Modélisation moléculaire des copolymères PMMA-PS
Texte intégral
Figure

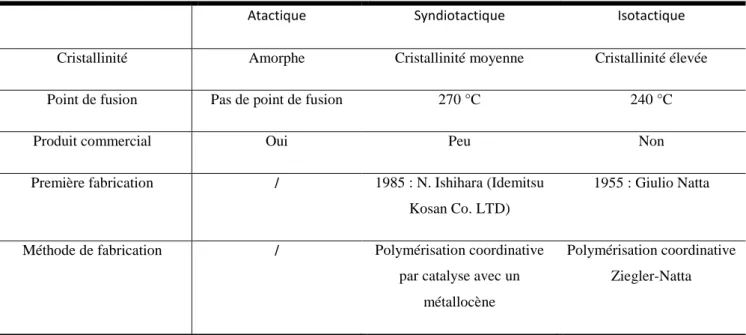
![Tab. I.2 Propriétés mécanique des polymères [2]](https://thumb-eu.123doks.com/thumbv2/123doknet/7781686.258594/22.892.74.847.401.711/tab-propriétés-mécanique-des-polymères.webp)

Documents relatifs
Hence, the effect of 364 dispersal on beta-diversity and regional productivity decreased at the end of the experiment, which 365 resulted in the absence of a
Dans le contexte actuel où il est important d’optimiser les antibiotiques à notre disposition et de rationaliser l’utilisation des combinaisons d’antibiotiques,
Chronic persistent HHV-6B infection after sulfasalazine-induced DRESS with demonstration of HHV-6 encoded small noncoding RNAs (sncRNAs) in Crohn’s-like colitis: Case report.
ont appliqué cette méthode en premier lieu à la cyclisation de poly(tétrahydrofurane) en partant d’un polymère linéaire porteur de groupes ammonium cycliques (N-phényl- ou
Les « réorganisations locales » réalisées lors d’un premier recuit sont effacées lorsque le matériau est soumis par la suite à des conditions favorisant la mobilité
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
On a porté la température pour laquelle le second coefficient de viriel s'annule en fonction de la composition du copolymère en montrant sur une deuxième
