Étude du mécanisme moléculaire d'incorporation
de la molécule de l'hôte ICAM-1 par le VIH-1
Thèse
Pascal Jalaguier
Doctorat en Microbiologie et Immunologie
Philosophiae Doctor (Ph. D.)
Québec, Canada
© Pascal Jalaguier, 2013
iii
Résumé
Le virus de l’immunodéficience humaine de type-1 (VIH-1) a été identifié comme étant l’agent responsable de la pandémie qui frappe actuellement notre civilisation. En une trentaine d’années le virus à causé la mort de plus de 35 millions de personnes. Une telle épidémie dans l’histoire de l’humanité n’est pas une première; rappelons-nous dans le passé, la peste noire ou encore la grippe espagnole qui ont fait elles aussi des millions de morts. Cependant, pour la première fois dans l’ère moderne, nous avons la possibilité de lutter contre ce fléau grâce à l’étude approfondie de la biologie du virus qui a permis, entre autres, l’avènement de la trithérapie. Le VIH-1 acquiert un nombre impressionnant de molécules cellulaires tout au long de son cycle réplicatif. En dépit de tous les efforts consacrés à l’étude des molécules de l’hôte, le(s) mécanisme(s) exact par lequel toutes ces molécules sont acquises n'est toujours pas connu. Néanmoins, dans le cas d'ICAM-1, une des molécules transmembranaires les plus étudié il apparaît que le précurseur viral Pr55Gag
est un candidat potentiel d'interaction avec ICAM-1. Par conséquent, nous avons étudié et caractérisé au niveau moléculaire le processus d'incorporation d'ICAM-1 en utilisant dans un premier temps un modèle de pseudo particules virales (VLPs) dérivé de Pr55Gag. La
substitution de plusieurs domaines de Pr55Gag tels que la nucléocapside, SP2 et p6 n'ont pas
eu d'effet sur son acquisition. Par la suite, nous avons démontré que la protéine de matrice (MA) est nécessaire à son incorporation. Nous avons confirmé ces résultats préliminaires en générant le mutant de matrice dans un clone moléculaire couramment utilisé en laboratoire (NL4.3). Des études complémentaires suggèrent que les deux tiers C-terminal de la MA sont importants et en particulier treize acides aminés présents dans l'hélice-α 5. De plus, en se basant sur la modélisation 3D des interactions protéines-protéines et en validant par la suite ces prédictions par immunocapture, nous avons trouvé qu'une série d'acides aminés acides de la MA interagit avec des acides aminés basiques présents sur le domaine cytoplasmique d'ICAM-1 et sont responsables de son incorporation. En résumé, nos résultats apportent de nouvelles connaissances dans le mécanisme moléculaire régissant l'acquisition d'ICAM-1, une molécule de l'hôte connu pour renforcer l'infectiosité virale et moduler la pathogénèse du VIH-1.
v
Table des matières
Résumé ... iii
Table des matières ... v
Liste des illustrations ... ix
Liste des tableaux ... xi
Liste des abréviations ... xiii
Avant-propos ... xvii
Remerciements ... xvii
Contributions scientifiques ... xix
Introduction ... 1
Chapitre 1 : Le virus de l’immunodéficience humaine de type 1 (VIH-1) ... 1
1.1. Origine des VIH et VIS ... 4
1.2. L'infection par le VIH-1 ... 5
1.3. Premiers cas documentés ... 5
1.4. Classification ... 6
1.5. Organisation génomique et morphologique ... 7
1.6. Les protéines virales ... 9
1.6.1. Group-specific antigen (Gag) ... 9
1.6.2. Matrice (MA) ... 10 1.6.3. Capside (CA) ... 11 1.6.4. Nucléocapside (NC) ... 13 1.6.5. Domaine tardif (p6) ... 14 1.6.6. Gag-Pol ... 14 1.6.7. Protéase (PR) ... 14 1.6.8. Transcriptase inverse (RT) ... 15 1.6.9. Intégrase (IN) ... 15 1.6.10. Enveloppe (Env)... 16
1.6.11. Protéines virales régulatrices ... 18
1.6.12. Protéines virales accessoires ... 18
1.7. Tropisme viral et corécepteurs ... 20
1.8. Le cycle réplicatif ... 23
vi
1.8.2. Les étapes tardives ... 29
1.9. Encapsidation du génome viral ... 34
1.10. Transport des composants viraux au site d'assemblage ... 36
1.11. Acquisition de l’enveloppe virale ... 39
1.12. Libération des particules virales ... 41
1.13. Modèle de fission membranaire ... 44
1.14. Maturation des virions ... 45
1.14.1. Protéolyse séquentielle de Gag ... 45
Chapitre 2 : La pathogenèse virale et traitement ... 49
2.1 La pathogenèse ... 49
2.2. Le traitement antirétroviral ... 52
Chapitre 3 : L’incorporation des molécules de l'hôte par le VIH-1 ... 55
3.1. Identification des protéines étrangères acquises par le VIH-1 ... 55
3.2. Mécanismes d’incorporation des molécules de l’hôte ... 58
3.3. Principales protéines de l'hôte incorporées par le VIH-1 ... 61
3.3.1. ICAM-1 ... 62
3.3.2. HLA-II ... 63
3.3.3. Autres molécules de surface ... 63
3.3.4. La protéine de choc thermique 70 (Hsp70) ... 64
3.3.5. INI1/HSNF5 ... 64
3.3.6. APOBEC3G ... 65
3.3.7. Cyclophiline A (CypA) ... 65
3.3.8. Le cytosquelette d'actine ... 65
3.3.9. Les tétraspanines ... 66
Chapitre 4 : L'interaction entre ICAM-1 et LFA-1 ... 67
4.1. Les Intégrines ... 67
4.1.1. Les gènes de LFA-1 ... 67
4.1.2. Structure de LFA-1 ... 68
4.2. La superfamille des Immunoglobulines ... 69
4.2.1. Gène ICAM-1 ... 70
4.2.2. Structure ICAM-1 ... 70
vii
4.2.4. La signalisation dépendante d'ICAM-1 ... 72
4.2.5. ICAM-1 et les radeaux lipidiques ... 74
4.3. Mécanisme et fonction de l'interaction ICAM-1/LFA-1 ... 75
4.4. Les pathologies associées aux molécules de surface LFA-1 et à ICAM-1 ... 77
Chapitre 5 : Hypothèses et objectifs ... 79
Chapitre 6 : La production efficace de pseudo particules virales du VIH-1 à partir d'un vecteur d'expression de mammifère nécessite la présence du domaine N-terminal de la capside. ... 83
Chapitre 7 : L'acquisition sélective d'ICAM-1 par le VIH-1 est dépendante du domaine de matrice... 123
Chapitre 8 : Discussion, perspectives et conclusion ... 165
8.1. Caractérisation fonctionnelle des VLPs ... 166
8.2. Capacité des VLPs à incorporer ICAM-1 ... 168
8.3. Passage du modèle de VLPs au VIH-1 ... 169
8.4. Stratégies d'entrée des peptides de pénétration cellulaire (CPP) ... 175
8.5. Conception d’un peptide inhibiteur d'ICAM-1 couplé à des CCP ... 178
Bibliographie ... 181
Annexe : L'expression de la forme activée du LFA-1 à la surface des cellules cibles permet l'infection des virus ICAM-1 positifs par un mécanisme indépendant du CCR5. ... 213
ix
Liste des illustrations
Figure 1. Microscopie électronique de virus produits à partir de lymphocytes. ... 3
Figure 2. Organisation génomique du VIH-1 ... 7
Figure 3. Organisation morphologique de virion ... 7
Figure 4. Le précurseur protéique Pr55Gag . ... 9
Figure 5. Structure 3D de la matrice du VIH-1. ... 10
Figure 6. Structure 3D de la capside du VIH-1 ... 11
Figure 7. Structure 3D de la nucléocapside... 13
Figure 8. Étapes précoces du cycle viral. ... 23
Figure 9. Résumé de l'entrée du VIH-1. ... 26
Figure 10. Étapes tardives du cycle réplicatif. ... 30
Figure 11. Structure et modélisation 3D de la capside mature. ... 33
Figure 12. Structure secondaire de la région 5' de l'ARN du VIH-1. ... 35
Figure 13. Transport et assemblage de Pr55Gag à la membrane. ... 38
Figure 14. Modèle du «myristyl switch». ... 40
Figure 15. Résumé de la machinerie de bourgeonnement ESCRT. ... 44
Figure 16. Représentation des étapes de maturation du VIH-1. ... 47
Figure 17. Cinétique d'infection. ... 50
Figure 18. Méthodes de détection de protéines cellulaires présentes sur le VIH-1. ... 57
Figure 19. Mécanismes d'incorporation potentielle des protéines de l'hôte par le VIH-1. ... 61
Figure 20. Les trois états conformationnels des intégrines. ... 69
Figure 21. Représentation schématique d'ICAM-1. ... 70
Figure 22. Modèle d'oligomérisation d'ICAM-1. ... 71
Figure 23. ICAM-1 et signalisation. ... 73
Figure 24. Structure simplifiée des radeaux lipidiques.. ... 74
xi
Liste des tableaux
Tableau 1. Corécepteurs du VIH-1, VIH-2 et VIS ... 22 Tableau 2. Liste non exhaustive des CPP ... 175
xiii
Liste des abréviations
ABCE1 ATP binding cassette E1
ADN Acide désoxyribonucléique
ADNc Acide désoxyribonucléique complémentaire
AP Activator protein
APOBEC3G Apolipoprotein B mRNA-editing enzyme-catalytic
Polypeptide-like-3G
ARN Acide ribonucléique
ARNt Acide ribonucléique de transfert
ARV AIDS-associated retroviruses
ATP Adénosine-5'-triphosphate
BCL-2 B-cell lymphoma 2
CA Capside
Ca2+ Ion calcium
CD4BS CD4 binding site
CDC Centers for disease control
CHMP Chromatin modifying proteins
CPA Cellule présentatrice d’antigène
cSMAC Central supramolecular activation clusters
CT C-tail
CTD C-terminal domain
CTL Cytotoxic T lymphocyte
CypA Cyclophiline A
DIS Dimerization initiation site
dNTP Désoxyribonucléotide
EF1-α Elongation factor-like alpha
EIF4G Eukaryotic translation initiation factor 4 gamma
Env Enveloppe
ESCRT Endosomal sorting complexes required for transport
Gag Group-specific antigens
GM1 Ganglioside 1
GRID Gay-related immune deficiency
HR Heptad repeat helice
HRS Hepatocyte growth factor-regulated tyrosine kinase
substrate
HSI Hematopoietic lineage cell-specific protein 1
HSP70 Heat shock protein 70
xiv
ICAM-1 Intercellular adhesion molecule type 1
IFNα Interféron alpha
IGSF Immunoglobulin superfamily
IN Intégrase
INI1 Integrase interactor 1
ITIM Immunoreceptor tyrosine based inhibition motif
JAM-1 Junctional adhesion molecule 1
kDa Kilodalton
LAD Leukocyte adhesion deficiency
LAV Lymphadenopathy associated virus
LEDGF Lens epithelium-derived growth factor
LFA-1 Leukocyte function associated-1 antigen
LTR Long terminal repeat
MA Matrice
Mac-1 Macrophage-1 antigen
MADCAM Mucosal vascular addressin cell adhesion molecule
MIDAS Metal ion dependent adhesion site
MHR Major homology region
MPER Membrane proximal external region
MSD Major splice donor
MVB Multivesicular bodies
NC Nucléocapside
NEF Negative regulatory factor
NFAT Nuclear factor of activated T cell
NF-κB Nuclear factor kappa B
NRE Negative regulatory element
NSI Non-syncytium-inducing
NTD N- terminal domain
PABP Poly(A)-binding protein
PBS Primer binding site
PECAM Platelet endothelial cell adhesion molecule
pH Potentiel hydrogène
PHA Phytohémaglutinine
PIC Pre-integration complex
PKC Protéine kinase C
PIP2 Phosphatidylinositol-biphosphate
PBMC Peripheral blood mononuclear cell
Pol Polymérase
PR Protéase
pSMAC Peripheral supramolecular activation clusters
xv
REV Regulator of expression of virion proteins
RMN Résonance magnétique nucléaire
RT Reverse transcription
RTC Reverse transcription complex
SH Src homology domain
SHP-2 SH2–containing protein tyrosine phosphatase 2
SI Syncytium-inducing
SIDA Syndrome de l’immunodéficience humaine acquise
SL Stem loop
SP1 Spacer peptide 1
SP2 Spacer peptide 2
SRC Sarcome de souris
STAT Stransducers and activators of transcription
TAF TBP (Tata binding protein) associated factor
TAR Tat responsive region
TAT Trans-activator of transcription
TCR T cell receptor
TEM Tetraspanin enriched microdomain
TI Transcriptase inverse
TIP-47 Tail-interacting protein 47
7TM 7 Transmembrane domain
TMD Transmembrane domain
TRIM Tripartite motif
TSG101 Tumor susceptibility gene 101
U3 Unique 3' sequence
UEV Ubiquitin E2 variant
UNG2 Nuclear uracil-DNA glycosylase
UTR Unstranslated regions
VCAM Vascular cell adhesion molecule 1
VIF Viral infectivity factor
VIH Virus de l’immunodéficience humaine
VIS Virus de l’immunodéficience simienne
VLP Virus-like particle
VPR Viral protein R
VPS Vacuolar protein sorting
VSV-G Vesicular stomatitis virus G glycoprotein
xvii
Avant-propos
Remerciements
Mes premiers remerciements sont adressés aux Drs. Benoit Barbeau, Louis Flamand et Caroline Gilbert pour avoir accepté d’évaluer ma thèse dans un délai relativement court.
Je souhaite remercier mon directeur de thèse le Dr. Michel J. Tremblay de m’avoir accueilli si gentiment durant ces cinq années. Merci pour votre disponibilité, votre efficacité et votre humanité. Je voulais associer ces remerciements à mon co-directeur de thèse le Dr. Réjean Cantin qui, en plus du temps qu’il m’a accordé tout au long de ma thèse, m’a fait partager sa passion pour le golf. Ce qui fut une de mes plus belles découvertes au Québec. Merci Redge d’avoir pris le temps de corriger ma thèse pendant ton congé parental.
Par la suite, et pour être sûr de n’oublier personne, un grand merci à l’ensemble des membres passés et présents de l’équipe MJT. Ainsi qu’aux personnels du CRI. Si je fais allusion au CRI que dire alors de mes amis du Bloc T. Un grand merci à mon ami de longue date Mathieu ainsi qu’à ses acolytes. J’ai dû venir au Canada pour trouver des Français qui pensent comme moi…
Une mention spéciale à mes amis de toujours Jo, Tony, Cyril et Jérémy qui m’ont manqué toutes ces années. Ne vous inquiétez pas les gars, je suis de retour.
Bien sûr, je veux remercier mes parents et ma sœur à qui je dois tout, et même plus, pour leur soutien inconditionnel, même s’ils n’ont jamais vraiment compris ce que je faisais à la fac et encore moins au doctorat. Vous avez toujours été là pour moi. Un grand merci à mes beaux-parents que je n’ai pas souvent vu ces dernières années, mais je suis ravi de faire partie de leur famille.
Finalement, mes derniers remerciements iront à ma fiancée Sandra pour avoir vécu loin des siens pendant mon doctorat et de m’avoir supporté au quotidien. Merci pour ce merveilleux cadeau que tu m’as fait en me donnant une fille. J’espère qu’elle sera un jour aussi fière de moi que je le suis d’elle. Je vous aime.
xix
Contributions scientifiques
Mes études de doctorat ont mené à la publication d'un article scientifique dans la revue PLOS One en 2011, d'une déclaration d'invention, suivi d'un dépôt de brevet et de deux autres papiers en premiers auteur soumis en 2013.
Le premier article «Efficient Production of HIV-1 Virus-Like Particles from a Mammalian Expression Vector Requires the N-Terminal Capsid Domain » publié en 2011. Dans cet article, j'ai réalisé et conçu l'essentiel des expérimentations à l'exception des mutants de Pr55Gag qui ont tous été réalisés par Karine Turcotte et Alexis Danylo. Réjean Cantin m'a suivi dans ma démarche scientifique sous la supervision de Michel J. Tremblay. Karine Turcotte a écrit la partie «Pr55Gag molecular construct» du matériel et méthode. J'ai
effectué l'analyse des résultats et rédigé l'article. Réjean Cantin et Michel J. Tremblay ont effectué la correction de l'article.
Le deuxième article «Selective acquisition of host-derived ICAM-1 by HIV-1 is a matrix-dependent process: consequences for subsequent course of infection and therapeutic approaches» a fait l'objet de la déclaration d’invention, d'un dépôt de brevet et est rédigé pour être potentiellement soumis à la revue scientifique PLOS Pathogens. Dans cet article, j'ai réalisé et conçu l'essentiel des expérimentations à l'exception de la partie de modélisation 3D et d'interactions des protéines réalisée par Halim Maaroufi. Réjean Cantin m'a suivi dans ma démarche scientifique sous la supervision de Michel J. Tremblay. Halim Maaroufi a réalisé la figure 5 ainsi que la description correspondante dans le matériel et méthode. J'ai effectué l'analyse des résultats et rédigé l'article. Réjean Cantin et Michel J. Tremblay ont effectué la correction de l'article.
Le troisième article «High affinity LFA-1 on HIV-1 target cells allows R5-tropic strains carrying cellular ICAM-1 to bypass CCR5 usage» a fait l'objet d'une soumission dans la revue scientifique Journal of Virology. Dans cet article, j'ai réalisé la totalité des expérimentations. Réjean Cantin m'a suivi dans ma démarche scientifique sous la supervision de Michel J. Tremblay. J'ai effectué l'analyse des résultats et rédigé l'article. Réjean Cantin et Michel J. Tremblay ont effectué la correction de l'article.
xxi
Citation «…»
1
Introduction
Chapitre 1 : Le virus de l’immunodéficience humaine de
type 1 (VIH-1)
À la fin des années 70 et au début des années 80, des médecins aux États-Unis ont noté une augmentation de consultation quelque peu inhabituelle, où des jeunes hommes précédemment en bonne santé venaient consulter pour des troubles immunologiques. Ce nouveau syndrome est caractérisé par une lymphadénopathie généralisée, des infections opportunistes (pneumonie à Pneumocystis carinii, rétinite associé au cytomégalovirus et méningite à cryptocoque) ainsi qu'une variété de cancers non conventionnels de type lymphome non Hodgkinien et sarcome de Kaposi [1]. Le dénominateur commun de ces malades était une déplétion massive des lymphocytes T CD4+ dans le sang périphérique. Ce
n'est que le 5 juin 1981 que commence officiellement l'épidémie du Syndrome de l’Immunodéficience Acquise (SIDA), lorsque le CDC note dans sa revue Morbidity and Mortality Weekly Report une recrudescence de cas de pneumocystose chez cinq hommes homosexuels à Los Angeles [2,3,4]. En raison du fait que la majorité des patients ont eu de nombreuses relations sexuelles, il est suggéré en juin 1982 qu'un agent infectieux transmis sexuellement pourrait être la cause de cette immunodépression, mais rien n'est vraiment sûr à ce moment [5,6]. Comme les premiers malades sont exclusivement homosexuels, le syndrome est appelé dans un premier temps le gay-related immunodeficiency disease (GRID), mais les autorités sanitaires se rendent compte rapidement que d'autres populations sont touchées, comme les hémophiles [7], les usagers hétérosexuels de drogues par injection intraveineuse, ou encore des immigrants haïtiens [8]. En vue d'abandonner cette dénomination erronée, le CDC créé le terme Acquired immune deficiency syndrome (AIDS) ou en français le SIDA [9]. Dès le départ, l'origine virale est privilégiée, eu égard aux modes de transmission alors identifiés (sanguinet sexuel). Robert Gallo et son équipe, qui ont découvert le premier rétrovirus humain, le HTLV-1 [10], pensent qu'un mutant de ce dernier est la cause du SIDA. Il justifie son hypothèse par le fait que le HTLV-1 fait
2
proliférer les T CD4+. Cet agent infectieux faisant l'inverse, une mutation peut donc en être la cause. Cette hypothèse est renforcée par le fait que certains des cas haïtiens sont positifs à un test de dépistage du HTLV-1. Cette positivité se révèlera être causée par un biais, car la prévalence du HTLV-1 était très élevée en Haïti.
À partir de 1982, avec les premiers cas identifiés en France, la recherche française débute sur ce sujet. Willy Rozenbaum, médecin à l'hôpital Bichat de Paris, veut inciter les chercheurs à étudier plus en amont le SIDA et à en trouver la cause. Par l'intermédiaire de Françoise Brun-Vézinet, une collègue médecin, Willy Rozenbaum contacte Jean-Claude Chermann, Françoise Barré-Sinoussi et Luc Montagnier, de l'unité d'oncologie virale de l'Institut Pasteur, qui disposaient des outils nécessaires pour étudier les rétrovirus. Ces derniers acceptent de commencer les recherches. En 1983, après l'échec de Robert Gallo dans sa tentative d'isoler le virus dans les échantillons sanguins de patients atteints du SIDA. Willy Rozenbaum pense alors que chez les malades du SIDA, la plupart des cellules infectées sont détruites et que c'est la raison du manque de résultats dans ces tentatives d'isolement du virus. Il a alors l'idée de chercher le virus dans un organe riche en lymphocytes, les ganglions lymphatiques de personnes malades mais qui ne sont pas encore en phase de SIDA. En janvier 1983, suite au prélèvement d'un échantillon d'un patient atteint d'une lymphadénopathie, pathologie identifiée comme une maladie opportuniste du stade pré-SIDA. Cet échantillon est mis en culture par Françoise Barré-Sinoussi qui découvre une activité de transcriptase inverse, confirmant la présence d'un rétrovirus. Une apoptose apparaît et l'adjonction de globules blancs à la mise en culture relance alors l'activité de transcriptase inverse. Un examen au microscope électronique a permis de visualiser, pour la première fois, le virus, le 4 février 1983 (figure 1)[11].
3 Figure 1. Microscopie électronique de virus produits à partir de lymphocytes.
Adaptée de [11]
Après une prise de contact avec Robert Gallo, pour un échange d'informations, l'équipe de l'Institut Pasteur confirme que le virus identifié chez le patient lymphadénopathique n'est pas le HTLV-1. Ce nouveau rétrovirus est alors appelé Lymphadenopathy Associated Virus (LAV) et les résultats sont publiés dans Science le 20 mai 1983 [11]. À ce stade, le lien entre le LAV et le SIDA n'est pas clairement établi par l'équipe de Luc Montagnier. David Klatzmann découvrent peu de temps après que le virus détruit les T CD4+ avec lesquels il est mis en culture [12]. On savait que le nombre de lymphocytes T CD4+ diminuait fortement chez les malades atteints du SIDA. Le LAV était donc sûrement l'agent provoquant cette maladie. L'équipe de Robert Gallo publie le 4 mai 1984, dans Science, les résultats de l'isolement d'un virus qu'elle considère comme responsable du SIDA et le nomme HTLV-3, qui s'avérera, bien plus tard, provenir d'un échantillon envoyé par l'Institut Pasteur [13]. L'équipe de Jay A. Levy à San Francisco fait de même le 24 août 1984 et trouve plusieurs rétrovirus, qu'elle nomme AIDS-associated retroviruses (ARV) [14]. Pendant un temps, les trois dénominations HTLV-3, LAV et ARV cohabiteront. En 1986, l’acronyme HIV (ou VIH en français) est choisi.
4
Le 18 juillet 1986, les résultats de l'étude d'un patient venant d'Afrique de l'Ouest sont publiés, dans Science, par l'équipe de Luc Montagnier, en collaboration avec des médecins portugais[15]. Les examens ont permis d'identifier un nouveau type de LAV, le LAV-2. Le séquençage du nouveau virus est réalisé l'année suivante, ainsi que la mise au point d'un test de dépistage. En 1986, le LAV (ainsi que les autres dénominations) est officiellement renommé en virus de l'immunodéficience humaine (VIH), le LAV-1 devient VIH-1 et le LAV-2, le VIH-2.
1.1. Origine des VIH et VIS
Les deux types de VIH (VIH-1 et VIH-2) infectant l'espèce humaine dérivent des virus de l'immunodéficience simienne (VIS), équivalents simiens des VIH [16]. Ainsi, certaines souches de VIH-2 sont impossibles à distinguer des souches VIS retrouvées chez les mangabeys de l'Ouest Africain et il existe une superposition parfaite des zones d'épidémies humaines et simiennes pour le VIH-2. En 1990, une équipe suggérait que le VIH-1 avait pour origine les populations de chimpanzés, se fondant sur l'organisation identique des génomes des souches VIH-1 et des VIS retrouvées chez les chimpanzés [17]. En 1999, l'origine simienne des souches humaines de VIH-1 était confirmée par la mise en évidence chez des patients camerounais de souches extrêmement proches des VIS circulant chez les chimpanzés de la même région.
L'analyse phylogénétique des lentivirus a confirmé le lien entre le VIS et le VIH. Cependant, les deux types de VIH (VIH-1 et VIH-2) sont assez éloignés l'un de l'autre ; et, alors que le VIH-1 est proche du VIScpz (infectant une sous-espèce de chimpanzés dits Pan
troglodytes troglodytes) [18], le VIH-2 est plus proche des VISsmm (infectant les mangabeys
couronnés) et des VISmac (infectant les macaques). Ainsi, le VIH serait issu de deux
5
L'infection par le VIH doit être considérée comme une zoonose, au même titre que d'autres maladies virales. Le réservoir de VIS est particulièrement important : on a recensé dix-huit espèces de singes infectés par des virus très différents sur le plan génomique et antigénique, ce qui implique que de nouvelles souches pourraient infecter l'espèce humaine [19].
1.2. L'infection par le VIH-1
Le passage des différentes souches de VIS, du singe à l'Homme, peut être expliqué par le fait que les singes sont souvent capturés pour servir de gibier ou d'animal de compagnie, et des expositions à du sang contaminé, lors de morsures ou par blessures lors du dépeçage des animaux peuvent expliquer comment ces virus ont infecté l'homme. C'est la théorie dite «du chasseur de viande de brousse», qui a été retenue par la communauté scientifique. Bien que généralement létal pour les virus, le franchissement de la barrière d'espèce, s'il réussit, peut permettre au virus de muter et ainsi de s'adapter à son nouvel hôte [19]. La datation du franchissement de la barrière des espèces n'est pas clairement définie, mais plusieurs études font remonter l'apparition du VIH au début du XXe siècle [20], voire avant, soit entre 1884 et 1924 [21].
1.3. Premiers cas documentés
Le premier signe documenté d'infection par le VIH chez l'homme remonte à 1959, année où une prise de sang est effectuée sur un homme à Léopoldville (l'actuelle Kinshasa), dans le Congo belge [22]. Suivent alors plusieurs patients atteints de maladies rares (notamment la maladie de Kaposi), aujourd'hui considérées comme maladies opportunistes dans les cas d'infections par le VIH. Des tests VIH ont par la suite confirmé la présence du virus. En 1969, aux États-Unis, un adolescent de quinze ans meurt à l'hôpital de Saint-Louis (Missouri) d'une forme particulièrement violente de maladie de Kaposi. Un test VIH est
6
effectué en 1987 par des chercheurs de l'université Tulane qui détectent la présence du VIH-1 dans le sang de l'adolescent, confirmant ainsi les soupçons apparus dès 1984 [23]. Lors de son entretien avec les médecins, le garçon avait déclaré être né à Saint-Louis et n'avoir jamais voyagé ou reçu de transfusion sanguine. Les médecins soupçonnaient le garçon d'être un prostitué, ce qui soutiendrait la thèse d'une contamination sexuelle et impliquerait l'existence d'un cas préalable aux États-Unis. En 1976, un matelot norvégien, sa femme et leur fille de neuf ans meurent des suites du SIDA. Le matelot avait présenté les premiers signes d'infection dès 1966, soit quatre ans après avoir séjourné dans des ports le long des côtes de l'Afrique de l'Ouest [24,25]. En 1977, une chirurgienne danoise, le Dr Grethe Rask, décède des suites du SIDA, après avoir séjourné au Congo dans les années
1970 [26].
1.4. Classification
Le VIH-1 est classé en quatre différents groupes (M, N, O et P). Chacun de ces groupes est le résultat d'événement de transmission entre espèces indépendantes. Le groupe M a été le premier à être découvert. Il représente la forme pandémique du VIH-1, responsable de l'infection de millions de personnes dans le monde. Il est virtuellement présent dans tous les pays. Le groupe O, a été découvert dans le années 1990 et il est moins prévalent que le groupe M [27,28]. En effet, il représente moins de 1% des infections au VIH-1. Il est présent au Cameroun, Gabon et dans les pays voisins[29,30]. Le groupe N a été identifié en 1998 et il est encore moins prévalent que le groupe O [31]. Actuellement, on a recensé uniquement 13 cas d'infection avec le groupe N d'individus vivant au Cameroun [32]. Finalement, le groupe P a été découvert en 2009 chez une Camerounaise vivant en France [33]. En dépit d'une recherche intensive, on a recensé uniquement qu'un autre cas de groupe P également au Cameroun[34]. Il existe neuf sous-types de VIH à l'intérieur du groupe M (A-D, F-H, J et K). À l'échelle mondiale, c'est le sous-type C qui est le plus répandu. Cependant, en Europe, en Amérique, au Japon et en Australie, c'est le sous-type B qui domine[35].
7
1.5. Organisation génomique et morphologique
Figure 2. Organisation génomique du VIH-1
Adaptée de [36]
Figure 3. Organisation morphologique de virion
8
Le VIH-1 est un virus enveloppé d’un diamètre variant de 120 à 200 nm contenant deux molécules d’ARN de polarité positive de 9200 nucléotides (figure 2) [38]. Il est composé d’une bicouche lipidique provenant de la membrane plasmique de la cellule productrice [39,40,41,42,43]. Toutefois, la composition de celle-ci est légèrement différente. En effet, l’analyse des phospholipides de l’enveloppe du VIH-1, comparée à ceux des cellules productrices, montre un enrichissement en cholestérol et en sphingolipides [44]. Dans cette bicouche sont insérées les glycoprotéines de l’enveloppe (Env). Ces dernières sont composées d’un trimère de sous-unités de gp120 et de gp41 reliées de façon non covalente. La quantité moyenne de protéines d’enveloppe à la surface des virions a été estimée par tomographie en microscopie électronique, à 14 trimères par particule [45]. Deux sortes de particules du VIH-1 sont observées en microscopie électronique : les particules immatures et les particules matures. Les particules VIH-1 sont produites par la cellule infectée sous forme immature. Ces particules virales sont non infectieuses. En microscopie électronique, elles se caractérisent par une couche dense aux électrons correspondant à l’accumulation de précurseurs Gag et Gag-Pol organisés de manière radiale sous l’enveloppe virale. Le nombre de précurseurs Gag est estimé à environ 2 500 pour un virion de 130 nm de diamètre [46]. A la fin du bourgeonnement ou peu de temps après leur libération dans le milieu extracellulaire, les particules du VIH-1 immatures sont converties en virus matures et infectieux. La protéase virale (PR) est activée et permet la libération de la transcriptase inverse (RT) et de l’intégrase (IN) à partir de la protéolyse du précurseur Gag-Pol. De plus, le précurseur Gag subit un clivage en 5 sites libérant les domaines de la matrice (MA), de la capside (CA), de la nucléocapside (NC) ainsi que les peptides SP1, SP2 et p6. Le virion subit alors un réarrangement de sa structure interne aboutissant à sa forme mature visible en microscopie électronique (figure 3). La face interne de la bicouche lipidique délimitant la particule est tapissée par les protéines MA. Celle-ci renferme une capside de forme conique formée par les protéines CA. L’analyse biochimique du contenu de la capside a permis de mettre en évidence une structure appelée nucléocapside. Elle est constituée des deux copies du génome ARN dimérisées liées aux protéines NC. La capside contient également les enzymes libérées par la protéolyse du précurseur Gag-Pol : PR, RT et IN. Outre les protéines de structures, le virion renferme également les protéines virales Vpr, Nef et Vif
9
[47], ainsi que de nombreux composants cellulaires dont les mieux caractérisés sont le ARNtLys3 et la cyclophiline A.
1.6. Les protéines virales
1.6.1. Group-specific antigen (Gag)
Le gène viral Gag code pour une polyprotéine de 500 acides aminés (figure 4) [48]. Cette protéine est conservée chez tous les rétrovirus et code pour les protéines structurales. Suite au bourgeonnement, la protéine subit un processus de maturation clivée en de multiples endroits par la protéase virale pour relâcher les protéines de structure du virus, décrites ici-bas [49]. Ce processus est absolument nécessaire pour la création de particules infectieuses matures [50]. Les virions immatures sont quant à eux incapables de procéder à la décapsidation nécessaire pour libérer le génome viral dans le cytoplasme.
10
1.6.2. Matrice (MA)
La MA est une protéine de 132 acides aminés composée de deux domaines de structure et de fonction distinctes. Le domaine N-terminal forme une tête globulaire constituée de quatre hélices-α qui intervient dans la liaison de la membrane plasmique grâce à son groupement myristyl positionné en N-terminal (figure 5). Comme le domaine MA du précurseur Gag, la partie N-terminale de la protéine MA mature est myristoylée par l’enzyme cellulaire N-myristyltransférase [52]. Pourtant, la capacité de liaison de la protéine MA à la membrane est moins efficace que celle du précurseur Gag. Ce phénomène s'explique par un changement de conformation du groupement myristyl qui lui permet d’être exposé dans le contexte du précurseur Gag et d’être séquestré dans le contexte de la protéine MA [53]. La protéine MA cytoplasmique a un rôle important lors des étapes précoces du cycle réplicatif du VIH-1, car elle assiste à des étapes d’import nucléaire du complexe de préintégration (PIC) [54]. Le domaine C-terminal de MA est formé d’une hélice-α projetée hors de la tête globulaire dont l’extrémité est non structuré [55]. Des analyses cristallographiques [53,56,57] ont montré que MA forme préférentiellement des trimères en solution et il semblerait que cette protéine s’assemble à la membrane sous forme d’hexamères de trimères [58]. Les résidus 42 à 77 sont impliqués dans la trimérisation de la protéine. Des mutations au niveau de ces résidus inhibent l’assemblage de la particule virale.
11
1.6.3. Capside (CA)
Les protéines CA sont les composants majeurs de la capside virale dans la particule mature. Celle-ci est composée d’environ 1500 molécules de CA [59] assemblées en réseau de 252 hexamères et de 12 pentamères [60]. La protéine CA monomérique est une protéine de 24 kDa composée de trois domaines : un domaine N-terminal (ou NTD), un domaine C-terminal (ou CTD) et un domaine «linker» flexible reliant les domaines NTD et CTD (figure 6). Le NTD, formé des résidus 1 à 145, est composé d’une structure en épingle à cheveux-β, de 7 hélices-α et d’une boucle riche en résidus proline intervenant dans la liaison de la protéine chaperon cellulaire cyclophiline A (CypA) [61].
Figure 6. Structure 3D de la capside du VIH-1
Adaptée de [62].
L’épingle à cheveux-β intervient comme un «interrupteur» moléculaire entraînant l’hexamérisation des protéines CA en se repliant dans l’interface formée par les hélices 1 à 3, aidant ainsi à placer le domaine NTD sous sa configuration mature [63,64]. La région linker, composée de six résidus, est non structurée dans CA monomérique. Toutefois, suite à l’oligomérisation de CA, elle se replie en hélice 310 [65]. Cette région permet le
12
[65,66]. Le CTD, composé des résidus 151 à 231, est structuré en 4 hélices-α. C’est un domaine très flexible [67,68] permettant à CA de modifier sa conformation sans déstabiliser sa structure en réseau [69]. La protéine CA mature est stabilisée par trois interfaces CA-CA intermoléculaires [70]. La première interface engage les trois premières hélices du domaine NTD pour former un réseau de 18 hélices au centre de l’hexamère (ou un réseau de 15 hélices pour les pentamères de CA). La deuxième interface lie les domaines CTD entre protéines de CA d’hexamères (ou pentamères) adjacents. La troisième interface permet la stabilisation des contacts des protéines de CA au sein d’un même hexamère (ou pentamère) grâce à l’interaction de la quatrième hélice du domaine NTD avec le domaine CTD de la protéine CA voisine. Les rôles respectifs de ces trois interfaces ne sont pas encore strictement établis dans la détermination de la structure conique et de la géométrie de la capside virale. Une simulation de la dynamique moléculaire assistée par informatique a montré que les interactions NTD-NTD seules aboutissent à la formation d’une capside tubulaire plate et que les interactions NTD-CTD sont indispensables à la structure en coque. Ainsi, la force des interactions NTD-NTD détermine le degré de courbure des extrémités de la capside [70]. Ce type d’interaction semble varier le long de son axe de symétrie (figure 6) [71]. La capside est une structure dont la stabilité est finement régulée lors des étapes précoces du cycle réplicatif. Le maintien de la morphologie de la capside virale est essentiel car les mutants ayant une capside dont la morphologie est aberrante perdent leur pouvoir infectieux lors des étapes précoces du cycle réplicatif [72,73]. De plus, les virions ayant subi des modifications de la région N-terminale de CA, au niveau de la boucle fixant la cyclophiline A (CypA), présentent un défaut des étapes d’initiation de la transcription inverse [74,75]. CypA est une cis-trans-peptidyl-propyl isomérase [76] possédant une activité de protéine chaperone dans les cellules [77]. Elle interagit avec la boucle riche en proline de la partie N-terminale du domaine CA du précurseur Gag. Il a été estimé qu’une protéine CypA est incorporée pour 10 précurseurs Gag [78,79,80]. CypA est un facteur indispensable au pouvoir infectieux lors des étapes précoces du cycle réplicatif. En effet, l’inhibition de l’incorporation de CypA par un traitement des cellules productrices à la cyclosporine A (un inhibiteur de CypA), ainsi que par des mutations dans le motif de fixation de CypA, entraîne une forte diminution du pouvoir infectieux des virus produits [78,80] caractérisée par un défaut de la synthèse des premiers intermédiaires de l’ADN
13
proviral [74]. Bien que CypA intervienne dans le repliement des protéines, elle ne semble pas être impliquée dans les étapes d’oligomérisation de Gag, ni dans la stabilité du virion immature. En effet, la visualisation de virions comportant des mutations au niveau du motif de fixation de CypA ne présentent aucun défaut de structure [81].
1.6.4. Nucléocapside (NC)
La protéine de nucléocapside (p7) est composée de 55 acides aminés et est fortement associée aux deux molécules d'ARN viral présent dans chaque virus (figure 7) [82]. NC facilite la dimérisation des molécules d'ARN virale ainsi que plusieurs étapes de la transcription inverse (association de l'ARNt [83], initiation et élongation de la transcription inverse, transfert de brin positif et négatif, déplacement de brin [84]) et de l'intégration. La protéine jouerait aussi un rôle protecteur pour l'ADN viral suite à la transcription inverse [85].
Figure 7. Structure 3D de la nucléocapside
14
1.6.5. Domaine tardif (p6)
La protéine p6 est composée de 52 acides aminés. Elle est située à la fin du précurseur Gag. Elle est impliquée dans l'incorporation de la protéine accessoire Vpr à l'intérieur de la particule virale [86] et dans le processus de bourgeonnement [87], notamment le recrutement du complexe ESCRT de par son interaction avec la protéine Tsgl0l et ALIX [88].
1.6.6. Gag-Pol
Cette protéine est produite suite à un changement de cadre de lecture du ribosome (-1) et code pour une polyprotéine de 1451 acides aminés codant à la fois pour les protéines structurales décrites précédemment et les protéines virales enzymatiques : protéase, transcriptase inverse et intégrase. Le changement de cadre de lecture est causé par une structure ARN secondaire et d'une «séquence glissante» composée de six résidus uridine [89,90]; la polyprotéine Gag-Pol est produite avec un rapport de 1 pour 20 comparativement à Gag.
1.6.7. Protéase (PR)
La protéase virale est constituée de 99 acides aminés. Elle doit se libérer par auto-catalyse du précurseur Gag-Pol avant de pouvoir cliver les autres composants enzymatiques et structuraux. Le dimère ainsi libéré engendre une réaction en chaîne, clivant les autres sites de clivage des précurseurs Gag-Pol présents à l'intérieur de la particule virale, menant à la formation rapide de nouveaux homodimères [91]. Le processus de maturation se poursuit jusqu'à ce que tous les précurseurs Gag soient clivés; la protéase est alors inhibée par l'accumulation du peptide p6 libéré de Gag. La protéase ne reconnaît pas ses cibles selon une séquence consensus, mais plutôt par des facteurs comme l'hydrophobicité, la disponibilité spatiale et la structure secondaire. La protéase virale peut également cliver certaines protéines cellulaires, dont des composantes du cytosquelette (ex. actine, laminine
15
et vimentine [92]), des protéines impliquées dans l'apoptose (Bcl-2 [93] et caspase 8 [94]) et dans la traduction des ARNm (EIF4G [95] et PABP [96]).
1.6.8. Transcriptase inverse (RT)
La transcriptase inverse est une enzyme de 560 acides aminés responsable de la conversion de l'ARN génomique viral en ADN double brin pour l'intégration dans le génome de la cellule hôte. Bien qu'étant libérée du précurseur Gag-Pol par la protéase virale suite au bourgeonnement de la particule virale, la conversion du génome ne s'effectue qu'après les étapes d'entrée et de décapsidation. La transcriptase inverse est en fait un hétérodimère composé de deux sous-unités : p66 et p51 [97]. Les deux sous-unités contiennent un domaine ADN polymérase ARN dépendant alors que p66 contient en plus un domaine RNaseH [98]. Ce domaine est responsable de la dégradation de la portion ARN contenue dans l'hybride ADN/ARN formé dans les étapes intermédiaires de la transcription inverse. La transcriptase inverse ne contient aucune activité d'édition 5'—»3', ce qui se traduit par une incapacité de corriger les erreurs introduites lors de la conversion du génome [99,100]. Par conséquent, le VIH-1 comporte un taux d'accumulation de mutations très élevé, pouvant atteindre 10-3 à 10-4, soit une ou deux mutation(s) par cycle de réplication. Cette
lacune constitue dans certains cas un avantage pour le virus qui peut ainsi rapidement développer les mutations nécessaires pour échapper au système immunitaire et aux drogues antirétrovirales.
1.6.9. Intégrase (IN)
L'intégrase virale est composée d'une chaîne de 288 acides aminés et catalyse l'insertion du génome viral (lorsque converti sous forme ADN double brin) avec le génome de la cellule hôte. Le choix du site d'intégration ne semble pas suivre de règles strictes. Toutefois, il est reconnu que les zones transcriptionnellement actives du génome sont favorisées [101]. L'intégrase est responsable de la catalyse de deux événements principaux, à savoir l'excision de deux nucléotides à chaque extrémité 3' du génome viral [102] et l’insertion de
16
celui-ci dans le génome cellulaire. La forme active de l'intégrase est un homodimère [103]; des formes d'ordre supérieur sont hypothétiquement possibles (tétramère, octamère). L'intégrase interagit avec la protéine cellulaire LEDGF pour se lier à la chromatine. L'événement d'intégration occasionne des dommages à l'ADN et provoque une réponse cellulaire en ce sens [104].
1.6.10. Enveloppe (Env)
Le gène Env code pour une polyprotéine de 856 acides aminés appelée gpl60 qui contient les deux composantes de l'enveloppe virale, gpl20 et gp41. Les protéines de l’enveloppe gp120 et gp41 sont libérées à partir du clivage du précurseur Env (ou gp160) synthétisé au niveau des ribosomes associés au réticulum endoplasmique. Le précurseur Env est tout d’abord synthétisé sous la forme d’une protéine de 90 kDa (p90) [105] possédant à son extrémité N-terminale un peptide signal éliminé une fois entré dans le RE. Lors de son transport jusqu’à la membrane plasmique, il subit d’importantes modifications. Il est glycosylé et oligomérisé en trimère; on parle alors du précurseur gp160. Il contient dans sa partie centrale un site de clivage reconnu par une enzyme cellulaire, la furine. La protéolyse de gp160 génère les glycoprotéines d’enveloppe matures : la glycoprotéine de surface gp120 et la glycoprotéine transmembranaire gp41. Ces deux protéines forment un complexe relié par des interactions non covalentes.
Glycoprotéine 120 (gp120)
La première composante issue de gpl60 est une protéine de 483 acides aminés nommée gpl20. Elle est responsable de l'interaction avec le récepteur CD4 [12,104] et corécepteurs CXCR4 et/ou CCR5 [106,107,108,109] à la surface de la cellule. La gp120 contient 5 domaines relativement bien conservés (C1-C5) et 5 boucles variables (V1-V5) nommées ainsi de part leur grande hétérogénéité génétique. Chacune de ces régions variables possèdent une structure en boucle formée par un pont disulfure à sa base à l'exception de V5. Ces boucles variables sont présentes à la surface de la gp120 et elles participent à l'évasion immunitaire et à l'interaction avec les corécepteurs (V3)[110,111]. La préférence
17
pour l'un ou l'autre des corécepteurs est déterminé par la boucle V3 et influence grandement le type cellulaire que le virus infecte. Cette région hypervariable est aussi la cible de la plupart des anticorps neutralisants, de par son exposition à la surface de la molécule [111]. Des études de tomographie ont démontré que la gpl20 est retrouvée sous forme de trimère [112] et est acquise à la particule virale par l'intermédiaire de gp41.
Glycoprotéine 41 (gp41)
La gp41 est une glycoprotéine transmembranaire d’environ 345 acides aminés organisée en trois domaines majeurs : le domaine extracellulaire (ectodomaine), le domaine transmembranaire (TMD pour Transmembrane Domain) et le domaine C-terminal appelé CT (C-Tail). Le domaine extracellulaire contient les régions impliquées dans les étapes de fusion dont la partie N-terminale hydrophobe appelée peptide de fusion [113], une région polaire et deux régions hydrophobes (HR1 et HR2) en hélice-α. HR1 et HR2 sont liées ensemble par la formation d’un pont disulfure avec une boucle hydrophile. Le peptide de fusion est normalement caché dans la structure quaternaire gp120/gp41. Lors des étapes de fixation des glycoprotéines d’enveloppe sur les récepteurs et corécepteurs cellulaires, la gp41 change de conformation et expose le peptide de fusion au niveau de la membrane cellulaire. Cette étape permet la formation du pore de fusion [112,114,115]. La région HR2 est suivie d’un domaine de 24 acides aminés, riche en tryptophane appelé Membrane Proximal External Region (MPER)[116,117]. Cette région très conservée est indispensable à la «fusogenécité» et donc au pouvoir infectieux du virus. Le TMD de la gp41 est une petite séquence très conservée de 25 acides aminés. Deux modèles topologiques de la gp41 coexistent actuellement. Dans le premier modèle, le TMD constitué d’hélice-α traverse la membrane plasmique une seule fois, plaçant le domaine CT dans la particule. Le deuxième modèle, basé sur la reconnaissance d’épitopes par des anticorps neutralisants, propose que le domaine TMD traverse trois fois la membrane et expose ainsi une partie du domaine CT du côté externe de la membrane [118]. Le domaine CT est constitué en son centre de trois segments amphiphiles en hélice-α appelés LLP-1, LLP-2 et LLP-3. La structure des segments LLP est très conservée chez les lentivirus.
18
1.6.11. Protéines virales régulatrices
Transactivator of Transcription (Tat)
La protéine Tat est une protéine de 14-kDa, constituée de 86 acides aminés qui peut être divisée en cinq domaines. Les domaines 1 à 3 sont essentiels pour son activité de transactivation. Le domaine 4 permet sa liaison à l'ARN viral et sa localisation nucléaire tandis que le domaine 5 contribue à l'infectiosité virale et est responsable des autres fonctions de Tat. Les mécanismes d'action de Tat sont décrits en de plus amples détails dans la partie du cycle réplicatif.
Regulator of expression of virion proteins (Rev)
La protéine Rev est une petite protéine de 13-kDa et de 116 acides aminés ayant deux domaines fonctionnels. Elle est responsable de l'export des ARNm non-épissés et mono-épissés [119,120]. Le domaine N-terminal est riche en arginine et permet la liaison spécifique des ARNm viraux et sa localisation nucléaire. De chaque côté se trouvent des séquences requises pour la multimérisation de Rev [121]. Le domaine C-terminal est riche en leucine et contient le signal d'export nucléaire [122].
1.6.12. Protéines virales accessoires
Viral infectivity factor (Vif)
La protéine Vif contient 192 acides aminés et a été rapidement reconnue comme essentielle pour la réplication du VIH-1 dans les lymphocytes T [123,124]. Toutefois, la permissivité de certaines lignées cellulaires à l'infection par des virus déficients pour Vif laissait supposer l'importance de l'interaction de Vif avec un facteur cellulaire [125,126]. Ce facteur est la molécule APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G) dont l'activité antivirale est neutralisée par Vif [127]. Pour ce faire,
19
Vif empêche l'incorporation d'APOBEC3G dans la particule virale en favorisant la polyubiquitination de la molécule qui sera ainsi dégradée par le protéasome [128,129].
Viral Protein R (Vpr)
Le gène Vpr code pour une protéine de 96 acides aminés qui est incorporée dans la particule virale [130,131] via son interaction avec p6 [132]. La protéine incorporée dans le virion ou nouvellement synthétisée dans la cellule infectée possèdent divers propriétés : i) par sa présence dans le PIC, Vpr facilite l'import nucléaire du PIC notamment lors de l'infection de cellule quiescente comme les macrophages [133,134], ii) l'incorporation dans le virion de l'uracil-DNA glycosylase 2 (UNG2) par Vpr diminue les erreurs induite par la transcriptase inverse [135], iii) Vpr induit la mort cellulaire par arrêt [136] ou non [137] du cycle cellulaire en phase G2 et iiii) Vpr participe à la trans-activation de gènes cellulaires et viraux induisant une immunosuppression [138,139,140].
Viral protein unique (Vpu)
La protéine Vpu est une protéine membranaire de 81 acides aminés composée de 3 hélices-α. Elle est exprimée à un stade tardif du cycle réplicatif et possède deux fonctions principales. La première est de séquestrer le récepteur viral CD4 à l'intérieur de la cellule infectée afin de le faire dégrader par le protéasome, empêchant ainsi la surinfection et facilitant le bourgeonnement des virus produits [141,142]. La seconde est de bloquer l'action de la tétherine qui empêche le bourgeonnement du virus [143]. La tétherine est une protéine transmembranaire qui retient les virions nouvellement formés à la surface cellulaire en absence de la Vpu [143,144]. Lorsque Vpu est présente, elle interagit avec la tétherine pour induire sa dégradation et favoriser alors la libération des virions [145,146].
20
Negative Regulatory Factor (Nef)
La protéine Nef est une protéine d’environ 206 acides aminés qui peut subir plusieurs modifications post-traductionnelles. Elle débute en N-terminal par une séquence très conservée reconnue par la N-myristyl transférase dont le motif est MGXXXXS/T [147]. Lors de la réaction de myristoylation, la méthionine de cette séquence est éliminée. Cette modification permet l’ancrage de la protéine Nef aux membranes de la cellule infectée. L’étude de la localisation de Nef par fractionnement cellulaire, dans des cellules transfectées, a montré que moins de 60% des protéines exprimées ont une localisation membranaire et que la portion restante a une localisation cytoplasmique [148]. La protéine Nef peut être clivée par PR au sein du virion, séparant le domaine d’encrage myristoylé du domaine central de la protéine [149]. Le domaine central est constitué d’une boucle flexible de 33 résidus en C-terminal qui permet des interactions avec différents facteurs cellulaires via différents motifs de Nef [150]. Nef participe à la régulation de l'expression de surface du CD4 et du CMH-I [151,152,153]. De plus malgré son nom, Nef augmente l'infectiosité des virions et modules certaines voies de signalisation cellulaire [154].
1.7. Tropisme viral et corécepteurs
Peu de temps après la découverte du CD4 comme étant le récepteur principal du VIH-1 et du VIS, de nombreuses évidences se sont accumulées et ont indiqué que le CD4 n'est pas le seul responsable pour que la fusion virale opère. En 1986, Maddon a montré que des cellules murines exprimant le CD4 à la surface permettaient la liaison du virus mais ne lui conféraient pas la possibilité d'entrée [155]. Les données de cette étude suggèrent alors que les cellules murines n'expriment pas un cofacteur ou un corécepteur nécessaire à l'entrée du VIH-1. Une observation majeure sur les différents isolats viraux ont permis de les classer en deux groupes distincts dépendamment de leurs propriétés biologiques. En effet, en 1986 Asjo a été le premier à décrire ces deux groupes en fonction de leurs taux de réplication dans les peripheral blood mononuclear cells (PBMC) (lent/faible vs rapide/élevé) [156]. Des rapports concordants en 1988 par Tersmette faisaient état de deux groupes de virus : des virus d'un côté capables d'induire des syncytia (SI) et de l'autre côté incapable d'induire
21
des syncytia (NSI) [157]. Les travaux de Gartner en 1986 ont également confirmé l’existence de virus infectant soit les macrophages (M-Tropique), soit les cellules T (T-Tropique) [158]. Les différences entre les types d'isolats sont localisées essentiellement sur la boucle V3 de la gp120 [159]. A partir de là, la communauté scientifique a admis que les deux groupes de virus utilisaient différents corécepteurs à la surface pour entrer dans les cellules cibles. En 1996, Berger et ses collaborateurs ont été les premiers à cloner le corécepteur du VIH-1, le CXCR4 [109]. La co-expression du CXCR4 avec le CD4 dans les cellules murines permettait la fusion des virus T-Tropique et SI mais pas ceux M-Tropique et NSI. Depuis, plusieurs groupes ont identifié simultanément le CCR5 comme étant le corécepteur pour les NSI [106,108,160]. Le CCR5 et le CXCR4 sont les deux corécepteurs majeurs du VIH-1 et toutes les souches virales peuvent utiliser soit un (virus R5 et X4) soit les deux (virus R5X4) pour rentrer dans les cellules T CD4+. Les virus R5 sont transmis dès
le début de l'infection et persistent tout le long de la maladie, alors que les virus X4 apparaissent tardivement dans la maladie et sont présent dans près de 50% des isolats chez les personnes sidéennes. Le CCR5 et le CXCR4 sont tous les deux des membres de la famille des récepteurs de chimiokine à 7 domaines transmembranaires (7TM). Plus d'une douzaine d'autres 7TM on été reportés comme étant des corécepteurs dans des lignées cellulaires T CD4+ pour des souches virales particulières (tableau 1). Ces corécepteurs sur
aussi des récepteurs de chimiokines ou apparentés. Actuellement, il existe peu d'évidence sur le fait que ces corécepteurs jouent un rôle in vivo.
22
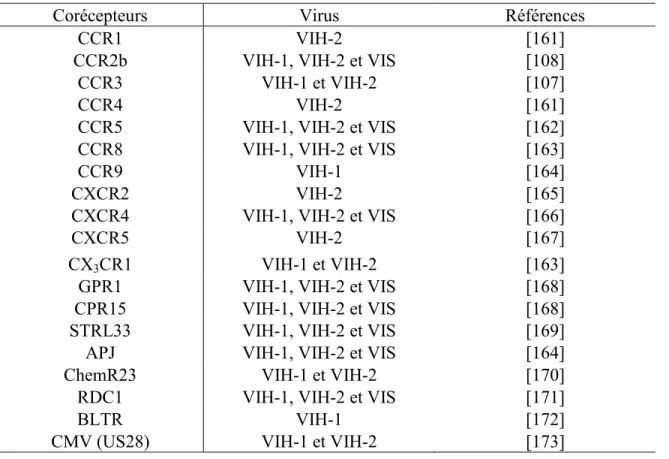
Tableau 1. Corécepteurs du VIH-1, VIH-2 et VIS
Corécepteurs Virus Références
CCR1 VIH-2 [161]
CCR2b VIH-1, VIH-2 et VIS [108]
CCR3 VIH-1 et VIH-2 [107]
CCR4 VIH-2 [161]
CCR5 VIH-1, VIH-2 et VIS [162]
CCR8 VIH-1, VIH-2 et VIS [163]
CCR9 VIH-1 [164]
CXCR2 VIH-2 [165]
CXCR4 VIH-1, VIH-2 et VIS [166]
CXCR5 VIH-2 [167]
CX3CR1 VIH-1 et VIH-2 [163]
GPR1 VIH-1, VIH-2 et VIS [168]
CPR15 VIH-1, VIH-2 et VIS [168]
STRL33 VIH-1, VIH-2 et VIS [169]
APJ VIH-1, VIH-2 et VIS [164]
ChemR23 VIH-1 et VIH-2 [170]
RDC1 VIH-1, VIH-2 et VIS [171]
BLTR VIH-1 [172]
CMV (US28) VIH-1 et VIH-2 [173]
Modifiée de [174].
Un certain nombre de corécepteurs alternatifs incluant CCR3, CCR8 et STRL33 sont utilisés efficacement par certaines souches virales pour infecter des lignées indicatrices [163,175,176]. STRL33 est exprimé dans le placenta et CPR15 dans le colon [177]. Par conséquent, ces deux corécepteurs ont le potentiel de jouer un rôle lors de la transmission maternelle et homosexuelle respectivement [176]. En effet, des isolats pédiatriques ont été reportés comme étant capable d'utiliser le STRL33. ChemR23 est quant à lui exprimé sur les cellules dendritiques et il est un candidat potentiel impliqué dans la transmission virale [170]. Une étude récente à identifié une nouvelle mutation du CCR5 chez le Mangabey qui lui permet d'utiliser le CXCR6, le GRP15 et GPR1 dans un contexte d'infection non pathogénique du VIS in vivo [178], suggérant un éventail plus large de cellules potentiellement cibles chez le VIS [179].
23
1.8. Le cycle réplicatif
1.8.1. Les étapes précoces
Figure 8. Étapes précoces du cycle viral.
Modifiée de [180].
Attachement et entrée
L'entrée du VIH-1 est la première étape du cycle réplicatif. Elle commence par l'adhésion du virus à la cellule hôte et finie avec la fusion des membranes virales et cellulaires qui aboutit à la libération de la coque virale dans le cytoplasme. Ces événements dépendent d'une série d'interaction protéines-protéines qui peut être divisée en plusieurs phases. Certaines d'entres elles sont essentielles et d'autres servent à moduler l'efficacité du processus d'entrée.
Tout d'abord, le virus doit se lier à sa cellule cible par l’intermédiaire de l'enveloppe virale ou bien par les protéines membranaires cellulaires qu'il a incorporées. C'est le cas avec la molécule ICAM-1 et son ligand lymphocyte function-associated antigen 1 (LFA-1), mais aussi de la sélectine-L et de son ligand CD62 exprimé à la surface des cellules endothéliales. L'attachement peut être relativement non spécifique, comme l'interaction d'Env avec les héparan sulfates chargés négativement présents à la surface cellulaire [181]. Elle peut être plus spécifique comme l'interaction entre Env et l'intégrine α4β7 ou le
24
dentritic cell-specific intercellular adhesion molecule 3-grabbing non-intergrin (DC-SIGN) [182,183]. L'attachement du VIH-1 à la cellule hôte via l'ensemble de ces facteurs va permettre à Env de s'approcher intimement du récepteur viral, le CD4 ainsi que des corécepteurs favorisant l'infection [184]. Cependant, les facteurs d'attachement diffèrent des récepteurs par le fait qu'ils ne sont pas essentiels à l'entrée. Bien qu'ils augmentent l'infection in vitro, leurs rôles physiologiques in vivo n'est pas encore clairement établis. La seconde étape de l'entrée du virus et la première absolument nécessaire à l'infection est la liaison d'Env sur son récepteur primaire, la protéine cellulaire, le CD4[155,185]. Env est un trimère glycoprotéique hautement glycosylé constitué d'un hétérodimère de gp120 et gp41. La sous unités gp120 est responsable de la liaison au CD4. Le CD4 est quant à lui un membre de la super famille des immunoglobulines qui a comme fonction physiologique le renforcement de la signalisation médiée par le T-cell receptor (TCR). Env va interagir avec CD4 de part son site de liaison au CD4 (CD4bs) présent sur gp120. Cette liaison entraîne un réarrangement de V1/V2 et par la suite de V3[186]. De plus, la liaison au CD4 entraine la formation d'un feuillet de relais ou «bridging sheet» qui est constitué de 4 feuillets β organisées par paires antiparallèles et spatialement non reliées entre elles[187]. Le bridging sheet et le repositionnement de la boucle V3 jouent un rôle critique dans l'accomplissement des étapes subséquentes: le réarrangement des corécepteurs [187,188].
La troisième étape de l'entrée du virus, est la liaison au corécepteur (voir la section sur le tropisme viral et les corécepteurs). C'est cette étape qui va activer la fusion membranaire. La quatrième étape est la migration du virus au site de fusion. Une série de nouvelles études ont montré que le virus détourne différentes voies de transport cellulaire pour atteindre des régions membranaires spécifiques favorisant ainsi une entrée efficace et une infection productive. Certains virus sont capables de glisser sur la membrane jusqu'à un site propice favorable à son entrée. Enfin, le virus a souvent besoin d'être internalisé par la cellule hôte par la machinerie d'endocytose pour permettre une fusion efficace [189,190,191]. On sait depuis longtemps que l'entrée du VIH-1 s'effectue de manière pH indépendante, ce qui a longtemps était perçu comme le signe d'une entrée par fusion à la surface membranaire
25
[192]. Toutefois le fait que le virus ne requiert pas une acidification du pH pour son entrée n'implique pas forcement que la fusion s'effectue à la surface cellulaire. Bien au contraire, l'endocytose du VIH-1 aurait un rôle majeur dans l'infection des T CD4+ [193]. En effet, malgré que la plupart des virus internalisés par endocytose dans les T CD4+ soient ultérieurement dégradés, la voie d'endocytose procure une voie d'entrée productive pour le virus dans ce type cellulaire [193]. Cette voie aurait été longtemps sous-estimée, comme le démontre l’étude de Miyauchi dans laquelle les auteurs concluent que la voie d'endocytose est une voie d'entrée majeure pour le virus dans ces cellules [194]. De plus, des études in vitro ont montré que la dissémination du VIH-1 d'une cellule à l'autre était plus efficace que la transmission avec des particules libres [195,196]. Si on a d'abord cru que cette observation était due à la formation de syncytia entre les cellules infectées et les cellules avoisinantes [196], il est maintenant établi que la grande efficacité de transmission du virus d'une cellule à l'autre serait plutôt due à la formation de synapses virologiques, des structures adhésives intercellulaires formées entre les lymphocytes T CD4+ infectés et non
infectés [197]. Il a été démontré que les virus transférés d'une cellule à l'autre seraient d'abord internalisés par endocytose [198] par une voie dépendante des molécules de clathrine et de dynamine [199]. Ce mécanisme de transfert donne de nombreux avantages au virus; il a notamment été démontré que celui-ci était beaucoup moins sensible à l'action des anticorps neutralisants lorsqu'il était transmis par ce mécanisme plutôt que sous sa forme libre [200].
La cinquième et dernière étape de l'entrée du virus est la fusion membranaire médiée par Env. L'interaction de la gpl20 avec son corécepteur produit un deuxième changement de conformation qui permet l'exposition de l'ectodomaine de la gp41 (forme fusogénique). Sous cette conformation, les domaines HR 1 et 2 de la gp41 sont exposés et le peptide de fusion est inséré dans la membrane plasmique. Cet intermédiaire est sensible à l'action des inhibiteurs de fusion. La gp41 effectue finalement un dernier changement conformationnel lui permettant d'adopter une structure en épingle. Il y a ensuite hémifusion et formation du pore de fusion (figure 9) [201,202].
26
Figure 9. Résumé de l'entrée du VIH-1. Adaptée de [202]. Décapsidation
Les mécanismes de la décapsidation sont encore peu documentés car difficiles à étudier. En effet, à cause de l’absence de détection de la capside dans le cytoplasme en microscopie électronique, et de l’absence de la protéine CA dans les complexes de transcription inverse (RTC reverse transcription complex) purifiés, il a tout d’abord été suggéré que la décapsidation avait lieu immédiatement après les étapes de fusion, à proximité de la membrane plasmique.
Un deuxième modèle de décapsidation a ensuite été proposé, indiquant que la capside reste intacte durant une courte période après son entrée dans le cytoplasme et que la décapsidation se déroule graduellement durant le transport et les étapes de transcription inverse. Des expériences de purification de RTC à partir de cellules infectées ont montré que ces complexes contiennent des quantités variables de protéines de CA [203,204,205,206]. La décapsidation serait déclenchée par des changements successifs de l’environnement cellulaire, permettant le contact avec différents facteurs cellulaires nécessaires à la transcription inverse.
Un dernier modèle de décapsidation propose que la capside reste intacte durant son trafic jusqu’au pore nucléaire, où auraient lieu les étapes de la transcription inverse. Ce modèle suggère que l’intégrité de la capside préserve l’échappement de RT du complexe viral et que la transcription inverse est facilitée au voisinage du pore nucléaire par une
![Figure 2. Organisation génomique du VIH-1 Adaptée de [36]](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/29.918.172.787.266.982/figure-organisation-génomique-vih-adaptée.webp)
![Figure 4. Le précurseur protéique Pr55 Gag . Adaptée de [51].](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/31.918.408.552.614.1031/figure-précurseur-protéique-pr-gag-adaptée.webp)
![Figure 5. Structure 3D de la matrice du VIH-1. Adaptée de [56].](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/32.918.329.551.730.1037/figure-structure-d-matrice-du-vih-adaptée.webp)
![Figure 6. Structure 3D de la capside du VIH-1 Adaptée de [62].](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/33.918.383.581.451.802/figure-structure-d-capside-du-vih-adaptée.webp)
![Figure 7. Structure 3D de la nucléocapside Adaptée de [48].](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/35.918.401.566.561.929/figure-structure-d-nucléocapside-adaptée.webp)

![Figure 9. Résumé de l'entrée du VIH-1. Adaptée de [202].](https://thumb-eu.123doks.com/thumbv2/123doknet/6726997.185186/48.918.121.760.116.323/figure-résumé-entrée-vih-adaptée.webp)


