Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITÉ DE POITIERS (Facultés des Sciences Fondamentales et Appliquées)
(Diplôme National-Arrêté du 7 août 2006)
Ecole Doctorale : Ingénierie Chimique, Biologique, et Géologique Secteur de recherche : Chimie organique, Minérale et Industriellle
Présentée par :
Jean Tallineau
*********************************
Organocériens, électrocyclisations et applications
à la synthèse de substances naturelles
********************************* Directeurs de thèse :
Jean-Marie COUSTARD, Professeur, Université de Poitiers Frédéric LECORNUÉ, Maître de Conférences, Université de Poitiers
********************************* Soutenue le 10 décembre 2009 devant la commission d’examen *********************************
Jury
Mme M.-C.VIAUD-MASSUARD
Professeur, Université de Tours, G.I.C.C. Rapporteurs
Mr J. OLLIVIER Chargé de Recherche HDR-CNRS, Université Paris Sud, LSOM - ICMMO
"
Mme C. GUILLOU Directeur de recherche CNRS, ICSN, Gif s/ Yvette Examinateurs
Mr P. KRAUSZ Professeur, Université de Limoges "
Mr J.-M. COUSTARD Professeur, SRSN, Université de Poitiers
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITÉ DE POITIERS (Facultés des Sciences Fondamentales et Appliquées)
(Diplôme National-Arrêté du 7 août 2006)
Ecole Doctorale : Ingénierie Chimique, Biologique, et Géologique Secteur de recherche : Chimie organique, Minérale et Industriellle
Présentée par :
Jean Tallineau
*********************************
Organocériens, électrocyclisations et applications
à la synthèse de substances naturelles
********************************* Directeurs de thèse :
Jean-Marie COUSTARD, Professeur, Université de Poitiers Frédéric LECORNUÉ, Maître de Conférences, Université de Poitiers
********************************* Soutenue le 10 décembre 2009 devant la commission d’examen *********************************
Jury
Mme M.-C.VIAUD-MASSUARD
Professeur, Université de Tours, G.I.C.C. Rapporteurs
Mr J. OLLIVIER Chargé de Recherche HDR-CNRS, Université Paris Sud, LSOM - ICMMO
"
Mme C. GUILLOU Directeur de recherche CNRS, ICSN, Gif s/ Yvette Examinateurs
Mr P. KRAUSZ Professeur, Université de Limoges "
Mr J.-M. COUSTARD Professeur, SRSN, Université de Poitiers
« J’ai vu plus loin que les autres parce que je me suis juché sur les épaules d’un géant » Sir Isaac Newton (1643-1727) « En essayant continuellement on finit par réussir. Donc : plus ça rate, plus on a de chances que ça marche » Devise Shadock
Avant d’exposer mes travaux qui ont conduit à ce mémoire, je tiens à remercier toutes celles et ceux qui ont permis, d’une manière ou d’une autre, sa réalisation.
Monsieur André Amblès, Professeur à l’Université de Poitiers, qui m’a accueilli dans son laboratoire « Synthèse et Réactivité des Substances Naturelles » - UMR CNRS 6514.
Monsieur Jean-Marie Coustard, Professeur de l’Université de Poitiers, pour m’avoir permis de rejoindre son équipe « Réactions en Milieu Super Acide » pour terminer cette thèse et pour le réel intérêt qu’il a porté à ce travail.
Je remercie très sincèrement Frédéric Lecornué, Maitre de Conférences à l’Université de Poitiers, pour avoir guidé ce travail durant ces trois années ponctuée de repas arrosés, d’humour et de très bonne musique (ou pas). Merci pour avoir conduit ces travaux lorsque l'ancien capitaine avait décidé que ce bateau n'était pas assez intéressant pour lui.
Je tiens également à exprimer toute ma reconnaissance à Madame Catherine Guillou, Directeur de recherche à l’Université de Gif s/ Yvette, qui m’a fait l’honneur de présider le jury de thèse et de juger ce travail.
Merci également à Madame Marie-Claude Viaud-Massuard, Professeur à l’Université de Tours, Monsieur Jean Ollivier, Chargé de recherche HDR-CNRS à l’Université de Paris Sud et Monsieur Pierre Krausz, Professeur l’Université de Limoges pour avoir accepté de juger et commenter ce travail.
de parties sans fin teintées de coup-bas, de mauvaise foi mais surtout de grands moments: Aurélien "Paulo" Lavaud, Benjamin "Kenny" Ovide, Nicolas "Gros Moche" Cimetière, Maxence "Maréchal des Logis" Vidal, Marie Fricaut, Emanuel "Manu" Rousselière, Eglantine "Glanglan" Bazeille. Merci pour tous ces moments qui ont rendu ces trois années extrêmement agréables.
Je tiens aussi à remercier Hervé "Roger" Rogeon: merci pour ces soirées Ligue Des Champions ainsi que pour nos "After Foot" ont largement contribués à rendre ces trois années de thèse inoubliables.
Courrage nos souffrances s'arreteront avec l'éviction du Christophe Rocancourt du Football en 2010. Prends ça Raymond Demonech !!! Une question reste cependant sans réponse : comment peut-on connaitre autant de choses sur autant de sports et supporter le PSG ?
Sébastion "Saul" Picard: merci pour ton acceuille dans le laboratoire, tu as largement participé à la réalisation de ces travaux par ta bonne humeur, ta bonne musique (ou pas ) ainsi que par ta gentillesse (sans oublier les cafés au saké).
Merci aux autres thésard encore en activité a l'heure ou j'écris ces mots: Marion Grinda, Thibaut Legigand, Fei Liu, Guillaume Compain, David Kpogbemabou, Céline Estournel et Marie-Anne Guglielmi.
Mes remerciement vont aussi à tous les permanents qu'il m'ai été donné de cotoyer durant ces trois années:
Sébastien Papot: merci pour ces discussions parfois constructives et parfois complètement hors de propos que se soit à propos de chimie ou bien de sujet dont je ne peut écrire un seul de tes mots sur cette page.
ton aide extrêmement précieuse lors que notre chère RMN 300 faisait des siennes.
Sans oublier Bruno Violeau, Sébastien Thibaudeau, Brigitte Renoux, Jean-François Chollet, Martine Mondon, Agnès Mingot, Hélène Carreyre, Jérôme Désiré, Laurent Grasset, Yves Blériot, Laurent Lemée, Joëlle et Francis Barbot, Claire Guichard, Lilian Ripault et Sylvie Liu. Merci à tous pour votre présence et votre bonne humeur qui ont permis à ces trois années de devenir inoubliables.
Les anciens qui naviguent maintenant sous d'autres horizons: Poulos, Coucouille, P'ti Ju, Vincent, Cédric, Grand Ju, Ouercho, Emilie, Sophie, Florian: merci pour vote acceuille et votre présence qui a emmaillé ces trois années de bons moments et de discussions qui nous ont plus ou moins permis de refaire le monde.
Ces remerciements ne seraient pas complet sans quelques mots pour ma famille qui m'a toujours soutenu et encouragé dans mon parcours universitaires mais aussi dans mon parcours personnel. Un grand remerciement pour Nathalie Robineau pour m'avoir elle aussi soutenu et supporter durant ces 3 années.
INTRODUCTION GENERALE 5
I. Les Lanthanides en synthèse organique 6
II. Préparation des organocériens 7
1. A partir d’organolithiens 7
2. A partir de réactifs de Grignard 8
3. Par énolisation 8
a. A partir d’énolates de lithium 8
b. A partir de cétones a-halogénées. 9
4. Structure des organocériens 10
a. Alkyl-, allyl-, alcynyl- et arylcériens 10
b. Enolates de cérium 10
c. Chlorure de cérium 11
III. Réactivité des organocériens 11
1. Réactions d’addition des organocériens sur des dérivés carbonylés 12
a. Addition sur les cétones. 12
b. Addition sur les cétones a!b-insaturées. 14
c. Addition sur les acides carboxyliques et leurs dérivés. 14
d. Addition sur les fonctions nitriles, imines et nitro. 16
e. Mise en œuvre des réactions 18
2. Réactivité des énolates de cérium 20
a. Addition sur les composés carbonylés 20
b. Addition sur les imines 21
IV. Application des organocériens en synthèse totale. 22
1. Synthèse totale du (±)-Pleurotine 22
2. Synthèse totale de la Roseophiline 22
3. Synthèse totale de (±)-Dihydro-epi-déoxyarteannium B 23
OBJECTIF DE THESE 25
CHAPITRE I : SYNTHESE D’ARYLCYCLOALCENES ET SYNTHESE TOTALE DU
(±)-LAUROKAMURENE B 26
1ERE PARTIE : SYNTHESE EN UN SEUL POT D’ARYLCYCLOALCENES 27
I. Introduction 28
II. Réactions de couplages 28
1. Réactions de couplages pallado-catalysés 28
a. Couplage de Heck 28
c. Couplage de Stille 32
d. Couplage de Negishi 32
2. Autres réactions métallo-catalysées 32
a. Le nickel 32
b. Le Fer 33
c. Le Cuivre 34
d. Le Colbalt 34
e. Mise en œuvre des réactions de couplages métallo-catalysées 35
3. Addition nucléophile / déshydratation 37
a. Addition nucléophile 37
b. Elimination 38
4. Synthèse d’arylcycloalcènes par réarrangement semi-pinacolique/fragmentation de Grob. 39 III. Synthèse en un seul-pot d’arylcycloalcènes. 40
1. Mise au point des conditions opératoires 41
2. Synthèse d’arylcycloalcènes 43
a. Influence de la taille de cycle. 44
b. Influence des substituants sur le dérivé carbonylé et/ou l’organocérien. 44
c. Substrats azotés 46
d. Synthèse d’hétéroarylcycloalcènes 48
2EME PARTIE : SYNTHESE TOTALE DU (±)-LAUROKAMURENE B 49
I. Introduction 50
II. Précédentes synthèses du Laurokamurène B 50
1. Synthèse totale du (±)-Laurokamurène B 50
2. Synthèse totale du (+)-Laurokamurène B 52
III. Application des organocériens à la synthèse du (±)-Laurokamurène B 53
1. Synthèse de la (±)-2,2,3-triméthylcyclopentanone 54 a. Rétrosynthèse 54 b. Voie A 54 c. Voie B 55 d. Voie C 56 e. Voie D 56
2. Addition d’organocérien sur la 2,2,3-triméthylcyclopentanone (±)-62 60
IV. Conclusion du chaptire I 61
CHAPITRE II : SYNTHESE DE BENZAZEPINES PAR ELECTROCYCLISATION-1,7
D’YLURES D’AZOMETHINES A,B;G,D-INSATURES 62
1ERE PARTIE : SYNTHESE DE CYCLOALCENYLBENZALDEHYDES 63
I. Introduction 64
1. Synthèses existantes des 2-cycloalcénylbenzaldéhydes 64
a. Synthèse par ouverture d’oxazolines 64
2. Réaction de cycloaddition 1,3-dipolaire 67
a. Généralités 67
b. Les dipôles-1,3 67
c. Les ylures d’azométhines stabilisés 69
d. Les ylures d’azométhines non-stabilisés 70
3. Electrocyclisation-1,7 d’ylures d’azométhines 71
a. Electrocyclisation-1,7 d’ylures d’azométhines non-stabilisés 72
b. Electrocyclisation-1,7 d’ylures d’azométhines stabilisés 73
II. Synthèse des 2-cycloalcénylbenzaldéhydes à 5, 6 et 7 chaînons 75
1. Etude rétrosynthétique 75
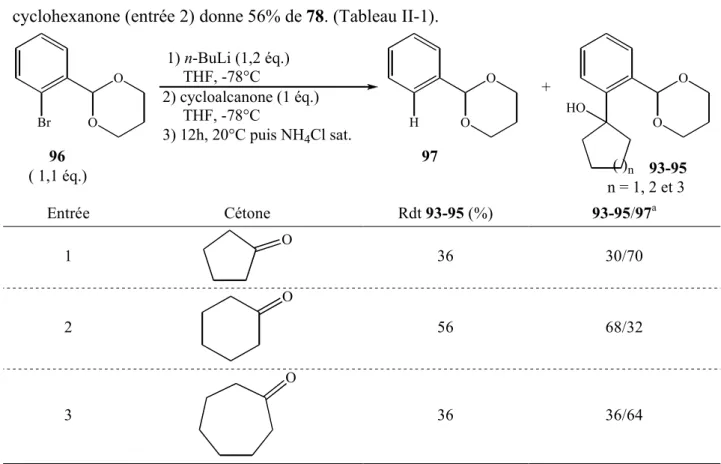
2. Synthèse des alcools tertiaires 93-95 76
a. Addition d’organolithiens 76
b. Utilisation d’organocériens 78
3. Obtention des cycloalcénylbenzaldéhydes 77-79 79
a. Elimination E2 de la fonction alcool 79
b. Déprotection de la fonction aldéhyde 80
c. Cyclisation intramoléculaire 82
4. Optimisation de la synthèse des aldéhydes 77-79 83
a. Synthèse des aldéhydes 77 et 78 à partir des alcools 93 et 94 83
b. Synthèse des aldéhydes 77 et 78 à partir de l’acétal 96 84
III. Conclusion 84
2EME PARTIE : SYNTHESE DE BENZAZEPINES PAR REACTION
D’ELECTROCYCLISATION-1,7 D’YLURES D’AZOMETHINES 86
I. Addition de dérivés d’acides aminés sur les aldéhydes 77-78 87
1. Préparation des dérivés d’acides aminés 87
2. Premiers résultats 87
3. Piégeage des ylures d’azométhines intermédiaires 90
4. Synthèse de diverse benzazépines substituées 91
a. Obtention des benzazépines substituées 91
b. Structure des benzazépines 93
II. Conclusion du chapitre II 95
CHAPITRE III : ESSAIS VERS LA SYNTHESE TOTALE DE LA (±)-MORPHINE 96
I. Introduction 97
1. Historique 97
2. Généralités sur la morphine 97
3. Synthèses précédentes de la morphine 98
a. Synthèse de Gates (1956) 98
b. Synthèse de Mulzer 101
c. Synthèse d’Ogasawara 104
II. Etude rétrosynthètique 106
1. Rappels 106
III. Essais vers la synthèse totale de la (±)-morphine 108
1. 1ère stratégie de synthèse : voie A 108
a. Bromation de l’isovanilline 108
b. Fonctionnalisation de la bromo-isovanilline 165 109
c. Synthèse des alcools tertiaires 110
d. Déprotection de la fonction aldéhyde et cyclisation intramoléculaire 113
2. 2ème stratégie de synthèse 115
a. Schéma rétrosynthétique envisagé 115
b. Synthèse du synthon 188 116
c. Synthèse de l’aldéhyde 164 117
d. Cyclisation intramoléculaire 118
3. 3ème stratégie de synthèse 120
a. Synthèse du 1,3-dithiane 199 121
b. Synthèse de l’aldéhyde 198 123
IV. Conclusion du chapitre III 126
PARTIE EXPERIMENTALE 133
INDICATION GENERALES 134
PARTIE EXPERIMENTALE DU CHAPITRE I 136
PARTIE EXPERIMENTALE DU CHAPITRE II 156
PARTIE EXPERIMENTALE DU CHAPITRE III 187
I. Les Lanthanides en synthèse organique
Depuis les années 1970, l’utilisation des lanthanides en synthèse organique est sans cesse croissante et décrite dans la littérature pour un grand nombre de réactions telles que des réactions d’oxydation et de réduction. Cependant, la possibilité d’accéder à des composés organolanthanides permettant de créer des liaisons C-C par additions nucléophiles reste l’attrait principal cette famille.1
Le lanthanide le plus employé à ce jour est le samarium, principalement utilisé sous la forme d’iodure de samarium(II) : SmI2. Depuis sa première utilisation en synthèse organique par
Kagan en 1977,2 l’iodure de samarium(II) est largement employé pour des réactions aussi variées qua la création de liaisons carbone-carbone ou des réactions de cyclisation (Schéma 1).3
R1 SmI2X R2 O R3 + OH R1 R3 R2 puis hydrolyse O O Br Ph O Me O O Ph Me OH H SmI2 THF, -78°C, 15 min 95% Schéma 1
Le nitrate d’ammonium cérique (CAN : (NH4)2Ce(NO2)6) est aussi utilisé en synthèse
organique en tant qu’agent d’oxydation doux.4
En 1982, Imamoto et coll. furent les premiers à préparer une série d’organocériens à partir d’organolithiens et de CeI3.5 Ils étudièrent les propriétés des organocériens et en particulier leur
addition nucléophile sur des cétones conduisant à la formation d’alcools tertiaires avec d’excellents rendements. Les organocériens sont maintenant largement utilisés quand l’utilisation de réactifs de Grignard ou d’organolithiens classiques ne permettent pas l’obtention de résultats satisfaisants.6
1 a) G. A. Molander, Chem. Rev. 1992, 92, 29
b) H. B. Kagan, J.-L. Namy, Lanthanides : Chemistry and Use in Organic Synthesis, Ed. S. Kobayashi, Springer-Veslag, 1999.
c) P. G. Steel, J. Chem. Soc., Perkin Trans. 1 2001, 2727. d) H. B. Kagan, Tetrahedron 2003, 59, 10351.
2 a) J. L. Namy, P. Girard, H. B. Kagan, Nouv. J. Chim. 1977, 1, 5.
b) J. L. Namy, P. Girard, H. B. Kagan, J. Am. Chem. Soc. 1980, 102, 2693.
3 D. J. Edmonds, D. Johnston, D. J. Procter, Chem. Rev. 2004, 104, 3371 et références citées. 4 T.-L. Ho, Synthesis 1973, 347.
5 T. Imamoto, T. Kusumoto, M. J. Yokoyama, J. Chem. Soc., Chem. Commun. 1982, 1042. 6 H.-J. Liu, K.-S. Shia, X. Shang, B.-Y. Zhu, Tetrahedron 1999, 55, 3803.
II. Préparation des organocériens
Les organocériens sont généralement préparés in-situ par transmétallation d’organomagnésiens ou d’organolithiens en présence de CeX3 (X = I, Cl). Ils sont utilisés
directement in-situ car ils ne peuvent pas être stockés du fait de la forte hygroscopie des halogénures de cérium.
1. A partir d’organolithiens
Le premier organocérien a été obtenu par Imamoto et al.5 en 1982 par traitement de n-butyllithium par du triiodure de cérium(III) préparé in-situ à partir de cérium métal et de diiode (Schéma 2).
n-BuLi + CeI3 THF, -78°C n-BuLi,CeI3
Schéma 2
Sur un plan pratique, une nouvelle procédure fut mise au point utilisant du chlorure de cérium(III) heptahydraté CeCl3.7H2O dont l’eau est éliminée par chauffage (140°C) sous
pression réduite (0,2 mmHg). Le chlorure de cérium(III) anhydre ainsi obtenu est alors mis en suspension dans le THF anhydre afin de créer le complexe CeCl3.nTHF. Ce dernier traité par un
organolithien conduit à la formation de l’organocérien désiré : cette méthode reste la plus utilisée à ce jour.7,8 Imamoto et al. ont ainsi obtenu la première série d’organocériens (Schéma
3).9
R-Li CeCl3
R = alkyle, allyle, alcyne alcényle et aryle
THF, -78°C RLi,CeCl
3
Schéma 3
Les organocériens obtenus à partir d’organolithiens sont peu stables thermiquement. Ceux possédant des hydrogènes en ! se décomposent à 0°C tandis que ceux n’en ayant pas peuvent s’avérer stables jusqu’à des températures avoisinant les 60°C. Pour ces raisons, les réactions impliquant l’utilisation des organocériens sont la plupart du temps menées à basse température (T < -60°C).
7 T. Imamoto, Y. Sugiura, N. Takiyama, Tetrahedron Lett. 1984, 25, 4233. 8 V. Dimitrov, K. Kostova, M. Genov, Tetrahedron Lett. 1996, 37, 6787.
2. A partir de réactifs de Grignard
Les réactifs de Grignard peuvent aussi être convertis en organocériens via une transmétallation avec CeCl3. La procédure est similaire à celle décrite précédemment en
effectuant l’addition du réactif de Grignard à une suspension de CeCl3 dans le THF à 0°C. Cette
méthode est applicable pour l’obtention d’une large gamme d’organocériens exceptés pour la formation d’alcénylcériums.10 Ces derniers n’étant pas stables à 0°C, ils sont générés par transmétallation à -78°C.11 Les organocériens préparés à partir d’organomagnésiens sont plus stables thermiquement que ceux issus d’organolithiens ce qui permet de les préparer vers 0°C (Schéma 4).
3. Par énolisation
a. A partir d’énolates de lithium
La méthode la plus courante pour obtenir un énolate de cérium consiste à effectuer une transmétallation entre un énolate de lithium et du chlorure de cérium(III). Cette méthode peut être appliquée à différents substrats énolisables tels que les cétones,12 les esters, les amides13 et les nitriles.14
Le substrat subit tout d’abord une déprotonation par une base lithiée (N,N-diisopropylamidure de lithium (LDA) ou bis(triméthylsilyle)amidure de lithium (LiHMDS)). L’addition de cet énolate à une suspension de chlorure de cérium(III) dans le THF à -78°C donne lieu à une transmétallation permettant d’obtenir l’énolate de cérium souhaité (Schéma 5).
10 a) T. Imamoto, N. Takiyama, K. Nakamura, Tetrahedron Lett. 1985, 26, 4763.
b) T. Imamoto, N. Takiyama, K. Nakamura, T.Hatajima, Y. Kamiya, J. Am. Chem. Soc. 1989, 111, 4392.
11 T. Imamoto, T. Hatajima, K. Ogata, Tetrahedron Lett. 1991, 32, 2787. 12 T. Imamoto, T. Kusumoto, M. Yokoyama, Tetrahedron Lett. 1983, 24, 5233. 13 H. J. Liu, N. H. Al-Said, Tetrahedron Lett. 1991, 32, 5473.
14 X. Shang, J. H. Liu, Synth. Commun. 1994, 24, 2485.
RMgX CeCl3 THF, 0°C RMgX,CeCl3
R = alkyle, alcyne et aryle
RMgX CeCl3 THF, -78°C RMgX,CeCl3
R = alcényle
CH3CO2Et Cl2CeCH2CO2Et
O OCeCl2
CH3CN Cl2CeCH2CN
CH3CONMe2 Cl2CeCH2CONMe2
a
a
b
c
a : LDA, -78°C, THF, puis CeCl3, -78°C ; b : LiHMDS, -78°C, THF, puis CeCl3, -78°C ; c :
LDA, -78°C, THF, puis CeCl3, -78°C à 20°C.
Schéma 5
b. A partir de cétones "-halogénées.
La formation d’énolates de cérium peut aussi être effectuée à partir d’"-bromocétones. Cette méthode nécessite l’utilisation d’iodure ou de chlorure de cérium(III) en présence d’une quantité stoechimétrique de NaI.15
Le mécanisme supposé de la formation est décrit par une réaction concertée durant laquelle le dérivé "-halogéné subit simultanément une chélation de l’atome d’oxygène par l’atome de cérium et une déhalogénation (Schéma 6).
Ph O Br THF 20°C CeI3 Ph O Br Ce I I I -IBr Ph OCeCl2 Ph O Br NaI, THF 20°C CeCl3 Ph O Br Ce Cl Cl Cl -IBr Ph OCeCl2 I Schéma 6
Cette dernière méthode n’est pas applicable aux esters "-halogénés. L’utilisation de benzènetelluronate de lithium (PhTeLi) en présence de chlore de cérium(III) est nécessaire afin
d’accéder de manière satisfaisante à l’énolate de cérium désiré (Schéma 7).16 EtO O Br PhTeLi (2 éq.) CeCl3 (1 éq.) Et2O, 0°C à 20°C EtO O Br PhTe CeCl3 EtO OCeCl2 -LiCl
PhTeBr PhTeLi PhTeTePh
Schéma 7
4. Structure des organocériens
a. Alkyl-, allyl-, alcynyl- et arylcériens
Bien que la chimie des organocériens ait éveillé l’intérêt de nombreux chimistes depuis le début des années 1980, il est important de noter que la structure réelle des organocériens n’a pu être déterminée avec précision. Comme décrit précédemment, les organocériens issus de réactifs de Grignard sont moins sensibles à la température que ceux issus d’organolithiens. Il est évident que l’ion métallique précurseur intervient dans la structure de l’organocérien, influençant sa stabilité, et donc qu’il est impératif de les différencier dans leur description. Pour cela, dans ce manuscrit, les organocériens seront toujours décrits en tenant compte de leurs « origines » (Schéma 8).
RLi RLi,CeCl3
RMgX CeCl3 RMgX,CeCl3
CeCl3
Organocériens issus d'organolithiens
Organocériens issus d'organomagnésiens
Schéma 8
b. Enolates de cérium
De la même manière, la structure des énolates de cérium n’a pu être déterminée avec précision. Les énolates de cérium peuvent être écrits de deux manières différentes si l’on considère que leur formation réside en la création d’une liaison carbone-cérium ou bien une liaison oxygène-cérium (Schéma 9).
LDA puis CeCl3 H3C O NMe2 H2C OCeCl2 NMe2 Cl2CeH2C O NMe2 ou Schéma 9 c. Chlorure de cérium
Le chlorure de cérium(III) nécessaire à l’obtention d’organocériens est décrit comme étant « anhydre » du fait que les organomagnésiens et organolithiens sont extrêmement sensibles à la présence d’eau dans le milieu. Cependant la procédure de déshydratation du chlorure de cérium(III) heptahydraté ne permet pas d’éliminer toutes les molécules d’eau.17 Il apparaît que le
chlorure de cérium(III) se présente sous la forme d’un complexe dans le THF dont l’unité monomère possède pour formule [Ce(µ-Cl)3(THF)(H2O)], c’est à dire avec une molécule d’eau
sous forme de ligand lié au cérium. On ne peut donc pas à proprement parler de chlorure de cérium(III) anhydre.
III.
Réactivité des organocériens
Ce chapitre va traiter des propriétés des organocériens. Pour mieux comprendre celles-ci, il est essentiel de parler de l’oxophilie du cérium. En effet, la réactivité et la régioseléctivité de ces composés peuvent s’expliquer par la chélation de l’atome de cérium avec les doublets non-liants de l’oxygène des dérivés carbonylés. Par analogie, lors de l’addition d’organocériens sur des imines ou des nitriles, le cérium se chélate aux doublets non-liants de l’atome d’azote (Schéma 10). R1 O R2 RLi,CeCl3 H3O R1 OH R R2 R1 N R2 R3 RLi,CeCl3 H3O R1 NHR3 R R2 O R1 R2 Cl2 Ce Cl Li R N R1 R2 Cl2 Ce Cl Li R R3 Schéma 10
1. Réactions d’addition des organocériens sur des dérivés carbonylés a. Addition sur les cétones.
Imamoto et al.5 furent les premiers en 1982 à démontrer que l’addition nucléophile d’organocériens sur des cétones conduit à la formation d’alcools tertiaires avec d’excellents rendements.
L’exemple le plus frappant est celui concernant l’addition de l’iodure de n-butylcérium sur la p-iodoacétophénone conduisant à la formation de l’alcool 1 de manière quantitative. A titre comparatif, l’addition de n-butyllithium ne permet ni d’obtenir le produit d’addition ni de récupérer le produit de départ en raison d’un échange lithium-iode conduisant après hydrolyse à la formation d’acétophénone (Schéma 11).
I Me O THF, -65°C, 3h n-BuLi,CeI3 : Rdt = 99% n-BuLi : Rdt = 0% I Me OH n-Bu 1 Schéma 11
Les organocériens permettent d’effectuer des réactions d’addition sur des cétones facilement énolisables, chose difficilement réalisable par des organolithiens. L’addition d’iodure de n-butylcérium sur la 1,3-diphénylpropan-2-one permet d’obtenir l’alcool 2 avec un rendement quantitatif alors que l’utilisation de n-butyllithium ne permet d’obtenir que 33% d’alcool 2 et de récupérer 61% de la cétone de départ, ceci étant dû à son énolisation (Schéma 12).5
Ph O Ph THF, -65°C, 3h Ph Ph n-Bu OH 2 n-BuLi,CeI3: Rdt = 98% n-BuLi : Rdt = 33% Ph O Ph H Ph OLi Ph H2O, H Ph O Ph Enolisation de la cétone - Butane n-Bu Li Schéma 12
Imamoto et al. ont aussi démontré que l’addition nucléophile d’un organocérien n’est pas ou que très peu influencée par l’encombrement stérique des substrats. Ainsi l’addition de s-butylcérium sur l’acétophénone conduit à l’alcool 3 avec un rendement quantitatif (Schéma 13).
O THF, -65°C, 3h Me s-Bu OH 3 s-BuLi,CeCl3 98% Schéma 13
Par la suite, Imamoto et al. ont montré que les organocériens obtenus par transmétallation avec du chlorure de cérium(III) possédaient la même réactivité que ceux obtenus à partir d’iodure de cérium(III). Dès lors, la préparation d’organocériens s’est principalement effectuée à partir de chlorure de cérium(III) heptahydraté, composé commercial et peu onéreux.
L’utilisation d’organocériens permet de limiter les réactions d’aldolisation et de réduction des cétones habituellement observées avec les réactifs de Grignard (Schéma 14). L’addition du chlorure d’isopropylmagnésium sur la cyclopentanone conduit à la formation d’un produit majoritaire 5 issu de la réaction d’auto-aldolisation, le poduit d’addition quant à lui n’est formé qu’à hauteur de 3%. La même réaction effectuée en présence de chlorure de cérium(III) permet d’obtenir 80% de l’alcool 4 souhaité en supprimant la réaction d’aldolisation.18 Ceci s’explique par la très faible basicité des organocériens qui n’est pas suffisante pour déprotonner la cétone en ". La réaction d’aldolisation est alors « bloquée » et la réaction d’addition nucléophile est favorisée. O HO O HO 4 5 THF, 0°C, 1h i-PrMgCl i-PrMgCl,CeCl3 3% 80% 88% traces Schéma 14
b. Addition sur les cétones ",!-insaturées.
L’addition d’organomagnésiens sur des cétones ",!-insaturées conduit à la formation de régioisomères issus de la compétition entre l’addition-1,2 et l’addition-1,4 alors que l’utilisation d’organocériens conduit majoritairement à la formation de produit d’addition-1,2. En effet, l’addition de chlorure d’isopropylmagnésium sur la (E)-4-phénylbut-3-ène-4-one donne les produits 6 et 7 avec des rendements respectifs de 12% et 53%. L’utilisation de l’organocérien correspondant permet d’obtenir le produit 6 à hauteur de 91% (Schéma 15).
Ph O THF, 0°C, 1h Ph Ph OH O i-PrMgCl i-PrMgCl,CeCl3 12% 91% 53% 5% 6 7 Schéma 15
Cette régiosélectivité peut s’expliquer à nouveau par l’oxophilie du cérium. Lors de l’addition d’un organocérien sur une cétone ",!-insaturée, le cérium vient se chélater sur l’oxygène du carbonyle favorisant ainsi l’addition-1,2.
c. Addition sur les acides carboxyliques et leurs dérivés.
L’addition d’organocérien sur des acides carboxyliques,19 des amides de Weinreb,20 des
chlorures d’acides,21 des esters22 et de cétènes23 permet d’obtenir facilement des cétones (Schéma 16).
19 Y. Anh, T. Cohen, Tetrahedron Lett. 1994, 35, 203.
20 G. Brenner-Weib, A. Giannis, K. Sandhoft, Tetrahedron 1992, 48, 5855. 21 N. R. Natale, S. G. Yocklovich, B. M. Mallet, Heterocycles 1986, 8, 2175. 22 K. Blades, T. P. Lequeux, J. M. Percy, Tetrahedron 1997, 53, 10623. 23 Y. Kita, S. Matsuda, S. Kitagaki, Y. Tsuzuki, S. Akai, Synlett 1991, 401.
OH O Ph PhLi (1 éq.), CeCl3 (2 éq.) THF, -78°C, 95% Ph Ph O NMe(OMe) O Ph n-BuMgBr,CeCl3 (2 éq.) THF, 0°, 66% n-Bu O Ph O N Li,CeCl3 N(i-Pr)2 O O N N(i-Pr)2 O Ph O THF, -78°C, 50% Me3SiHC C O PhLi,CeCl3 (1,5 éq.) THF, -78°C puis NH4Cl 79% Ph O SiMe3 PhCOCl (1 éq.) i-Pr
CO2Et (EtO)2P(O)CF2LiCeCl3 (1 éq.)
THF, -78°C 71% i-Pr O CF2PO(OEt)2 Schéma 16
L’utilisation de conditions adéquates permet l’obtention d’alcools tertiaires et d’allylsilanes à partir d’esters8,24 de lactones25 et de chlorures d’acides (Schéma 17).26
24 a) B. A. Narayama, W. H. Bunnelle, Tetrahedron Lett. 1987, 28, 6261.
b) W. H. Bunnelle, B. A. Winterfeldt, Org. Synth. 1990, 69, 89.
25 a) T. V. Lee, J. A. Channon, C. Cregg, J. R. Porter, F. S. Roden, H. T. L. Yeoh, Tetrahedron 1989, 45, 5877.
b) T. V. Lee, J. R. Porter, F. S. Roden, Tetrahedron Lett. 1988, 29, 5009.
PhCO2Me OH Ph i-PrMgCl,CeCl3(2 éq.) THF, 0°C 97% O O TMSCH2MgCl,CeCl3 (2 éq.) THF, -70°C à t° amb. 74% HO TMS C9H19COCl TMSCH2Li,CeCl3 (4 éq.) THF, -78°C 87% C9H19 TMS Schéma 17
La formation des allysilanes s’explique par une double addition de l’organocérien suivie d’une élimination de triméthylsilanol (Schéma 18).
R1 O X X = groupement partant TMSCH2Li,CeCl3 R1 OH TMS - HOSiMe3 (2 éq.) TMS R1 TMS Schéma 18
d. Addition sur les fonctions nitriles, imines et nitro.
Les amines peuvent être obtenues par addition nucléophile d’organométalliques sur des imines ou leurs dérivés (oximes, hydrazones, etc…). Cependant, le faible caractère électrophile de la liaison C=N ne permet pas l’obtention de bons rendements et l’emploi de ces méthodes en synthèse organique reste donc limité.
En 1986, Lipshutz et al. ont décrit la l’addtition d’un organocérien sur l’imine 8 pour conduire à la formation du bisamide 9 avec un bon rendement (Schéma 19).27
O N i-Pr O NHOMe n-BuLi,CeCl3 THF, -78°C 75% O N H O NHOMe 8 9 n-Bu i-Pr Schéma 19
Par la suite, Denmark et al.28 ont publié une nouvelle méthode de synthèse d’amine chirale. L’addition d’organocériens sur la SAMP-hydrazone 10 permet la création d’un centre stéréogène avec un bon rendement et une très bonne diastéréosélectivité en composé 11. L’addition d’un organocérien sur la SAMP hydrazone ",!-insaturée 12 conduit à nouveau à la formation préférentielle du produit d’addition-1,2 13 tout en conservant un très bon excès diastéréoisomérique (Schéma 20). Ph N H N OMe Ph N Me N OMe H O MeO 1) MeLi,CeCl3, THF, -78°C 2) ClCO2Me 81% (ed = 96%) 10 N H N OMe N Me N OMe H O MeO 1) MeLi,CeCl3, THF, -78°C 2) ClCO2Me 82% (ed = 92%) 12 11 13 Schéma 20
L’addition d’un organocérien issu d’un organolithien sur une fonction nitrile conduit à la formation d’une amine avec un très bon rendement. Dans le cas d’un nitrile ",!-insaturé, la régiosélectivité des organocériens pour l’addition-1,2 est à nouveau observée.29 De plus, on observe une très bonne chimiosélectivité des organocériens vis-à-vis des cétones et aldéhydes par rapport à d’autres fonctions telles que les nitriles (Schéma 21).30
28 S. E. Denmark, T. Weber, D. W. Piotrowski, J. Am. Chem. Soc. 1987, 109, 2224. 29 a) E. Giganek, J. Org. Chem. 1992, 57, 4521.
b) J. Limanto, B. Dorner, P. N. Devine, Synthesis 2006, 24, 4143.
N CN Ph(H2C)2 n-BuLi,CeCl3 (3 éq.), THF -65°C à 25°C, 2h 90% N Ph(H2C)2 n-Bu n-Bu NH2 Ph CN CH3Li,CeCl3 (3 éq.), THF -65°C à 25°C, 2h 77% Ph NH2 N CN CHO OBn H CeCl3,Li TMS Bu THF, -78°C, 71% N CN OBn H OH TMS Bu Schéma 21
Les hydroxylamines N,N-disubstituées peuvent être obtenues avec d’excellents rendements par l’addition d’organocériens issus de réactifs de Grignard sur des composés nitro (Schéma 22).31 NO2 1) n-PrMgBr,CeCl3 (2 éq.), -40°C, THF 2) 10% CH3CO2H N OH n-Pr 90% Schéma 22
e. Mise en œuvre des réactions
La mise en œuvre de ces réactions d’addition est généralement effectuée par l’emploi d’un excès d’organocériens conduisant à l’obtention de milieux réactionnels fortements hétérogènes.32
Il existe plusieurs manières de mettre en oeuvre ces réactions d’addition nucléophile d’organocériens sur des dérivés carbonylés.
La première procédure (Imamoto) consiste à effectuer l’addition d’un réactif de Grignard ou un organolithien à une suspension de chlorure de cérium(III) dans le THF pour former
31 G. Bartoli, E. Marcantoni, M. Petrini, J. Chem. Soc., Chem. Commun. 1993, 1373. 32 a) X. Li, S. M. Singh, F. Labrie, Tetrahedron Lett. 1994, 8, 1157.
b) N. Greeves, L. Lyford, J. E. Pease, Tetrahedron Lett. 1994, 2, 285. c) N. Greeves, L. Lyford, Tetrahedron Lett. 1992, 33, 4759.
l’espèce réactive. Le dérivé carbonylé est ensuite additionné à cette solution d’organocérien pour effectuer l’addition nucléophile. Cette méthode reste la plus employée à ce jour.
La deuxième méthode (Dimitrov) consiste à activer le dérivé carbonylé en l’additionnant à la suspension de chlorure de cérium. L’addition nucléophile est ensuite effectuée lors de l’addition de l’organolithien à cette solution de dérivé carbonylé « activé » (Schéma 23).33
R1 R2 O THF CeCl3 R1 R2 O CeCl3 1) R3-Li 2) H3O R2 R1 OH R3 R1 O R2 Cl2 Ce Cl Li R3 Schéma 23
Une étude comparative de ces deux méthodes est décrite dans le tableau 1.34
n-Bu
HO O
n-BuM ou n-BuM,CeCI3
M = Li ou MgBr
Entrées Réactifs Méthodes Rdt (%)
1 n-BuLi 26 2 n-BuLi,CeI3 Imamoto 97 3 n-BuLi,CeI3 Dimitrov 80 4 n-BuMgBr 28 5 n-BuMgBr,CeI3 Imamoto 96 6 n-BuMgBr,CeI3 Dimitrov 92 Tableau 1
L’utilisation de la méthode d’Imamoto ou de Dimitrov permet systématiquement d’améliorer les rendements (entrées 1,2,3 et entrées 4,5,6) par rapport à la procédure classique d’addition d’organométallique sur des dérivés carbonylés.
33 V. Dimitrov, S. Bratovanov, S. Simova, K. Kostova, Tetrahedron Lett. 1994, 36, 6713. 34 N. Takeda, T. Imamoto, Organic Synth. 2004, Coll. Vol. 10, 200.
Entrées Réactifs de Grignard Cétones Produits Rdt (%) Imamoto (Dimitrov) Rdt (%) CeCl3.2LiCl 1 i-PrMgCl O i-Pr HO 72 (80) 94 2 i-PrMgCl O HO i-Pr 73 98 3 i-PrMgCl Ph Ph O Ph Ph i-Pr HO - 95 4 MeMgCl Me Me Me Me O Me Me Me OH Me Me 47 65 Tableau 2
Récemment Knochel et al. a décrit la préparation d’une solution de CeCl3.2LiCl dans le
THF (0,3-0,5 M) et emploie le terme d’additif quant au rôle du cérium.35 A cette solution, est ajoutée à 0°C la cétone. Après 1h d’agitation, l’organomagnésien est ajouté goutte-à-goutte. Dans cette étude, Knochel compare l’efficacité de sa procédure avec celle d’Imamoto et de Dimitrov (Tableau 2).
Les rendements selon la procédure de Knöchel sont généralement supérieurs. Cependant, l’inconvénient de cette procédure est la préparation relativement longue de la solution de CeCl3.2LiCl dans le THF.
2. Réactivité des énolates de cérium
a. Addition sur les composés carbonylés
La création de liaison C-C peut aussi être effectuée par la réaction d’aldolisation. Imamoto et al.ont publié en 1982 une méthode utilisant des énolates de cérium. Ces nouvelles conditions d’aldolisation limitant les réactions d’énolisation conduisent à de meilleurs rendements que lors de l’utilisation d’énolates de lithium (Schéma 24).12
Ph OM O OH O Ph -78°C, THF M = Li : Rdt = 28% M = LiCeCl3 : Rdt = 79% Schéma 24
L’adidition d’énolates de cérium sur des cétones ",!-insaturées conduit préférentiellement à l’obtention du produit d’addition-1,2 comme décrit précédemment pour l’addition d’organocériens.36
b. Addition sur les imines
L’addition d’énolates de lithium sur des imines facilement énolisables ne donne pas de bons rendements.37 De plus, afin de créer la liaison C-C, il est nécessaire que l’imine soit suffisamment électrophile pour subir l’addition de l’énolate. Dans le cas d’imines substituées par un groupement électrodonneur, l’addition de l’énolate peut ne pas être observée, l’énolate de lithium agissant comme une base en déprotonant l’imine.
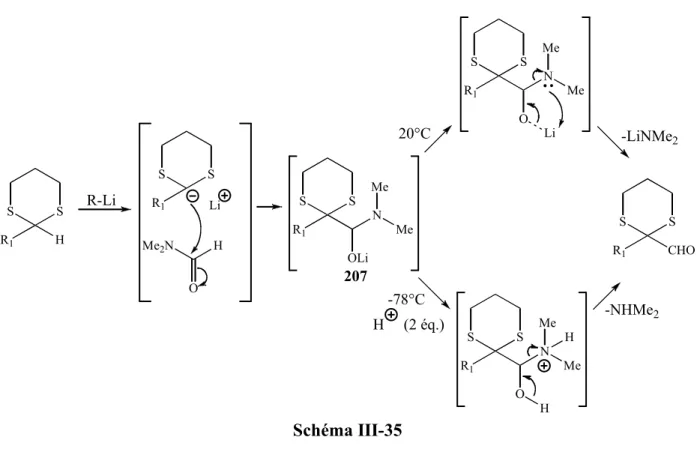
L’utilisation d’énolates de cérium pallie cette faible électrophilie des imines ; De plus, il a été montré que la réaction d’addition est systématiquement suivie d’une réaction de cyclisation conduisant à la formation de lactames à 4 chaînons. Un mécanisme pouvant expliquer la formation de ces lactames est décrit dans le schéma 25.38
MeO OLi,CeCl3 R N R2 R1 N Ce O R1 R2 H R MeO Cl Cl N MeO O Ce Cl Cl R R1 R2 -MeOCeCl2 N O R2 R R1 énolate E Schéma 25
36 H.-J. Liu, B.-Y. Zhu, Can. J. Chem. 1991, 69, 2008. 37 T. Iimori, Tetrahedron Lett. 1985, 26, 1523.
IV. Application des organocériens en synthèse totale.
Depuis les années 1980, le champ d’application des organocériens en synthèse organique n’a eu de cesse de s’étendre. Leur oxophilie, leur potentiel nucléophile, leur régiosélectivité ainsi que leur faible basicité en ont fait un outil puissant pour la synthèse de molécules complexes.
1. Synthèse totale du (±)-Pleurotine
En 1988, Hart et al. ont publié la synthèse totale du (±)-Pleurotine39 17 en effectuant l’addition de l’organocérien 15 issu du chlorure de 2,2-diméthoxyphénylmagnésium sur la cétone bicyclique 14 pour conduire au produit d’addition 16 avec un excellent rendement (Schéma 26). O CO2Me H Me OTBDMS OMe MeO H CO2Me Me OTBDMS OH THF, -78°C 92% OMe MeO 14 15 16 O Me H O O O O H 17 MgCl,CeCl3 + (±)-Pleurotine Schéma 26
2. Synthèse totale de la Roseophiline
En 1998, Fürstner et al. ont publié la première synthèse totale de la Roseophiline 21,40 un
39 D. J. Hart, H. C. Huang, J. Am. Chem. Soc. 1988, 110, 1634. 40 A. Fürstner, H. Weintritt, J. Am. Chem. Soc. 1998, 120, 2817.
agent antibiotique isolé à partir de Streptomyces griseoviridis.41 L’étape clé consiste à synthétiser le composé 20 par addition nucléophile d’un synthon pyrrolique 18 sur la cétone macrocyclique 19. Là encore, l’utilisation d’un organocérien a permis l’obtention du composé 20 avec un bon rendement permettant de poursuivre la synthèse jusqu’à l’obtention du chlorydrate de Roseophiline (Schéma 27).
N SEM Me Me OH O NSi(i-Pr)3 MeO Cl N SEM Me Me O O NSi(i-Pr)3 MeO Cl Li,CeCl3 62% N Me Me O NH MeO Cl THF, -78°C 21 18 19 20 .HCl Schéma 27
L’addition de l’organocérien 18 s’effectue sur la face la plus accessible du carbonyle de la cétone pour donner le composé 20.
3. Synthèse totale de (±)-Dihydro-epi-déoxyarteannium B
Plus récemment, Dudley et al. ont publié la synthèse de la (±)-Dihydro-epi-déoxyarteannium B 24.42 Cette synthèse totale passe par l’obtention de la molécule 23 par
41 Y. Hayakawa, K. Kawakami, H. Seto, K. Furihata, Tetrahedron Lett. 1992, 33, 2701.
addition de l’organocérien issus du bromure de vinylmagnésium sur la cétone 22 (Schéma 28). O SiMe3 SiMe3 OTIPS OTIPS OH MgBr,CeCl3 THF, >91% O O 22 23 24 (+)-dihydro-epi-deoxy-arteannuin B Schéma 28
L’ensemble des réactions décrites précédemment montre la grande utilité des organocériens quant à effectuer des réactions d’additions nucléophiles sur des groupements carbonylés et leurs dérivés. Les propriétés décrites précédemment seront employées dans la suite de ce manuscrit afin d’effectuer des réactions d’additions nucléophiles sur des cétones cycliques.
Objectif de thèse
Ce manuscrit se composera de trois chapitres.
Le premier chapitre traitera de la mise au point d’une méthode de synthèse en un seul pot d’arylcycloalcènes. Cette méthode consistera en l’addition d’organocériens sur des cétones cycliques pour conduire à la formation d’alcoolates intermédiaires qui seront ensuite mis en présence d’agents de sulfonation pour subir une élimination E2 à l’aide de différentes bases. Une
fois les conditions opératoires mises au point et appliquées à différents substrats, cette méthode sera appliqué à la synthèse d’un sesquiterpène d’origine naturelle : le (±)-Laurokamurène B.
Le deuxième chapitre sera consacré à la mise au point et à l’optimisation d’une voie de synthèse permettant d’accéder aux 2-cycloalcènylbenzaldéhydes à 5, 6 et 7 chaînons. Ces aldéhydes seront ensuite mis en présence d’acides aminés et de leurs dérivés permettant d’accéder à des benzazépines de manière efficace et rapide par réaction d’électrocyclisation-1,7 d’ylures d’azométhines.
Le troisième chapitre décrira trois stratégies vers la synthèse totale de la (±)-morphine mettant en œuvre une réaction clé de cyclisation intramoléculaire acido-catalysée observée lors de la synthèse des 2-cycloalcénylbenzaldéhydes décrite dans le deuxième chapitre.
Chapitre I : Synthèse
d’arylcycloalcènes et synthèse totale
du (±)-Laurokamurène B
1
ère
partie : Synthèse en un seul pot
d’arylcycloalcènes
I. Introduction
La voie d’accès la plus efficace pour l’obtention d’arylcycloalcènes consiste en la création de la liaison C-C entre les groupements aryles et les synthons vinyliques cycliques (Schéma I-1).
La création de cette liaison C-C peut s’effectuer selon deux procédés. Le premier consiste à effectuer une réaction de couplage métallo-catalysé, le second consiste à effectuer l’addition nucléophile d’un organométallique sur des cétones cycliques (Y = OH) conduisant à la formation d’un alcool tertiaire qui peut ensuite subir des réactions d’élimination.
II. Réactions de couplage
Les réactions de couplages métallo-catalysé sont très utilisées en synthèse organique pour la préparation d’arylcycloalcènes. Les réactions de couplage dépendent du métal utilisé comme catalyseur. Ainsi, les catalyseurs au palladium et au nickel sont les plus employés à ce jour ; le cuivre , le cobalt et plus récemment le fer peuvent aussi être utilisés.
1. Réactions de couplage pallado-catalysé
Les catalyseurs les plus employés en synthèse organique pour la synthèse d’arylcycloalcènes sont les catalyseurs au palladium. Les réactions les plus connues sont les réactions de Heck43, de Suzuki-Miyaura44, de Stille45, de Negishi46 et de Kumada.
a. Couplage de Heck
Le couplage de Heck est la réaction la plus simple à mettre en œuvre. Cependant la synthèse d’arylcycloalcènes via cette méthode peut conduire à la formation de régioisomères difficilement séparables (Tableau I-1). 47
43 Pour une revue sur le couplage de Heck voir : I. P. Beletsakaya, A. V. Cheprakov, Chem. Rev. 2000, 100, 3009. 44 Pour une revue sur le couplage de Suzuki-Miyaura voir : N. Miyaura, A. Suzuki, Chem. Rev. 1995, 95, 2457. 45 Pour une revue su le couplage de Stille voir : P. Espinet, A. M. Echavarren, Angew. Chem. Int. Ed. 2004, 43,
4704.
46 Pour une revue sur le couplage de Negishi voir : V. B. Phapale, D. J. Cardenas, Chem. Soc. Rev. 2009, 38, 1598. 47 C. G. Hartung, K. Köhler, M. Beller, Org. Lett. 1999, 5, 709.
R ( )n R' R X ( )n R' Y + Schéma I-1
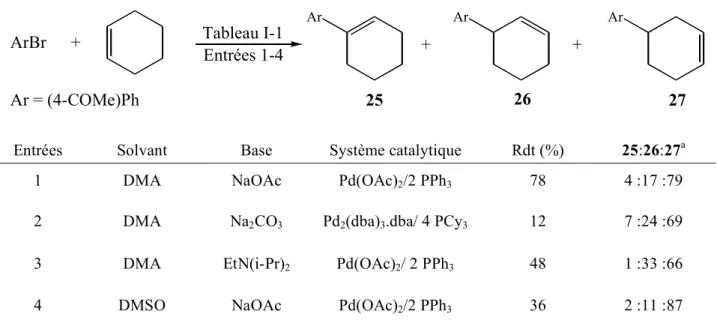
Tableau I-1 Entrées 1-4 Ar = (4-COMe)Ph ArBr + Ar Ar Ar + + 25 26 27
Entrées Solvant Base Système catalytique Rdt (%) 25:26:27a
1 DMA NaOAc Pd(OAc)2/2 PPh3 78 4 :17 :79
2 DMA Na2CO3 Pd2(dba)3.dba/ 4 PCy3 12 7 :24 :69
3 DMA EtN(i-Pr)2 Pd(OAc)2/ 2 PPh3 48 1 :33 :66
4 DMSO NaOAc Pd(OAc)2/2 PPh3 36 2 :11 :87
a : rapport déterminé par analyse des aires des pics obtenus en GC.
DMA : N,N-diméthylacetamide
Tableau I-1
Dans cette étude, Beller et al. montrent que la sélectivité du couplage de Heck vis-à-vis de l’un ou l’autre des régioisomères ainsi que le rendement sont très dépendant des conditions expérimentales (solvant, base, système catalytique) (entrées 1-4).
La formation de régiosiomères peut être expliquée par le cycle catalytique de la réaction de Heck décrit dans le schéma I-2. L’addition oxydante du catalyseur sur le dérivé bromé et insertion du complexe palladié sur l’oléfine conduit à la formation du complexe 28a à partir duquel la !-élimination d’hydrure conduit à la formation du composé 25 et 26. Cependant, la réinsertion du catalyseur sur le produit 26 conduit à la formation du complexe 29 qui par !-élimination d’hydrure conduit au régioisomère 27 (Schéma I-2).
Schéma I-2
En 2006, Fochi et al. ont publié un synthèse d’arylcycloalcènes par couplage de Heck en employant des o-benzénedisulfimides d’arénediazonium.48 L’o-benzènedisulfimidure permet d’obtenir des sels de diazoniums stables et joue le rôle de base pour la régénération du catalyseur après !-élimination d’hydrure (Schéma I-3).
O2 S N S O2 Ar N2 Pd(OAc)2 1 mol % EtOH 95%, 20°C + o-benzènedisulfimidure Ar Ar + X X = XH + Schéma I-3 b. Couplage de Suzuki-Miyaura
Plusieurs séries d’arylcycloalcènes ont été synthétisées par le couplage d’acides boroniques avec des triflates d’énoles,49 d’halogènes,50 ou de phosphates d’énols (Schéma I-4).51
Ar X + Y R X = , B B(OH)2; Y = OTf, Br, Cl O O Pd(0), Base reflux Ar R Schéma I-4
Keay et al. ont par ailleurs publié la synthèse d’arylcycloalcènes par réaction combinée en un seul pot de Shapiro-Suzuki à partir de trisylhydrazone (cette réaction sera décrite plus précisément dans le chapitre II) (Schéma I-5).52
N NHTris n-BuLi (3 éq.), hexane/TMEDA, 10 min-78°C à 0°C, 5 min
puis B(Oi-Pr)3, 1h
Evaporation de l'hexane, toluène, Na2CO3
5% mol Pd(OAc)2, ArBr, PPh3 10% mol reflux
R
Ar
Schéma I-5
49 a) J.-M. Fu, V. Snieckus, Tetrahedron Lett. 1990, 12, 1665.
b) T. Oh-e, N. MIyaura, A. Suzuki, Synlett 1990, 221. c) D. J. Wustrow, L. D. Wise, Synthesis 1991, 993. d) P. Wipf, M. Furegati, Org. Lett. 2006, 9, 1901.
e) P. C. Stanislawski, A. C. Willis, M. G. Banwell, Org. Lett. 2006, 10, 2143.
50 C. Song, Y. Ma, Q. Chai, C. Ma, W. Jiang, M. B. Andrus, Tetrahedron 2005, 61, 7438. 51 Y. Nang, Z. Yang, Tetrahedron Lett. 1999, 40, 3321.
c. Couplage de Stille
En 2001, Fu et al. ont publié la synthèse d’arylcyclopentènes par réaction de couplage de Stille par emploi de chlorures vinyliques53 ou de triflates d’énols (Schéma I-6).54
Ar SnBu3 + Cl Pd2(dba)3 1,5% mol ligands 3,5% mol CsF (2,2 éq.) dioxane, 100°C, 40h (1,1 éq.) Ar Ar = phényl 2-thiophène 2-furane Rdt = 91% Rdt = 83% Rdt = 93% Schéma I-6 d. Couplage de Negishi
La réaction de couplage de Negishi a été employée avec succès pour la synthèse d’arylcycloalcène à partir de chlorures vinyliques55 ou des triflates d’énols (Schéma I-7).56
ZnCl R Cl + Pd(Pt-Bu)3)2 2% mol (1,5 éq.) THF, NMP 100°C R R = 4-OMe R = 2-Me R = 2,2,4-Me Rdt = 82% Rdt = 94% Rdt = 96% Schéma I-7
2. Autres réactions métallo-catalysées
Bien que les réactions de couplages soient le plus souvent mises en œuvre sous catalyse au palladium, le nickel, le fer, le cuivre ainsi que le cobalt peuvent effectuer ces réactions.
a. Le nickel
Les catalyseurs au nickel semblent être une bonne alternative à l’emploi de catalyseur au palladium dont le coût peut devenir parfois problématique. Deux séries d’arylcycloalcènes ont été obtenues par couplage d’oraganomagnésiens et de phosphates57 ou de triflates d’énols58 en
53 W. Su, S. Urgaonkar, P. A. McLaughlin, J. G. Verkade, J. Am. Chem. Soc. 2004, 126, 16433. 54 V. Farina, B. Krishnan, D. R. Marshall, G. P. Roth, J. Org. Chem. 1993, 58, 5434.
55 C. Dai, G. C. Fu, J. Am. Chem. Soc. 2001, 123, 2719. 56 R. McCague, Tetrahedron Lett. 1987, 6, 701.
présence de Nickel (Schéma I-8). OMe OTf PhMgBr (2 éq.) 2h, 20°C NiCl2dppe ; Rdt = 76% Ni(acac)2 ; Rdt = 80% OMe Ph OPO(OPh)2 PhMgBr (1,5 éq.) NiCl2(dppe)2 1% mol
20°C ( )n Ph ( )n R1 R1 n = 1 ou 2; R1 = (CH2)3OTBS Rdt = 68-92% Schéma I-8 b. Le Fer
Les premiers travaux traitant de réactions de couplage catalysées par des complexes du Fer(III) sont dus à Kochi et al. au début des années 1970.59 Cependant, cette méthode restait
limitée au couplage d’organomagnésiens avec des halogénures vinyliques. Il aura fallu attendre les années 2000 pour que le fer soit considéré comme une alternative économique et écologique au palladium.
Fürstner et al.60 ont publié la synthèse d’arylcycloalcènes par réaction de couplage entre des réactifs de Grignard et des triflates d’énols cycliques en présence d’une quantité catalytique de tris(acétoacétate) de fer(III) (Schéma I-9).
TfO R PhMgBr (1,3 éq.) Fe(acac)3 (5% mol) THF / NMP -30°C Ph R
R = Me, CO2Et, 1,3-dioxolane
Rdt = 47-74%
Schéma I-9
58 C. A. Busacca, M. C. Eriksson, R. Fiaschi, Tetrahedron Lett. 1999, 40, 3101. 59 a) J. K. Kochi, Acc. Chem. Res. 1974, 7, 251.
b) S. M. Neumann, J. K. Kochi, J. Org. Chem. 1975, 5, 599. c) J. K. Kochi, J. Organomet. Chem. 2002, 653, 11.
c. Le Cuivre
Les diarylcuprates de lithium en excès peuvent donner des réactions de couplage avec les triflates d’énols permettant d’accéder à des arylcycloalcènes (Schéma I-10).61 Le couplage peut aussi être effectué avec un léger excès de réactif de Grignard en présence d’une quantité catalytique de CuI, sans observer d’isomérisation du système diènique.62
TfO tBu Ph2CuLi (3,5 éq.) THF, -15°C, 12h Rdt = 75% Ph tBu TfO ArMgBr (1,5 éq.) Ar CuI (10% mol) THF, 0°C n = 1; 86-92% n = 2; 87-94% ( )n ( )n Schéma I-10 d. Le Colbalt
Une série d’arylcycloalcènes a pu être synthétisée par couplage d’acétates de cyclopentényles avec des halogénures d’aryles en présence de manganèse et d’une quantité catalytique de Co(II)Br2 (Schéma I-11).63
X
AcO
CoBr2 5% mol, Mn excès BPy 5% mol DMF, Pyridine (v/v = 15/2) ! CF3CO2H, 50°C X = Br => R = OCOMe : Rdt = 16% R = OMe : Rdt = 39% X= Cl => R = OCOMe : Rdt = 79% R = CN : Rdt = 23% R R Schéma I-11
Le cobalt introduit est sous la forme de bromure de Cobalt(II), or seuls les cobalt(I) et (0) présentent une activité catalytique. Un excès de manganèse est nécessaire afin d’amorcer la réaction par réduction du bromure de cobalt(II) en cobalt(I) ou (0). En fin de cycle catalytique, le cobalt se trouve à un état d’oxidation (II) ou (III) ; à nouveau le manganèse intervient en tant que
61 J. E. McMurry, W. J. Scott, Tetrahedron Lett. 1980, Vol. 21, 4313.
62 A. Sofia, E. Karlström, M. Rönn, A. Thorarensen, J.-E. Bäckvall, J. Org. Chem. 1998, 63, 2517. 63 M. Amatore, C. Gosmini, J. Périchon, Eur. J. Org. Chem. 2005, 989.
réducteur pour régénérer les espèces actives du cobalt (Schéma I-12).
Schéma I-12
e. Mise en œuvre des réactions de couplages métallo-catalysées
La mise en œuvre des réactions de couplage métallo-catalysées de Suzuki-Miyaura, Stille, Negishi et Kumada64 nécessite la préparation de synthons vinyliques. Les bromures et chlorures vinyliques sont obtenus à partir des cétones correspondantes ;65 les iodures vinyliques à partir des bromures par échange brome-iode (Schéma I-13).66
64 J. A. Miller, Tetrahedron Lett. 2002, 43, 7111.
65 A. Spaggiari, D. Vaccari, P. Davoli, G. Torre, F, Prati, J. Org. Chem. 2007, 72, 2216. 66 A. Klapars, S. L. Buchwald, J. Am. Chem. Soc. 2002, 124, 14844.
R O R' R X R' P(OPh)3 (1,1 éq.) X = Cl; Rdt = 75-96% X = Br; Rdt = 50-99% X2 (1,1 éq.) Et3N (1,2 éq.) R Br
R' CuI (5% mol)NaI, n-BuOH
NHMe NHMe 10% mol + R I R' Schéma I-13
Cependant les halogénures vinyliques réagissant lentement, les triflates d’énols correspondant leur sont donc le plus souvent préférés.
Les triflates d’énols sont eux aussi accessibles à partir des cétones correspondantes en milieu basique en présence de N-Phenylbis(trifluorométhanesulfonimide). Les triflates d’énols sont plus réactifs que les halogénures vinyliques, mais leur purification est rendue difficile par la présence de sous produits de réactions difficilement séparables (Schéma I-14).67
O R 1) i-Pr2NLi (1,1 éq.), THF, -78°C 2) PhNTf2 (1,5 éq.), 20°C TfO R Schéma I-14
Les phosphates d’énols quant à eux sont obtenus par traitement d’une cétone en milieu basique par du chlorophosphate de diphényle (Schéma I-15).68,64,57
O 1) NaHMDS, THF, -78°C 2) (Ph)2P(O)Cl, THF, -78°C OPO(Ph)2 Schéma I-15
67 Pour une revue voir : K. Ritter, Synthesis 1993, 735.
L’énolate formé par action de la base s’additionne sur le phosphore, la réaction est suivie de l’élimination de l’halogène pour donner le phosphate d’énol.
3. Addition nucléophile / déshydratation
Une autre approche de la synthèse d’arylcycloalcènes vise à créer la liaison C-C via l’addition nucléophile d’un organométallique (organomagnésien ou organolithien) sur des cétones cycliques conduisant à la formation d’alcools tertiaires.69 Ces alcools intermédiaires peuvent ensuite être déshydratés en milieu acide70 ou estérifiés par un agent de sulfonation71 afin
de subir une élimination E2 par une base, conduisant à la formation des arylcycloalcènes désirés
(Schéma I-15). ( )n Elimination R R' ( )n R R' OH R ( )n R' Br O + Addition nucléophile Schéma I-15 a. Addition nucléophile
L’addition nucléophile d’organolithiens ou de réactifs de Grignard sur des cétones est la méthode la plus employée permettant d’obtenir des alcools tertiaires. Les réactifs de Grignard et les organolithiens sont facilement accessible à partir des dérivés bromés correspondants (Schéma I-16). R ( )n R' M O M = MgBr, Li + H3O ( )n R R' OH R X X = Br, Cl Mg, Et2O, 0°C n-BuLi, THF, -78°C ou Schéma I-16
Cependant, l’addition de ces organométalliques fortement basiques sur des cétones cycliques aisément énolisables occasionne des réactions d’énolisation, de réduction (en cas
69 Pour une revue voir : M. Hanato, K. Ishihara, Synthesis, 2008, 1647.
70 R. C. Larock, Comprehensive Organic Transformations, 2nd ed ; Wiley-WCH, 1999, 291. 71 R. C. Larock, Comprehensive Organic Transformations, 2nd ed ; Wiley-WCH, 1999, 294.
d’utilisation de magnésiens possédant des hydrogènes en !), d’auto-aldolisation72 et de couplage pinacolique (Schéma I-17).
( )n O énolisation H R M RH ( )n O + + M O ( )n aldolisation O hydrolyse HO ( )n ( )n ( )n Mg Br O H R R -( )n OH Réduction ( )n O 2 Mg O O Mg ( )n ( )n couplage pinacolique O O Mg ( )n ( )n H OH OH ( )n ( )n Schéma I-17
Malgré ces réactions secondaires, l’addition de réactifs de Grignard ou d’organolithiens sur des cétones reste la méthode la plus employée pour la création de liaison C-C.
b. Elimination
Les arylcycloalcènes peuvent être obtenus à partir des alcools tertiaires par réactions d’éliminations. Les deux principales méthodes d’élimination sont les déshydratations et les éliminations de sulfonates.
La première méthode permettant d’accéder aux arylcycloalcènes à partir des alcools tertiaires consiste à effectuer une déshydratation en milieu acide par protonation de l’alcool puis élimination de type E1 (Schéma I-18).
H OH OH2 H -H -H2O élimination E1 Ar Ar Ar Ar Schéma I-18
Cependant cette déshydratation en milieu acide peut être problématique si les arylcycloalcènes possèdent des fonctions acido-sensibles (acétals, alcools…). De plus, ces conditions acides peuvent conduire à la formation de régioisomères de double liaison C=C.
Une autre méthode consiste à estérifier un l’alcool tertiaire. Cet ester peut ensuite subir une élimination E2 en milieu basique pour conduire à la formation de l’arylcycloalcène désiré
(Schéma I-19). OH OSO2R H RSO2Cl Base B R = CF3, Me, Et élimination E2 Ar Ar Ar Schéma I-19
Afin de pouvoir observer la réaction d’élimination E2, les 2 groupements (OSO2R et H)
doivent être anti-périplanaires (influence de la taille du cycle ainsi que de sa conformation). L’utilisation de ces conditions basiques permet la synthèse d’arylcycloalcènes fonctionnalisés.
4. Synthèse d’arylcycloalcènes par réarrangement semi-pinacolique/fragmentation de Grob.
Récemment, la synthèse d’arylcycloalcènes a été décrite grâce à la fragmentation de Grob à partir d’alcools allyliques.73 Dans cette réaction, le N-bromosuccinimide réagit avec la double
liaison de l’alcool allylique pour former un ion bromonium, qui se réarrange par migration de l’aromatique pour donner l’aldéhyde !-bromé cis. L’addition de OH- sur la fonction aldéhyde
est suivie de la perte de HCO2H. L’arylcycloalcène est issu de la perte de Br- du carbanion
précédemment obtenu (Schéma I-20).
R1 Ph O H N O O Br R1 Br Ph O H R1 Br Ph O H R1 Br Ph OH O NaOH R1 Br Ph R1 Ph -HCO2H -NaBr DME 20°C 83-91% Schéma I-20
Malgré les bons rendements affichés (75%-92%), cette méthode nécessite la synthèse préliminaire des alcools.
III.
Synthèse en un seul-pot d’arylcycloalcènes.
La préparation d’alcools tertiaires par addition nucléophile d’organocériens sur des cétones cycliques conduit à la formation d’alcools par une réaction propre. Pour ces raisons, il paraissait intéressant d’effectuer l’addition nucléophile d’organocériens sur des cétones cycliques afin de former des alcoolates intermédiaires qui seront estérifiés in-situ puis mis en présence d’une base afin de subir une élimination E2, conduisant ainsi à la formation des arylcycloalcènes désirés
(Schéma I-21).
En premier lieu, les conditions opératoires ont été mises au point afin d’optimiser les rendements mais aussi les quantités de réactifs utilisées. Une fois les conditions opératoires déterminées, une série d’arylcycloalcènes a été synthétisée.
R 1) n-BuLi R 2) CeCl3 Bases (3 éq.) Br OM 1,1 éq. M= Li,CeCl3 -30°C, agents estérifiants (3 éq.) puis 20°C, 12h R alcoolates intermédiaires 3) Cycloalcanones Schéma I-21
1. Mise au point des conditions opératoires
La mise au point des conditions opératoires a été effectuée sur la synthèse de l’arylcycloalcène 31 (Tableau I-2). Après échange lithium-brome entre le n-butyllithium et le 4-bromoanisole à -78°C dans du THF anhydre, l’organolithien ainsi obtenu est alors additionné à une suspension de chlorure de cérium(III) anhydre dans le THF à -78°C afin de former l’organocérien correspondant. La cyclohexanone est ensuite additionnée à l’organocérien, à -78°C, pour donner le produit d’addition nucléophile, sous la forme de l’alcoolate 30. Cet alcoolate est ensuite mis en présence d’une base puis, à -30°C d’un agent de sulfonation afin de procéder à la séquence estérification/élimination E2 pour donner l’arylcycloalcène 31.
1) n-BuLi (1,2 éq.) 2) CeCl3 (x éq.)
1) Base (3 éq.) Br
OLi,CeCl3 2) Agent de sulfonation (3 éq.) 3) Cyclohexanone (1 éq.) 30 (1,1 éq.) OMe OMe THF, -78°C THF, - 30°C 31 OMe
Entrée CeCl3 ( x éq.) Bases (3 éq.) Agents sulfonants (3 éq.) Rdt (%)
31 1 1,6 Et3N CH3SO2Cl 60 2 1,3 Et3N CH3SO2Cl 73 3 1,3 Et3N (CF3SO2)2O 70 4 1,3 DBU (CF3SO2)2O 73 5 1,3 DBU CH3CH2SO2Cl 62 6 1,3 DBU CH3SO2Cl 87 7 1,3 DABCO CH3SO2Cl 22 8 1,3 Pyridine CH3SO2Cl 15 9 1,3 DIPEA CH3SO2Cl 28