SNP calling from RNA-seq data without a reference genome: identification, quantification, differential analysis and impact on the protein sequence
Texte intégral
Figure
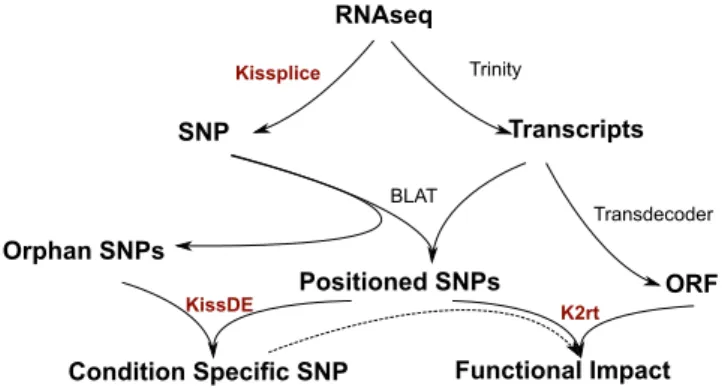
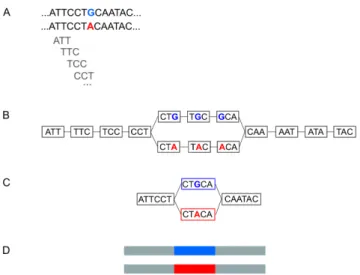
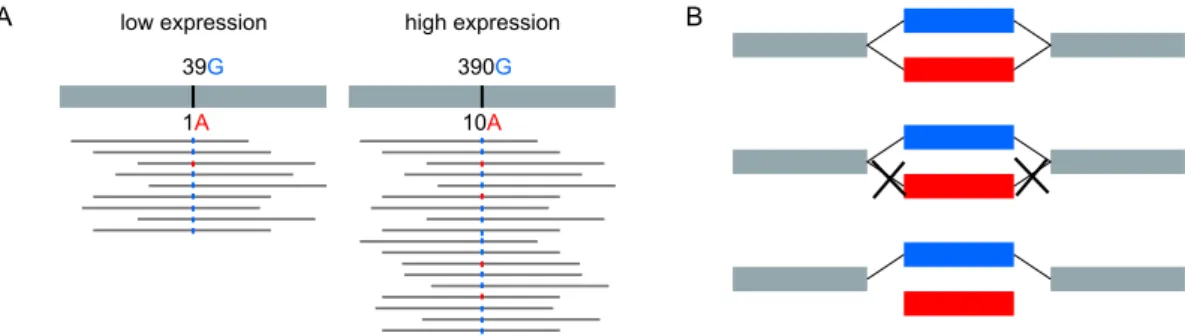

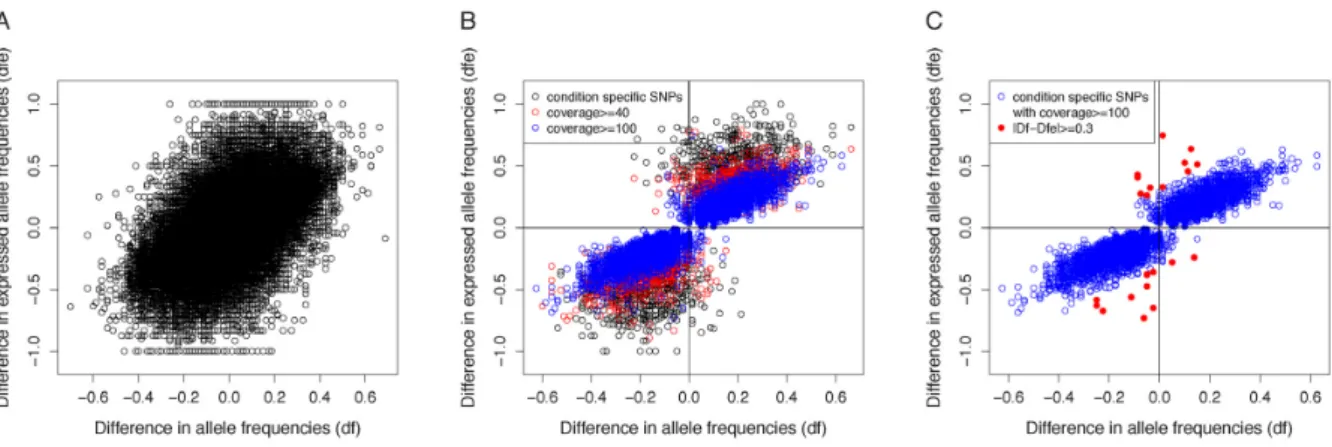

Documents relatifs
Our hypothesis that genes underlying the production of cuticular pheromones and mating responses at least partly differ, is also supported by the divergent dominance
robustness of using genome modification of Drosophila to characterize resistance mechanisms 252. identified in
In order to maintain consistency between related terms, we are reviewing existing classes by making use of their current classification into 11 different organ systems, such
For this purpose, we generated, by polymerase chain reaction ampli fi cation, one or two gene probes from each scaffold assigned to the chromosome II arms and mapped these probes to
Differential expression studies were until now restricted to gene level studies, i.e., ignoring the transcript level information, to cases where the set of expressed transcripts
Os mecanismos de silenciamento por meio das vias de piRNA e siRNAs se mostraram complexos em fêmeas e machos híbridos, uma vez que TEs que tinham piRNAs e
Neuronal expressions of ∆ Np53 (p53A) or p53 (p53B), 2 protein isoforms of Drosophila p53, are both efficient inducers of autophagy but each isoform induces a differential regulation
Collect female and/or male flies from fly cross in beginning step 2g (Generating Drosophila stocks), which express mito-GFP, mCherry-Atg8a and the desired GAL4 driver.. Maintain
