this section tend to confirm that the low Ru loading is responsible for the modest biological activity observed.
Table 5. 1O2 quantum yields (Φ( 1
O2)) in air saturated CH3CN and aqueous solution determined
by direct and indirect method upon excitation at 450 nm. Average of three independent measurements, +-10%. n.d.=not detectable. * = containing 5% DMSO.
Compound CH3CN direct CH3CN indirect D2O* direct PBS* indirect MSN-TRI-Ru 62 59 n.d. 1% Ru-complex 92 94 n.d. 3% 4. Conclusions
We have synthesised and characterised by different methods various mesoporous silica nanoparticles functionalised with the photoactive ruthenium(II) complex Ru(bipy)2
-dppz-7-hydroxymethyl][PF6]2. We have observed that the covalent binding of the ruthenium complex
can be achieved with modest functionalisation rates (between ca. 2-3% wt. Ru) by its reaction with chloropropyl- or isocyanatopropyl-functionalised nanoparticles via ether or urethane formation with the addition of different bases. In addition, physical adsorption of the ruthenium(II) complex leads to higher functionalisation rates of ca. 7% when using polyamine ligands attached to the mesoporous silica nanoparticles. The preliminary biological results on HeLa cervical cancer cells showed that the low loading of ruthenium complexes hampered phototoxicity. Future work will be in the preparation of higher-loader material.
Supplementary Materials: The supplementary material contains the main scheme of the reactions carried
out in this study; experimental description of additional reactions for incorporation of ruthenium complex with low functionalization rates; pore size distribution of the studied materials; DR-UV spectra of the studied materials, FT-IR spectra of the studied materials, MSN and ruthenium complex, nitrogen adsorption-desorption isotherm of MSN-TRI and table with the comparison of physical parameters of MSN,
MSN-TRI, MSN-TRI-Ru measured by N2 adsorption-desorption isotherm and emission and lifetime spectra of MSN-TRI-Ru.
Acknowledgments: We gratefully acknowledge financial support from the former Ministerio de Economía
y Competitividad-FEDER (current Ministerio de Ciencia, Innovación y Universidades) Spain (Grant no. CTQ2015-66164-R and CTQ2017-90802-REDT), the Swiss National Science Foundation (Professorships No. PP00P2_133568 and PP00P2_157545 to G.G), the University of Zurich (G.G) and the ERC (Consolidator Grant PhotoMedMet (GA 681679) to G.G.). We would also like to thank Universidad Rey Juan Carlos and Banco de Santander for supporting our Research Group of Excellence QUINANOAP (S.G.). This work has received support under the program «Investissements d’Avenir » launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G.G.). We thank Dr. Philippe Goldner for access to a state-of-the-art photophysical apparatus.
Ethical statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
References
2 K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563.
3 U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des. Devel. Ther., 2017, 11, 599–616. 4 L. Astolfi, S. Ghiselli, V. Guaran, M. Chicca, E. Simoni, E. Olivetto, G. Lelli and A. Martini,
Oncol. Rep., 2013, 29, 1285–1292.
5 M. Wenzel and A. Casini, Coord. Chem. Rev., 2017, 352, 432–460.
6 S. Spreckelmeyer, C. Orvig and A. Casini, Molecules, 2014, 19, 15584–15610.
7 G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar,
Cancers, 2014, 6, 1769–1792.
8 X. Dong and R. J. Mumper, Nanomedicine, 2010, 5, 597–615.
9 T. Kiss, É. A. Enyedy and T. Jakusch, Coord. Chem. Rev., 2017, 352, 401–423.
10 W. Wani, S. Shreaz, S. Prashar and S. Gómez-Ruiz, Coord. Chem. Rev., 2016, 312, 67– 98
11 Y. Ellahioui, S. Prashar and S. Gómez-Ruiz, Curr. Med. Chem., 2016, 23, 4450–4467. 12 D. Pérez‐Quintanilla, S. Gómez‐Ruiz, Ž. Žižak, I. Sierra, S. Prashar, I. del Hierro, M.
Fajardo, Z. D. Juranić and G. N. Kaluđerović, Chem. Eur. J., 2009, 15, 5588–5597.
13 G. N. Kaluđerović, D. Pérez-Quintanilla, I. Sierra, S. Prashar, I. del Hierro, Ž. Žižak, Z. D. Juranić, M. Fajardo and S. Gómez-Ruiz, J. Mater. Chem., 2010, 20, 806–814.
14 G. N. Kaluderović, D. Pérez-Quintanilla, Z. Zizak, Z. D. Juranić and S. Gómez-Ruiz, Dalton
Trans, 2010, 39, 2597–2608.
15 A. García-Peñas, S. Gómez-Ruiz, D. Pérez-Quintanilla, R. Paschke, I. Sierra, S. Prashar, I. del Hierro and G. N. Kaluđerović, J. Inorg. Biochem., 2012, 106, 100–110.
16 J. Ceballos‐Torres, P. Virag, M. Cenariu, S. Prashar, M. Fajardo, E. Fischer‐Fodor and S. Gómez‐Ruiz, Chem. Eur. J., 2014, 20, 10811–10828.
17 J. Ceballos-Torres, S. Prashar, M. Fajardo, A. Chicca, J. Gertsch, A. B. Pinar and S. Gómez-Ruiz, Organometallics, 2015, 34, 2522–2532.
18 S. Gómez-Ruiz, A. García-Peñas, S. Prashar, A. Rodríguez-Diéguez and E. Fischer-Fodor,
Materials, 2018, 11, 2.
19 I. del Hierro, S. Gómez-Ruiz, Y. Pérez, P. Cruz, S. Prashar and M. Fajardo, Dalton Trans. 2018, in press, DOI: 10.1039/C8DT02011F
20 D. Díaz-García, D. Cenariu, Y. Pérez, P. Cruz, I. del Hierro, S. Prashar, E. Fischer-Fodor and S. Gómez-Ruiz, Dalton Trans. 2018, in press, DOI: 10.1039/C8DT01677A
21 M. Z. Bulatović, D. Maksimović-Ivanić, C. Bensing, S. Gómez-Ruiz, D. Steinborn, H. Schmidt, M. Mojić, A. Korać, I. Golić, D. Pérez-Quintanilla, M. Momčilović, S. Mijatović and G. N. Kaluđerović, Angew. Chem. Int. Ed. Engl., 2014, 53, 5982–5987.
22 C. Bensing, M. Mojić, S. Gómez-Ruiz, S. Carralero, B. Dojčinović, D. Maksimović-Ivanić, S. Mijatović and G. N. Kaluđerović, Dalton Trans, 2016, 45, 18984–18993.
23 M. Appold, C. Mari, C. Lederle, J. Elbert, C. Schmidt, I. Ott, B. Stühn, G. Gasser and M. Gallei, Polym. Chem., 2017, 8, 890–900.
24 C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686.
25 L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804.
26 F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736.
27 G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138.
28 J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126. 29 C. Mari and G. Gasser, Chimia, 2015, 69, 176–181.
30 B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. de Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095–17101.
31 M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem.
Soc., 2013, 135, 11274–11282.
32 R. B. Sears, L. E. Joyce, M. Ojaimi, J. C. Gallucci, R. P. Thummel and C. Turro, J. Inorg.
Biochem., 2013, 121, 77–87.
33 L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. Le Dévédec and S. Bonnet, Angew. Chem. Int. Ed. Engl., 2017, 56, 11549–11553.
34 A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgenträger, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292.
35 C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem. Eur. J., 2014, 20, 14421–14436.
36 H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew.
Chem. Int. Ed. Engl., 2015, 54, 14049–14052.
37 S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519.
38 R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161– 17175.
39 P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol.
Sci., 2016, 15, 481–495.
40 C. S. Burke, A. Byrne and T. E. Keyes, J. Am. Chem. Soc., 2018, 140, 6945–6955.
41 Health Canada Approves Clinical Trial Application for Anti-Cancer Drug, http://theralase.com/pressrelease/health-canada-approves-clinical-trial-application-anti-cancer-drug/, (accessed June 4, 2018).
42 S. Rojas, F. J. Carmona, E. Barea and C. R. Maldonado, J. Inorg. Biochem., 2017, 166, 87–93.
43 D. Edeler, S. Arlt, V. Petković, G. Ludwig, D. Drača, D. Maksimović-Ivanić, S. Mijatović and G. N. Kaluđerović, J. Inorg. Biochem., 2018, 180, 155–162.
44 L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y.-S. Wong and T. Chen, Adv. Funct. Mater., 2014, 24, 2754–2763.
45 D. Zhang, Z. Wu, J. Xu, J. Liang, J. Li and W. Yang, Langmuir, 2010, 26, 6657–6662. 46 J. Wen, H. Yan, P. Xia, Y. Xu, H. Li and S. Sun, Sci. China Chem., 2017, 60, 799–805. 47 M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y.
Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603–11613.
48 S. Lechevallier, R. Mauricot, H. Gros‐Dagnac, S. Chevreux, G. Lemercier, E. Phonesouk, M. Golzio and M. Verelst, ChemPlusChem, 2017, 82, 770–777.
49 N. Ž. Knežević, V. Stojanovic, A. Chaix, E. Bouffard, K. E. Cheikh, A. Morère, M. Maynadier, G. Lemercier, M. Garcia, M. Gary-Bobo, J.-O. Durand and F. Cunin, J. Mater.
Chem. B, 2016, 4, 1337–1342.
50 R. Chouikrat, A. Seve, R. Vanderesse, H. Benachour, M. Barberi-Heyob, S. Richeter, L. Raehm, J.-O. Durand, M. Verelst and C. Frochot, Curr. Med. Chem., 2012, 19, 781–792. 51 J. Hess, H. Huang, A. Kaiser, V. Pierroz, O. Blacque, H. Chao and G. Gasser, Chem. Eur.
J., 2017, 23, 9888–9896.
52 C. Mari, H. Huang, R. Rubbiani, M. Schulze, F. Würthner, H. Chao and G. Gasser, Eur. J.
Inorg. Chem., 2017, 2017, 1745–1752.
53 C. Mari, R. Rubbiani and G. Gasser, Inorg. Chim. Acta, 2017, 454, 21–26.
54 Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398– 8400.
55 K. Nakamaru, Bull. Chem. Soc. Japan 1982, 55, 1639–1640.
56 C. Mari, V. Pierroz, A. Leonidova, S. Ferrari and G. Gasser, Eur. J. Inorg. Chem. 2015, 3879–3891.
57 I. E. Kochevar and R. W. Redmond, Methods in Enzymology, 2000, 319, 20–28.
58 D. García‐Fresnadillo, Y. Georgiadou, G. Orellana, A. M. Braun and E. Oliveros, Helv.
Chim. Acta 1996, 79, 1222–1238.
59 V. S. Bollu, A. K. Barui, S. K. Mondal, S. Prashar, M. Fajardo, D. Briones, A. Rodríguez-Diéguez, C. R. Patra and S. Gómez-Ruiz, Mater. Sci. Eng. C Mater. Biol. Appl., 2016, 63, 393–410.
60 S. Sánchez-Muñoz, S. Gómez-Ruiz, D. Pérez-Quintanilla, S. Morante-Zarcero, I. Sierra, S. Prashar, R. Paschke and G. N. Kaluđerović, ChemMedChem, 2012, 7, 670–679.
61 M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodríguez-Reinoso and J. Rouquerol, Pure Appl. Chem., 2015, 87, 1051–1069.