© Bastien Paré, 2019
Culture tridimensionnelle de fibroblastes dermiques,
dérivés de patients, pour l'étude de la Sclérose Latérale
Amyotrophique et l’identification de biomarqueurs
Thèse
Bastien Paré
Doctorat en médecine expérimentale
Philosophiæ doctor (Ph. D.)
Culture tridimensionnelle de fibroblastes dermiques, dérivés de patients, pour
l'étude de la Sclérose Latérale Amyotrophique et l’identification de
biomarqueurs
Thèse
Bastien Paré
Sous la direction de :
Dr François Gros-Louis, directeur de recherche
Dr François Berthod, codirecteur de recherche
iii
Résumé
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative hétérogène, incurable et sans traitement efficace qui se caractérise principalement par une dégénérescence sélective des neurones moteurs de la moelle épinière et du cerveau. Cette maladie se présente généralement par une faiblesse musculaire progressive, une hypertonie spastique, de la dysphagie, l’apparition de fasciculations, une paralysie presque totale et le décès de 3 à 5 ans après l’apparition des premiers symptômes. La SLA se présente sous deux formes distinctes : la SLA de type familial (SLAF) et la SLA de type sporadique (SLAS). La SLAF est associée à des mutations génétiques précises et est transmise de façon autosomale dominante dans la très grande majorité des cas. Quant à la SLAS, elle se distingue par son côté sporadique, sans cause génétique associée, et représente de 90 à 95 % de tous les cas de SLA.
La peau est considérée par certains comme le plus grand organe du corps humain. Elle joue un rôle important dans la thermorégulation ainsi que dans la synthèse de la vitamine D et agit comme barrière naturelle contre l’environnement. Ses couches principales, l’épiderme et le derme, sont principalement et respectivement formées de kératinocytes et de fibroblastes. Le concept du « non-cell autonomous toxicity » stipule qu’une cellule ne présentant pas de mutation associée à une maladie donnée peut présenter un phénotype pathologique. Dans l’étude de la SLA, ce concept s’applique tant aux cellules neuronales qu’aux cellules non neuronales, comme les cellules endothéliales ou les fibroblastes de peau. Hors du système nerveux central, les fibroblastes de peau pourraient représenter une source importante et non invasive d’échantillons biologiques pour l’étude de la SLA.
Les exosomes sont des vésicules extracellulaires de 30 à 200 nm de diamètre. Ils sont sécrétés par tous les types cellulaires et représentent un moyen de communication cellulaire important grâce au transport de diverses molécules, dont des protéines et de l’ARN. Ces vésicules représentent une potentielle source de biomarqueur pour l’étude de diverses maladies neurodégénératives, dont la SLA.
Dans le cadre du projet de recherche présenté dans cette thèse, les travaux réalisés ont permis de démontrer que l’utilisation de cellules de peau, dont des fibroblastes et des kératinocytes de patients atteints de SLA, permet d’étudier certains aspects de la pathologie de la maladie. En effet, l’utilisation d’un protocole de production de peau reconstruite en laboratoire par génie tissulaire à partir de cellules de patients atteints de
iv
SLA a permis de détecter différentes anomalies de la matrice extracellulaire en plus d’une délocalisation de la protéine TDP-43, précédemment détectée uniquement dans le système nerveux central de patients atteints de SLA. Les fibroblastes de peau ont aussi été démontrés comme étant une source d’intérêt pour la découverte de biomarqueurs associés à la maladie. Le sécrétome - le matériel biologique sécrété par une cellule – provenant des fibroblastes peut être purifié à l’aide d’une technique de précipitation protéique qui permet d’obtenir un culot pur, exempt d’impuretés et de sels provenant du milieu de culture. Parmi les éléments identifiés chez les protéines, les exosomes ont été démontrés d’intérêt et importants dans leur culture. En effet, lorsque cultivés en trois dimensions, les exosomes dérivés de fibroblastes de peau 3D contiennent différentes molécules augmentant la prolifération ainsi que la migration cellulaire. De plus, ces vésicules extracellulaires contiennent une grande quantité de protéines de la matrice extracellulaire, démontrant leur importance dans la sécrétion et l’assemblage de celle-ci en culture 3D. Ces exosomes ont de plus la capacité d’améliorer le processus de guérison de plaies dans un modèle de peau reconstruite en laboratoire formé de fibroblastes et de kératinocytes. Finalement, la protéine SOD1, associée au développement de certains types de SLA familiale, a pu être démontrée comme étant un biomarqueur neuropathologique possible de la SLA sporadique. Au même titre que des patients présentant une mutation du gène SOD1, des patients atteints de SLA sporadique présentent certains aspects pathologiques associés à la maladie, dont la présence d’agrégats cytoplasmiques de la protéine mal repliée dans les neurones moteurs du système nerveux central.
Globalement, mes travaux démontrent que les cellules de peau représentent un échantillon biologique important dans l’étude de la SLA et qu’elles pourraient constituer un outil novateur dans la découverte de nouveaux biomarqueurs de la maladie. Les exosomes sécrétés par les fibroblastes de peau en culture 3D ont été démontrés comme étant importants dans la prolifération, la migration cellulaire et la sécrétion de protéines de la matrice extracellulaire. Ces vésicules présentent un potentiel énorme dans la découverte de biomarqueurs associés à la maladie. La présence d’agrégats cytoplasmiques de la protéine SOD1 dans les neurones moteurs du système nerveux central de patients atteints de SLA sporadique permet de croire que cette protéine pourrait devenir un biomarqueur important dans le diagnostic de la maladie.
v
Abstract
Amyotrophic lateral sclerosis (ALS) is a heterogenous neurodegenerative disease. Presently, it is an incurable disease without any effective treatment and is characterised by selective degeneration of motor neurons in the central nervous system. The symptoms that most patients display include cramps, weakness and muscle atrophy of the hands and feet progressing to the forearms, shoulders and legs, eventually leading to complete paralysis. Nearly 90% of all ALS cases are sporadic, with no known cause. The other 10% of cases represent familial ALS and are associated to ALS-linked genes, such as SOD1, FUS/TLS, TARDBP, and
C9ORF72.
The skin is considered by some to be the biggest organ of the human body. It plays an important role in thermoregulation as well as vitamin D synthesis. Skin also acts as a natural barrier against environmental threats. It is comprised of the epidermis and the dermis, which are made of keratinocytes and fibroblasts, among other things. The non-cell autonomous toxicity paradigm in ALS has been well established. Outside of the central nervous system, skin fibroblasts could potentially be an important source of biomarkers.
The work presented in this thesis demonstrates that skin cells, such as fibroblasts and keratinocytes, derived from ALS patients, allow for the study of different pathological aspects of the disease. The use of a tissue-engineered skin from ALS patients skin cells allows for detection and observation of extracellular matrix structure abnormalities, as well as mislocalization of TDP-43, previously only detected in the motor neurons of patients. Results from experiments associated with this study shed more light on skin fibroblasts, which appear to be a potential source of novel biomarkers. Their secretome can be purified using an optimized protocol leading to pure proteins without salt contamination coming from the cell culture media. As a result, exosomes are of great interest for the discovery of novel biomarkers for the diagnosis of ALS, for following its progression, and for the culture of fibroblast cells. When cultivated in a 3D-fashion, the secreted exosomes contain molecules enhancing cell proliferation and migration, as well as high amounts of extracellular matrix proteins. These extracellular vesicles also help to enhance wound healing in a tissue-engineered model made of skin fibroblasts and keratinocytes. Finally, the SOD1 protein, which is associated with the development of some familial ALS cases, should be considered a potential neuropathological biomarker of sporadic ALS. Cytoplasmic aggregates of the misfolded protein were detected in the motor neurons of sporadic patients, alongside familial ALS patients who were carriers of an SOD1 mutation.
vi
Overall, this work shows that skin cells represent an important and minimally invasive biological sample in the study of ALS. These cells are also of interest in the discovery of novel ALS biomarkers. Exosomes secreted by skin fibroblast cells in a 3D culture are important in cell proliferation and migration. They play a crucial role in extracellular matrix protein secretion. The results of this study show that exosomes, proven to be secreted by dermal fibrobasts when cultivated in a 3D fashion, may become as a primary source of biomarkers in ALS. Cytoplasmic aggregates of misfolded SOD1 in motor neurons of sporadic ALS patients could lead to the development of diagnostic tests with SOD1.
vii
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des abréviations ... xiii
Liste des figures ... xvii
Liste des tables ... xix
Remerciements ... xx
Avant-propos ... xxi
Introduction, problématiques de recherche, hypothèses et objectifs ... 1
La sclérose latérale amyotrophique ... 2
La SLA familiale... 3
Superoxide dismutase 1 (SOD1)... 6
Physiopathologie de la SOD1 dans la SLAF ... 7
Transactive response DNA binding protein 43 kDa (TARDBP/TDP-43) ... 11
Physiopathologie de TDP-43 dans la SLAF ... 12
C9ORF72 ... 14
Physiopathologie de C9ORF72 dans la SLAF ... 15
RNA-binding protein FUS/TLS (Fused in Sarcoma/Translocated in sarcoma) ... 17
Physiopathologie de FUS/TLS dans la SLAF ... 18
Les modèles d’étude de la SLAF ... 21
Les modèles d’étude murins de la SLA ... 21
Le poisson zèbre ... 26
La mouche à fruits ... 29
Le nématode Caenorhabditis elegans ... 31
La SLA sporadique ... 32
Facteurs de susceptibilité génétique découverts par « genome-wide association studies »... 32
viii
Facteurs environnementaux ... 34
Physiopathologie de la SLA sporadique... 35
Modèles in vitro de la SLAS ... 36
L’importance des biomarqueurs dans l’étude des maladies neurodégénératives ... 39
Les biomarqueurs : définition générale et utilités ... 39
Les biomarqueurs de la SLA ... 41
La peau ... 53
Description de la peau chez l’humain... 54
Problématiques de recherche, hypothèse et objectifs ... 56
Chapitre 1: Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients... 60
1.1 Avant-propos ... 61
1.2 Résumé ... 63
1.3 Abstract ... 64
1.4 Introduction ... 65
1.5 Materials and methods ... 66
1.5.1 Patients ... 66
1.5.2 Skin biopsies and cell extraction/culture ... 67
1.5.3 Production of tissue-engineered skin equivalents ... 68
1.5.4 Autopsy procedure ... 68
1.5.5 Immunohistochemistry (IHC) ... 69
1.5.6 Masson’s trichrome staining ... 69
1.5.7 Immunofluorescence (IF) ... 70
1.5.8 Subcellular fractionation of TES and Western blotting ... 70
1.6 Results ... 70
1.6.1 Structural abnormalities detected in ALS-TES by IHC ... 70
1.6.2 TDP-43 cytoplasmic inclusion detected in ALS-TES derived skin ... 71
1.6.3 TDP-43 cytoplasmic inclusion detected in native skin biopsies and in post-mortem CNS tissue collected from ALS patients ... 76
ix 1.8 Conclusion ... 79 1.9 Competing Interest ... 79 1.10 Acknowledgements ... 79 1.11 Author Details ... 80 1.12 References ... 80
Chapitre 2: An Optimized Approach to Recover Secreted Proteins from Fibroblast Conditioned-Media for Secretomic Analysis... 90
2.1 Avant-propos ... 91
2.2 Résumé ... 93
2.3 Abstract ... 94
2.4 Introduction ... 95
2.5 Materials and methods ... 96
2.5.1 Sample selection and collection ... 96
2.5.2 Skin Biopsies and Fibroblast Cell Extraction/Culture ... 97
2.5.3 Fibroblast Induction and Protein Precipitation ... 98
2.5.4 Protein Conjugation ... 99
2.5.5 Electrophoresis ... 100
2.6 Results ... 103
2.6.1 Comparison of Protein Extraction Methods and the Effect of the Confluence Level and Collection Time on the Protein Recovery Yield ... 103
2.6.2 Effect of Cyanine Dye Conjugation on the Recovered Protein ... 104
2.6.3 Visualization of Whole Secreted Protein Resolved with 2D-DIGE ... 106
2.7 Discussion ... 108 2.8 Conclusion ... 111 2.9 Author Contributions ... 111 2.10 Funding ... 112 2.11 Acknowledgments ... 112 2.12 Supplementary Material ... 112
2.13 Conflict of Interests Statement ... 112
x
Chapitre 3: Misfolded SOD1 pathology in sporadic ALS ... 120
3.1 Avant-propos ... 121
3.2 Résumé ... 124
3.3 Abstract ... 125
3.4 Introduction ... 126
3.5 Materials and methods ... 127
3.4.1 Animals ... 127
3.5.2 Human samples and neuropathological examination ... 127
3.5.3 Immunohistochemistry ... 128
3.5.4 Immunocapture ... 128
3.6 Results ... 129
3.6.1 Specific staining of misSOD1 in FALS ... 129
3.6.2 Widespread staining of misSOD1 in SALS ... 131
3.6.3 Distinct patterns of misSOD1 in SALS ... 135
3.6.4 Immunocapture of misSOD1 from SALS spinal cord ... 139
3.7 Discussion ... 141 3.8 Author contributions ... 144 3.9 Funding ... 144 3.10 Acknowledgments ... 144 3.11 Supplementary figures ... 145 3.12 References ... 153
Chapitre 4: Dermal fibroblasts cultured in a 3D environment release exosomes promoting extracellular matrix remodelling ... 159
4.1 Avant-propos ... 160
4.2 Résumé ... 162
4.3 Abstract ... 163
4.4 Introduction ... 164
4.5 Material and Methods ... 165
xi
Cell isolation and culture ... 165
2D culture model ... 166
3D culture model ... 166
RNA extraction ... 166
Total protein extraction from culture media ... 167
Next-generation RNA sequencing and gene expression profiling ... 167
Quantitative mass spectrometry analysis ... 168
Exosomes production and isolation... 170
Quantification of isolated exosomes and exosomal proteins... 170
Exosomes uptake by fibroblast cells ... 172
Measurement of matrix metalloproteinases (MMPs) expression activity ... 173
Cell migration and proliferation assay ... 174
Enzyme-linked immunosorbent assay ... 175
Wound healing assay using 3D tissue-engineered skins ... 175
Statistical analyses ... 176
4.6 Results ... 176
RNA-sequencing and quantitative MS analyses show differencial gene and protein expression for each culture method ... 176
Skin fibroblasts cultured in a 3D fashion release large amount of exosomes ... 177
Dermal fibroblast-deribed exosomes have the capacity to travel from one cell to another ... 179
Exosomal proteins analyses revealed increased MMP expression and enzymatic activity ... 179
Exosomes, derived from 3D dermal fibroblast culture, induce cellular proliferation and migration ... 179
Dermal fibroblast-derived exosomes induced wound healing in self-assembled tissue-engineered skins ... 180
4.7 Discussion ... 181
4.8 Figures and Tables ... 182
4.9 Acknowledgment ... 193
4.10 References ... 193
Chapitre 5: Discussion ... 196
xii
5.1.1 Les modèles d’études de la SLA ... 198
5.1.2 La découverte de biomarqueurs dans la SLA – Les exosomes ... 201
5.1.3 La découverte de biomarqueurs dans la SLA – Tissus post-mortem ... 206
5.2 Perspectives de recherche ... 209
Conclusion générale ... 214
Bibliographie ... 216
Annexe ... 259
xiii
Liste des abréviations
• Acide desoxyribonucléique; ADN • Acide ribonucléique; ARN
• Alsin rho guanine nucleotide exchange factor; ALS2
• Amyotrophic lateral sclerosis 3 (Autosomal Dominant); ALS3 • Amyotrophic lateral sclerosis 7; ALS7
• Amyotrophic lateral sclerosis frontotemporal degeneration; ALS-FTD • Angiogénine; ANG
• Apolipoprotein E; ApoE
• Apurinic endonuclease, DNA repair enzyme; APEX • Ataxie cérébelleuse de type 2; SCA2
• Ataxin 2; ATXN2
• Calcium-responsive transactivator; CREST • Charged multivesicular body protein 2B; CHMP2B • Cholinergic receptor nicotinic alpha 3 subunit; CHRNA3 • Cholinergic receptor nicotinic alpha 4 subunit; CHRNA4 • Cholinergic receptor nicotinic beta 4 subunit; CHRNB4 • Chromosome 9, open reading frame 72; C9ORF72 • Chromosome 19, open reading frame 12; C19ORF12
• Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated proteins; CRISPR/Cas
• Coiled-coil-helix-coiled-coil-helix domain containing 10; CHCHD10 • Counts-per-million reads; CPM
• Cuivre; Cu
• D-amino acid oxidase; DAO • Deux-dimensions; 2D
• Differentially expressed in normal and neoplastic cells; DENN • Dipeptide repeat proteins; DPR
xiv • Dipeptidyl-peptidase 6; DPP6
• Dynactin; DCTN1
• Elongator complex protein 3; ELP3
• Endosomal sorting complex required for transport III; ESCRT-III • Encéphalopathies spongiformes transmissibles; EST
• Ewing sarcoma breakpoint region 1; EWSR1/EWS • Fast fatigable; FF
• FIG4 phosphoinositide 5-phosphatase; FIG1
• FGGY carbohydrate kinase domain containing; FGGY
• Fragments per kilobase of exon per million fragments mapped; FPKM • Fused in sarcoma/translated in liposarcoma; FUS/TLS
• Genome-wide association studies; GWAS • Glial cell-line derived neutrophic factor; GDNF
• Heterogeneous nuclear ribonucleoprotein A1; hnRNPA1 • Heterogeneous nuclear ribonucleoprotein A2/B1; hnRNPA2B1 • Homeostatic iron regulator; HFE
• Human embryonic kidney 293 cells; HEK 293 • Induced Pluripotent Stem Cells; iPSc
• Inositol 1, 4, 5-triphosphate receptor 2 gene; ITPR2 • Kilodaltons; kDa
• Kinesin associated protein 3; KIFAP3 • Kinesin associated protein 3; KINAP3 • Knock-in; KI
• Knockout; KO
• Kruppel-like factor 4; KLF4
• Label-Free Mas Spectrometry; LF-MS • Liquide céphalorachidien; LCR • Matrice extracellulaire; MEC
xv • Matrin 3; MATR3
• Matrix métalloprotéinase 2; MMP-2 • Matrix métalloprotéinase 9; MMP-9
• Metabotropic glutamate receptor 2; mGLUR2 • Neurofilament light chain; NEFL
• Neurofilament heavy chain; NEFH • Neuropathy target esterase; NTE • Nuclear localization sequence; NLS
• Octamer-binding transcription factor3/4; oct3/4 • Optineurine; OPTN
• Oxygène; O2
• Paraxonase; PON1, 2, 3
• Peripheral blood mononuclear cells; PBMC • Peripherin; PRPH
• Péroxyde d’hydrogène; H2O2 • Principal component analysis; PCA • Profilin 1; PFN1
• Reactive oxygen species; ROS
• Receptor tyrosine protein kinase ErbB-4; ERB4 • Repeat-associated non-AUG; RAN
• Sclérose latérale amyotrophique; SLA
• Sclérose latérale amyotrophique de type familiale; SLAF • Sclérose latérale amyotrophique de type sporadique; SLAS • Senataxin; SETX
• Sequestosome 1; SQSTM1
• Sigma non-opiod intracellular receptor 1; SIGMAR1 • Single nucleotid polymorphisms; SNP
xvi • Spatascin; SPG11
• Superoxyde Ddsmutase; SOD • Superoxyde dismutase Cu-Zn; SOD1
• Superoxyde dismutase 2, mitochondriale; SOD2 • Superoxyde dismutase Cu-Zn extracellulaire; SOD3 • Survival motor neuron; SMN
• Système nerveux central; SNC • TANK binding kinase 1; NEK1
• TAR DNA-binding protein 43; TDP-43/TARDBP • TATA-box binding protein associated factor 15; TAF15 • Transcription factor P64; c-myc
• Translocated in liposarcoma/Ewing’s sarcoma/TATA-binding protein-associated factor 15; TET • Trois-dimensions; 3D
• Tubulin alpha 4a; TUBA4A • Ubiquilin-2; UBQLN2 • Ultraviolet; UV
• UNC-13 homologue A; UNC13A • Upstream Activating Sequence; UAS • Valosin containing protein; VCP
• Vascular endothelial growth factor; VEGF
• Vascular endothelial growth factor receptor 2; VEGFR2
• Vesicle-associated membrane protein-associated protein B/C; VAPB • Virus de l'immunodéficience humaine 1; VIH-1
xvii
Liste des figures
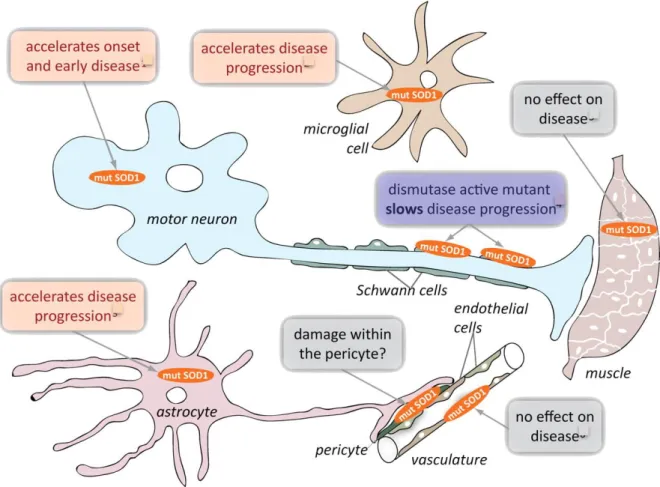
Figure 1 | Toxicité associée au mauvais repliement de la protéine SOD1 9 Figure 2 | Contribution de plusieurs types cellulaires dans le développement de la SLA 11
Figure 3 | Modèle simplifié de la protéinopathie de TDP-43 14
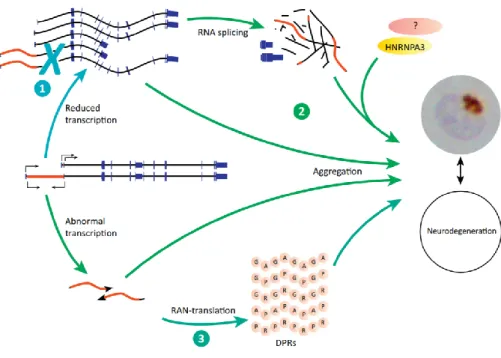
Figure 4 | Principaux phénomènes cytotoxiques associés à la répétition GGGGCC du gène C9ORF72 16
Figure 5 | Principaux phénomènes cytotoxiques associés au gène FUS/TLS (Gain et perte de fonction) 19
Figure 6 | Contribution des mutations de SOD1 dans différents types cellulaires 23
Figure 7 | Développement du modèle de peau reconstruite en laboratoire du Laboratoire d’organogénèse
expérimentale 55
Figure 1.1 | Structural abnormalities detected in ALS-derived tissue engineered skins 71 Figure 1.2 | Cytoplasmic TDP-43 accumulation detected in SALS- and C9orf72 FALS-derived tissue-engineered
skins 73
Figure 1.3 | Cellular counts and Western blots quantification of TDP-43 cytoplasmic accumulation 74 Figure 1.4 | Nuclear TDP-43 expression in C9orf72 fibroblasts cultured cells 75 Figure 1.5 | Cytoplasmic TDP-43 detection in native skin biopsies and corresponding CNS tissues in SALS
patients 76
Figure 1.6 | Familial C9orf72-linked ALS pedigree 85
Figure 1.7 | Tissue-engineered skin equivalent using self-assembly method 86 Figure 1.8 | Cytoplasmic TDP-43 accumulation detected in SALS-derived tissue-engineered skins 87 Figure 1.9 | Original Western blots and specificity of the protein fractionation method 89 Figure 1.10 | Cytoplasmic TDP-43 inclusions are detected in the fibroblast cells of SALS native skins 90 Figure 2.1 |Protein recovery yield in function of precipitation methods, collection times, and confluence levels
measured by the Bradford assay 104
Figure 2.2 | One dimension SDS-PAGE gel of secreted proteins precipitated from fibroblast conditioned-medium
and conjugated with CyDyes 105
Figure 2.3 | Visualization of whole secreted protein and quantification of the protein spots resolved with 2D-DIGE
at the optimal induction condition 107
Figure 2.4 | Visualization and quantification of the protein spots of different tested induction conditions and
precipitation methods 108
Figure 3.1 | Human SOD1 protein schematic representation with highlighted misSOD1 epitope mapping regions 129 Figure 3.2 | Immunodetection of misSOD1 accumulation in spinal cord of A4V SOD1 FALS patient 130 Figure 3.3 | Detected misSOD1 immunostaining patterns MisSOD1 immunostaining patterns detected in SALS
xviii
Figure 3.4 | Detection of misSOD1-positive ring-like structure in the SALS and control individuals 136 Figure 3.5 | Immunodetection of misfolded SOD1 aggregates detected in brain tissue sections of SALS patients by immunohistochimestry using the misfolded SOD1/conformational-specific 4–20Ra, 24–39Ra, 57–72Ra,
C4F6 and 131–153Ra antibodies 138
Figure 3.6 | Immunocapture of misSOD1 in SALS spinal cord tissue samples using the C4F6 and 24–39Ra
misSDO1 specific antibodies 140
Figure 3.7 | Immunodetection of misfolded SOD1 species detected in post-mortem tissue sections from SALS patients by immunohistochemistry using the misfolded SOD1-specific 4-20Ra antibody 145
Figure 3.8 | Immunodetection of misfolded SOD1 species detected in post-mortem tissue sections from SALS patients by immunohistochemistry using the misfolded SOD1-specific 24-39Ra antibody 146 Figure 3.9 | Immunodetection of misfolded SOD1 species detected in post-mortem tissue sections from SALS patients by immunohistochemistry using the misfolded SOD1-specific 57-72Ra antibody 147
Figure 3.10 | Immunodetection of misfolded SOD1 species detected in post-mortem tissue sections from SALS patients by immunohistochemistry using the SOD1 conformation specific C4F6 antibody 148 Figure 3.11 | Immunodetection of misfolded SOD1 species detected in post-mortem tissue sections from SALS patients by immunohistochemistry using the misfolded SOD1-specific 131-153Ra antibody 149 Figure 3.12 | Hematoxylin & Eosin stainings of corpora amylacea in sporadic ALS patients spinal cord sections 150 Figure 4.1 | Skin fibroblast RNA sequencing shows exosomes-associated gene expression is favorized in
3D-cultures cells 182
Figure 4.2 | Skin fibroblast cultured in a 3D fashion show an enhanced exosomes secretion 183 Figure 4.3 | Skin fibroblast secretome analysis shows ECM-related protein secretion is enhanced in a 3D culture
model 184
Figure 4.4 | 3D-cultured skin fibroblasts derived exosomes have the capacity to enter fibroblast cells and are
processed by the proteasome 185
Figure 4.5 | Skin fibroblasts exosomes contain active MMPs and enhance collagen digestion 186 Figure 4.6 | 3D-derived exosomes from skin fibroblasts enhance migration and proliferation 187 Figure 4.7 | 3D-derived exosomes from skin fibroblasts enhance wound healing in a tissue-engineered skin model and show higher concentration of internal KGF when derived from a 3D-culture 188 Figure 4.8 | ECM-related proteins are more present in 3D-derived skin fibroblasts exosomes 190 Figure 4.9 | Heatmap of the 75 most varying genes after RNA-sequencing analysis 192
xix
Liste des tables
Tableau 1 | Principaux gènes mutés et identifiés chez les patients atteints de SLAF 3
Tableau 2 | Gènes associés à la SLA sporadique 33
Tableau 3 | Possibles biomarqueurs de la SLA découverts dans la peau 47 Tableau 1.1 | Data information on ALS patients and controls recruited in the study 67
Tableau 1.2 | Revised El Escorial criteria for diagnosing ALS 85
Tableau 1.3 | Abnormal structural skin features detected in ALS-tissue engineered skin 87 Tableau 2.1 | Detailed clinical information of patients and controls enrolled in this study 97 Tableau 2.2 | Detailed protocol for gel and electrophoresis experiments 102 Tableau 2.3 | Detailed migration protocol for the 2D gel electrophoresis 103 Tableau 2.4 | Available 2D gel analysis software and spot quantification 111 Tableau 3.1 | Summary of the studied ALS patients and control individuals 131 Tableau 3.2 | Immunohistochemistry conditions using the Ventana BenchMark ULTRA 137 Tableau 3.3 | Studies describing the presence or absence of misSOD1 in human SALS post-mortem tissues
141
Tableau 3.4 | Summary of the proposed guidelines 141
Tableau 3.5 | Summary of misSOD1 accumulation in human patient spinal cords by following standardized
immunohistochemistry 151
Tableau 3.6 | Detailed description of the proposed guidelines 152
Tableau 4.1 | Data information on patients recruited in this study 189
Tableau 4.2 | Human MMP antibody array map 191
xx
Remerciements
Je tiens premièrement à remercier le Dr François Gros-Louis, mon directeur de recherche, pour son aide et sa disponibilité, ainsi que le Dr François Berthod, mon co-directeur de recherche, pour ses bons conseils ainsi que sa grande disponibilité.
Merci à notre équipe de recherche, plus particulièrement à Lydia Touzel Deschênes, Audrey Labarre, Christian Martel, Rémy Lamontagne, Aurélie Louit, Thiéry De Serres-Bérard, Marie-Josée Beaudet et Sabrina Bellenfant. Que ce soit pour votre aide, vos bons conseils ou simplement pour votre bonne humeur, vous aurez fait de mon passage au laboratoire un moment agréable.
Un merci tout spécial au Dr Nicolas Dupré pour ses innombrables projets et son positivisme constant. J’espère avoir l’occasion de collaborer avec vous dans les années à venir.
Papa, maman et Mérédith, un énorme merci pour votre support constant dans mes études. Vous avez toujours été là dans les moments difficiles et cette thèse, je vous la dois en partie. Merci Anna pour tes encouragements, ta bonne humeur et ton appui constant dans les derniers moments de mes études doctorales. Je ne pourrai jamais assez vous remercier.
Merci à Cassandra R. Goulet pour ton aide dans les derniers moments de mon doctorat. Dr. Goulet, j’en serai toujours reconnaissant. Un énorme merci à mes amies, Cassandra et Isabelle Lorthois, avec qui j’ai eu un plaisir fou.
Un merci tout special à Marc-André Rhéaume pour tes excellents commentaires concernant le français de ma thèse. Ton travail est très apprécié!
I would also like to thank Dr. David Taylor and ALS Canada for their great help during my graduate studies and for the PhD Training Award they granted me.
xxi
Avant-propos
Manuscrits publiés à titre de premier auteur faisant partie intégrante de cette thèse
Chapitre 1 : Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients
Bastien Paré1,2, Lydia Touzel-Deschênes2, Rémy Lamontagne2, Marie-Soleil Lamarre2, François-Dominique Scott1,2, Hélène T Khuong1,3, Patrick A Dion4, Jean-Pierre Bouchard3, Peter Gould5, Guy A Rouleau4, Nicolas Dupré3, François Berthod1,2 and François Gros-Louis1,2*
1. Department of Surgery, Faculty of Medicine, Laval University, Québec, Canada.
2. CHU de Québec Research Center, LOEX-Hôpital de l’Enfant-Jésus, 1401, 18e rue, Quebec G1J 1Z4, Canada.
3. ALS Clinic, Department of Neurological Sciences, CHU de Québec and the Faculty of Medicine, Laval University, Québec, Canada.
4. Montreal Neurological Institute and Hospital, Department of Neurology and Neurosurgery, McGill University, Montréal, Canada.
5. Department of Medical Biology, Division of Anatomic Pathology and Neuropathology, CHU de Québec, Hôpital de l’Enfant-Jésus, Québec, Canada.
*Correspondance : francois.gros-louis@fmed.ulaval.ca
Acta Neuropathol Commun. 2015 Jan 31;3:5. doi: 10.1186/s40478-014-0181-z
Cet article scientifique a été publié le 31 janvier 2015 dans le journal Acta Neuropathologica Communications. Celui-ci traite du développement d’un modèle de peau reconstruite en laboratoire par génie tissulaire dans le but d’étudier la Sclérose Latérale Amyotrophique. Cet article est directement associé à la première problématique de recherche présentée et les objectifs qui y sont associés.
Cet article a été réalisé sous la supervision du Dr François Gros-Louis. Je suis nommé premier auteur puisque j’ai réalisé la culture cellulaire associée au développement et la mise au point du modèle de peau pour l’étude de la SLA, l’analyse et l’interprétation des colorations histologiques, les immunofluorescences ainsi que les immunohistochimies et immunobuvardages de type Western associés à la caractérisation de la délocalisation de la protéine TDP-43 dans ce nouveau modèle. Lydia Touzel Deschênes, Rémy Lamontagne, Marie-Soleil
xxii
Lamarre et François-Dominique Scott ont aidé dans la réalisation des manipulations à titre d’assistants de recherche, étudiants ou stagiaires. Hélène T. Khuong, Patrick A Dion, Jean-Pierre Bouchard, Peter Gould, Guy A Rouleau et Nicolas Dupré ont agis en tant que cliniciens et collecté les échantillons biologiques. François Berthod et François Gros-Louis ont agi en tant que superviseurs principaux dans cette partie de mon projet de thèse et élaboré le design expérimental. François Gros-Louis détenait la principale ressource financière associée à ce projet.
Chapitre 2 : An optimized approach to recover secreted proteins from fibroblast conditioned-media for secretomic analysis
Bastien Paré1, Lydia Touzel Deschênes1, Roxane Pouliot1,4, Nicolas Dupré3 and Francois Gros-Louis1, 2 1. Laval University Experimental Organogenesis Research Center / LOEX, Division of Regenerative Medicine, CHU de Quebec Research Center – Enfant-Jésus Hospital, Quebec, Canada
2. Department of Surgery, Faculty of Medicine, Laval University, Québec, Canada
3. Neuroscience Division of the CHU de Québec and Department of Medicine of the Faculty of Medicine, Laval University, Québec, Canada
4. Faculty of Pharmacy, Laval University, Québec, Canada
Front Cell Neurosci. 2016 Mar 31;10:70. doi: 10.3389/fncel.2016.00070. eCollection 2016.
Cet article scientifique a été publié le 31 mars 2016 dans le journal Frontiers in Cellular Neuroscience. Celui-ci traite de l’optimisation de la culture de fibroblastes ainsi que de la récupération des protéines sécrétées par ces cellules, le sécrétome. Cet article est directement relié aux objectifs de cette thèse associés à la découverte de nouveaux biomarqueurs associés à la SLA.
Cet article a été réalisé sous la supervision du Dr François Gros-Louis. Je suis nommé premier auteur puisque j’ai réalisé la culture cellulaire associée à ce papier, la récupération du surnageant de culture, la précipitation du sécrétome ainsi que les analyses sur gel 1D et 2D. J’ai de plus réalisé l’analyse et l’interprétation des résultats. Lydia Touzel Deschênes et Roxane Pouliot ont aidé dans la réalisation des manipulations à titre d’assistants de recherche et de chercheure. Nicolas Dupré a agi en tant que cliniciens et collecté les échantillons biologiques. François Gros-Louis a agi en tant que superviseur principaal dans cette partie de mon projet de thèse et aidé à
xxiii
l’élaboration du design expérimental. François Gros-Louis détenait la principale ressource financière associée à ce projet.
Chapitre 3 : An optimized approach to recover secreted proteins from fibroblast conditioned-media for secretomic analysis
Bastien Paré1,2, Manuela Lehmann3, Marie Beaudin4, Ulrika Nordström3, Stephan Saikali5, Jean-Pierre Julien6,7, Jonathan D. Gilthorpe3, Stefan L. Marklund8, Neil R Cashman9, Peter M. Andersen3, Karin Forsberg10, Nicolas Dupré4, Peter Gould5, Thomas Brännström10 and François Gros-Louis 1, 2
1. Laval University Experimental Organogenesis Research Center / LOEX, Division of Regenerative Medicine, CHU de Québec Research Center – Enfant-Jésus Hospital, Québec, Canada
2. Department of Surgery, Faculty of Medicine, Laval University, Québec, Canada
3. Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden
4. Neuroscience Division of the CHU de Québec and Department of Medicine of the Faculty of Medicine, Laval University, Québec, QC, Canada
5. Department of Medical Biology, Division of Anatomic Pathology and Neuropathology, CHU de Québec, Hôpital de l'Enfant-Jésus, Québec, Canada
6. Department of Psychiatry and Neuroscience, Laval University, Québec City, Québec, Canada 7. Centre de Recherche CERVO, Québec City, Québec, Canada
8. Department of Medical Biosciences, Clinical Chemistry, Umeå University, Umeå, Sweden
9. Department of Medicine (Neurology), Brain Research Center, University of British Columbia, Vancouver, BC, Canada
10. Department of Medical Biosciences, Pathology, Umeå University, Umeå, Sweden
Sci Rep. 2018 Sep 21;8(1):14223. doi: 10.1038/s41598-018-31773-z.
Cet article scientifique a été publié le 21 septembre 2018 dans le journal Scientific Reports. Celui-ci traite de l’automatisation et l’amélioration de la technique d’immunohistochimie pour l’analyse de SOD1 et SOD1 mal-repliée dans des coupes de cerveau et de moelle épinière humaines dans l’étude de la SLA sporadique. Cet
xxiv
article est directement associé à l’objectif de recherche de cette thèse qui est la découverte de biomarqueurs associés à la SLA, dans ce cas-ci la SLAS.
Cet article a été réalisé sous la supervision des Dr François Gros-Louis, Dr Thomas Brännström et Dr Peter Andersen. Je suis premier auteur puisque j’ai réalisé les colorations immunohistochimiques, la prise de photos ainsi que l’analyse des colorations obtenues. Manuela Lehmann et Ulrika Nordström ont aidé à la réalisation des immunoprécipitations et immunobuvardages de type Western. Stephan Saikali, Peter V. Gould et Thomas Brännström ont aidé à la préparation des échantillons pathologique et l’interprétation des résultats. Jonathan D. Gilthorpe, Stefan L. Marklund, Neil R. Cashman, Karin Forsberg et Nicolas Dupré ont aidé à l’écriture de la publication et à l’interprétation de certains résultats. François Gros-Louis, Thomas Brännström et Peter Andersen ont agi en tant que superviseurs principaux de cet aspect du project de recherche. François Gros-Louis et Peter Andersen détenaient la principale ressource financière associée à ce projet.
Annexe : Potential skin involvement in ALS: revisiting Charcot’s observation – a review of skin abnormalities in ALS
Auteurs:
Bastien Paré et François Gros-Louis
Contribution des auteurs :
J’ai rédigé l’ensemble de la première version du manuscrit et collaboré à la version finale. François Gros-Louis a revu et corrigé le manuscrit.
Article publié dans :
Reviews in the Neurosciences 2017; 28(5): 551–572
xxv
Manuscrit à être soumis pour publication à titre de premier auteur faisant partie intégrante de cette thèse
Chapitre 4 : Dermal fibroblasts cultured in a 3D environment release exosomes promoting extracellular matrix remodelling
Bastien Paré (1,2), Cassandra R. Goulet (1,2), Thiéry De Serres-Bérard (1,2), Stéphane Bolduc (1,2), François
Berthod (1,2) and François Gros-Louis (1,2)*
1. Department of Surgery, Faculty of medicine, Laval University, Québec, QC, Canada
2. Division of Regenerative Medicine, Laval University Experimental Organogenesis Research Center/LOEX, CHU de Québec Research Center – Enfant-Jésus Hospital, Québec, QC, Canada
*To whom correspondence should be addressed
Cet article scientifique est sous sa forme finale pour une tentative de soumission au journal Nature Biotechnologies. Celui-ci traite de la différence d’expression génique et protéine de fibroblastes de peau entre la technique de culture 2D et 3D. Cet article est directement associé à l’objectif de recherche de cette thèse qui est la découverte de biomarqueurs associés à la SLA grâce aux fibroblastes de peau dérivés de patients atteints de la maladie. Les résultats de cet article scientifique pourront être utilisés comme base dans la découverte de nouveaux biomarqueurs de la SLA dérivés de cellules de peau de patients, grâce à l’analyse et la compréhension des mécanismes associés à la sécrétion d’exosomes en culture 3D, qui mimique plus fidèlement la peau native humaine.
Cet article a été réalisé sous la supervision des Dr François Gros-Louis et Dr François Berthod, mon co-directeur de recherche. Je suis co-premier auteur avec Cassandra R. Goulet puisque nous avons réalisé la grande majorité des expérimentations et interprétation des résultats. Thiéry De Serres-Bérard a grandement aidé à la réalisation des expérimentations de fermeture de plaies et l’analyse des résultats. Tous les auteurs de cet article scientifique ont participé de près ou de loin à la correction de l’article. Le séquençage nouvelle génération ainsi que les analyses subséquentes ont été réalisées par Génome Québec. La spectrométrie de masse et les analyses associées ont été réalisées par la plateforme de protéomique du CHU de Québec. François Gros-Louis et François Berthod détenaient la principale ressource financière associée à ce projet.
1
Introduction, problématiques de recherche, hypothèses
et objectifs
2
La sclérose latérale amyotrophique
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative hétérogène, incurable et sans traitement palliatif ou curatif efficace qui se caractérise principalement par une dégénérescence sélective des neurones moteurs de la moelle épinière et du cerveau. Apparaissant dans la cinquantaine, la SLA est principalement associée à la dégénérescence des neurones moteurs supérieurs et inférieurs, qui sont des acteurs primordiaux dans la communication entre les muscles et le système nerveux central. Cette maladie neurodégénérative se présente, pour la plupart des patients qui en sont atteints, par une faiblesse musculaire progressive, une hypertonie spastique, de la dysphagie, l’apparition de fasciculations, une paralysie presque totale et le décès de 3 à 5 ans après l’apparition des premiers symptômes, souvent par détresse respiratoire (Ilieva et al., 2009; Mulder et al., 1986; Robberecht and Philips, 2013; Tandan and Bradley, 1985). L’incidence et la prévalence de la SLA sont respectivement de 1-2 et de 4-6 pour 100 000 chaque année à travers le monde (Gros-Louis et al., 2006; Pasinelli and Brown, 2006). Dans la grande majorité des cas, la SLA se veut spinale, associée à l’apparition des premiers symptômes dans les bras et les jambes (Statland et al., 2015). Environ 30% des cas de SLA sont considérés bulbaires, caractérisés principalement par des problèmes de parole et de déglutition apparaissant tôt dans le développement de la maladie. Les problèmes de déglution chez les patients mènent généralement à une perte de poids importante ainsi qu’une cachexie (Hillel and Miller, 1989). De plus, la SLA de type bulbaire est généralement associée à une survie plus courte des patients, une qualité de vie réduite de façon importante (Chio et al., 2009; Daghlas et al., 2018; Hecht et al., 2002; Mitsumoto and Bene, 2009).
La SLA se présente sous deux formes distinctes : la SLA de type familial (SLAF) et la SLA de type sporadique (SLAS). Comme son nom l’indique, la SLAF est associée à des mutations génétiques précises et celles-ci sont transmises de façon autosomale dominante dans la très grande majorité des cas. Elle pourrait être transmise de façon récessive chez certains patients présentant une mutation dans les gènes ALS2 et SOD1 (D90A) (Hadano et al., 2001; Pasinelli and Brown, 2006). Près de 5 % de tous les cas de SLA sont associés à des mutations génétiques et plus de 50 % des cas de SLAF peuvent être attribués à des mutations dans les gènes SOD1, TARDBP, FUS et C9ORF72 (Chen et al., 2013; Gros-Louis et al., 2006; Leblond et al., 2014). Quant à la SLAS, elle se distingue par son côté sporadique et représente de 90 à 95 % de tous les cas de SLA (Gros-Louis et al., 2006; Leblond et al., 2014).
3
La SLA familiale
La SLA familiale représente environ 10 % de tous les cas de SLA. La recherche sur la SLAF telle qu’on la connait aujourd’hui a pris son envol lors de la découverte de premières mutations dans le gène SOD1 en 1993 (Rosen et al., 1993) confirmé en 1994 par Gurney et ses collaborateurs (Gurney et al., 1994a). Dans les dernières années, des mutations dans d’autres gènes ont été associées au développement de la maladie. Les plus importants sont TARDBP, FUS/TLS et C9ORF72, mais près de 50 gènes sont présentement associés à la SLAF (Ghasemi and H. Brown Jr, 2017). Une liste des gènes associés à la SLAF en date de mars 2017 est présentée ci-dessous (tableau 1) (Ghasemi and H. Brown Jr., 2017). Seulement les gènes les plus importants et étudiés dans les chapitres subséquents sont décrits et abordés.
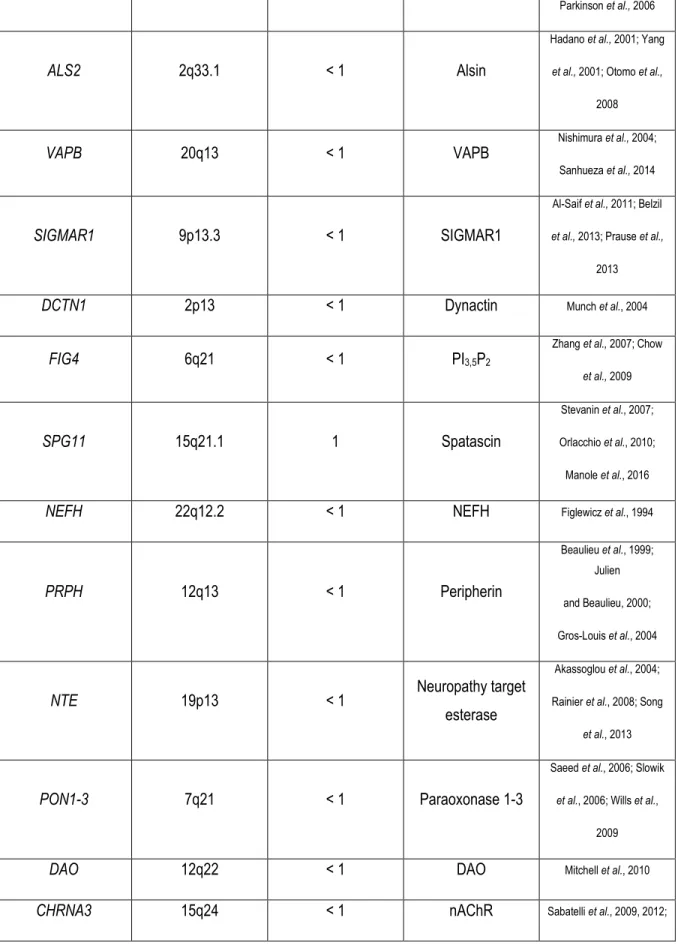
Tableau 1 Principaux gènes mutés et identifiés chez les patients atteints de SLAF
Gène Locus Portion de SLAF
(%) Protéine encodée Références
C9ORF72 9p21.3 40-50 C9ORF72 DeJesus-Hernandez et al., 2011; Renton et al., 2011; Cooper-Knock et al., 2012; Diekstra et al., 2014 SOD1 21q22 20-25 SOD1 Rosen et al., 1993; Watanabe et al., 2001; Forsberg et al., 2011 TARDBP 1p36.2 4-5 TDP-43 Rutherford et al., 2008; Sreedharan et al., 2008; Neumann, 2009 FUS 16p11.2 4-5 FUS Kwiatkowski et al., 2009; Vance et al., 2009; Huang
et al., 2010
OPTN 10p13 2-3 Optineurin Maruyama et al., 2010
4
VCP 9p13 1-2 VCP/p97
Forman et al., 2006; Kimonis et al., 2008; Johnson et al., 2010
ANG 14q11.2 1-2 Angiogenin Greenway et al., 2006;
Thiyagarajan et al., 2012
TUBA4A 2q35 1 Tubulin 4A Smith et al., 2014
UBQLN2 Xp11 < 1 Ubiquilin 2 Deng et al., 2011; Ugwu
et al., 2014
TAF15 17q11 < 1 TAF15 Couthouis et al., 2011
EWSR1 22q12.2 < 1 EWSR1 Couthouis et al., 2012
hnRNPA1 12q13 < 1 hnRNPA1
Bekenstein and Soreq, 2013; Kim et al., 2013; Le Ber
et al., 2014
hnRNPA2B1 7p15 < 1 hnRNPA2/B1 Kim et al., 2013; Le Ber et
al., 2014
SETX 9q34.13 < 1 Senataxin Chen et al., 2004
CREST 20q13.3 < 1 SS18L1 Chesi et al., 2013; Teyssou
et al., 2010
MATR3 5q31.2 < 1 Matrin 3 Johnson et al., 2014b;
Millecamps et al., 2014
ATXN2 12q24 1-2 Ataxin 2 Elden et al., 2010; Baumer
et al., 2014
ELP3 8p21.1 < 1 ELP3 Simpson et al., 2009; Fujita
et al., 2014
SQSTM1 5q35 < 1 P62/SQSTM1
Gal et al., 2009; Fecto et
al., 2011; Rubino et al.,
2012
5
Parkinson et al., 2006
ALS2 2q33.1 < 1 Alsin
Hadano et al., 2001; Yang
et al., 2001; Otomo et al.,
2008
VAPB 20q13 < 1 VAPB Nishimura et al., 2004;
Sanhueza et al., 2014
SIGMAR1 9p13.3 < 1 SIGMAR1
Al-Saif et al., 2011; Belzil
et al., 2013; Prause et al.,
2013
DCTN1 2p13 < 1 Dynactin Munch et al., 2004
FIG4 6q21 < 1 PI3,5P2 Zhang et al., 2007; Chow
et al., 2009
SPG11 15q21.1 1 Spatascin
Stevanin et al., 2007; Orlacchio et al., 2010; Manole et al., 2016
NEFH 22q12.2 < 1 NEFH Figlewicz et al., 1994
PRPH 12q13 < 1 Peripherin
Beaulieu et al., 1999; Julien and Beaulieu, 2000; Gros-Louis et al., 2004
NTE 19p13 < 1 Neuropathy target
esterase
Akassoglou et al., 2004; Rainier et al., 2008; Song
et al., 2013
PON1-3 7q21 < 1 Paraoxonase 1-3
Saeed et al., 2006; Slowik
et al., 2006; Wills et al.,
2009
DAO 12q22 < 1 DAO Mitchell et al., 2010
6
CHRNA4 20q13 < 1 Moriconi et al., 2011
CHRNB4 15q24 < 1 ERB4 2q34 < 1 Receptor tyrosineprotein kinase ErbB-4 Takahashi et al., 2013 CHCHD10 22q11 < 1 Mitochondrial protein Bannwarth et al., 2014; Chaussenot et al., 2014;
Johnson et al., 2014a
C19orf12 9q12 < 1 Mitochondrial
protein Deschauer et al., 2012
ALS3 18q21 < 1 Disulfide redox
protein Hand et al., 2002
ALS7 20p13 < 1 Inconnu Hand et al., 2002
ALS6-21 6p25, 21q22 < 1 Inconnu Butterfield et al., 2009
ALS-FTD 16p12 < 1 Inconnu Dobson-Stone et al., 2013
NEK1 4q33 Non applicable NIMA-related
kinase 1 P. Kenna et al., 2016
TBK1 12q14.2 Non applicable TANK Binding
Kinase 1
A. Freischmidt et al., 2015; E. T. Cirulli et al., 2015
Tableau tiré, adapté et modifié de Ghasemi and H. Brown Jr., 2017.
Superoxide dismutase 1 (SOD1)
La superoxyde dismutase cuivre/zinc, ou SOD1, fait partie de la famille des superoxydes dismutases (SOD) et est une enzyme dont le rôle principal est d’être une protéine antioxydante. Cette famille comporte trois protéines : la SOD cuivre/zinc cytoplasmique (SOD1), la SOD-manganèse, retrouvée dans les mitochondries (SOD2), ainsi que la SOD cuivre/zinc extracellulaire (SOD3). La SOD1 est une enzyme capable de catalyser la
7
dismutation des superoxydes en peroxyde (O2- → H2O2) [2 superoxydes + 2 H+ = O2 + H2O2] (Valentine et al., 2005). La protéine SOD1 est un homodimère présentant un ion cuivre et un ion zinc pour chaque monomère. Plus de 150 mutations ont été découvertes dans le gène SOD1 et elles représentent entre 12 et 15 % des cas de SLAF et environ 1 % des cas de SLAS (Renton et al., 2014). Le gène SOD1 code la protéine du même nom. Ce gène est situé sur le chromosome 21q22.1 et comprend 5 exons. La protéine codée comporte 153 acides aminés et possède un poids moléculaire de 16 kilodaltons (kDa). Cette protéine se présente sous forme d’homodimère ayant un cœur cuivre-zinc.
À la différence de la SLAS, la SLAF associée au gène SOD1 présente un phénotype clinique distinct. On lui associe entre autres un âge d’apparition des premiers symptômes plus précoce que la SLAS ainsi qu’une durée plus longue de la maladie à l’exception de certaines mutations comme la A4V, dont l’évolution se veut très rapide, généralement en moins de 1 an (Corcia et al., 2012).
C’est en 1993 que la première identification de mutations du gène SOD1 a été publiée (Rosen et al., 1993). Cette découverte représentait en fait la première cause reconnue de SLAF et a permis de paver la voie à la découverte de plusieurs aspects physiopathologiques et génétiques de la SLA. Parmi les mutations les plus étudiées, on trouve D90A, A4V, G37R, G93A et G93C, qui sont toutes des mutations non-sens.
Physiopathologie de la SOD1 dans la SLAF
La protéine SOD1 est principalement associée au stress oxydatif puisqu’elle fait partie de la grande famille des antioxydants. Sur le plan cellulaire, le stress oxydatif représente un problème dans la régulation des radicaux libres et des espèces d’oxygène réactives par les molécules et enzymes antioxydantes. Parmi les espèces réactives, on trouve les « reactive oxygen species » ou ROS. La SOD1 est capable d’interagir directement avec les ROS, ce qui permet à la cellule de limiter le stress oxydatif. La structure de la protéine joue un rôle important dans l’efficacité de son activité enzymatique puisque son cœur cuivre-zinc se trouve à être le site actif de la protéine (Perry et al., 2010; Trumbull and Beckman, 2009). Le cuivre présent dans le cœur cuivre-zinc a la capacité de passer du Cu2+ au Cu+ (cuivre) afin d’obtenir une activité enzymatique optimale (Trumbull and Beckman, 2009). Bien que le zinc ne participe pas aux réactions enzymatiques, il est important dans la stabilité du site actif et de la protéine (Trumbull and Beckman, 2009). Les mutations dans le gène SOD1 vont avoir des répercussions importantes tant dans les fonctions enzymatiques de la protéine que sur sa structure, ce qui peut mener à son mauvais repliement ainsi qu’à un changement majeur dans son activité enzymatique
8
(gain ou perte de fonction) (Deng et al., 2006; Ip et al., 2011; Saccon et al., 2013). Plusieurs études chez la souris ont permis de démontrer un gain de fonction toxique associé à certaines mutations, dont certaines (G37R, par exemple) conférant une activité enzymatique augmentée (Julien and Kriz, 2006; Kim et al., 2016; Philips and Rothstein, 2016, 2015). Plusieurs théories ont été émises quant à la toxicité de la SOD1 dans la SLAF. Certaines études soutiennent entre autres la théorie selon laquelle la protéine mutée présenterait une activité enzymatique aberrante (Beckman et al., 1993; Bowling et al., 1993; Hayward et al., 2002; Sau et al., 2007); d’autres soutiennent que la protéine pourrait avoir des propriétés caractéristiques d’un prion (Chattopadhyay et
al., 2008; Münch et al., 2011; Münch and Bertolotti, 2011, 2010) ou qu’elle pourrait induire un dysfonctionnement
mitochondrial (Carrì and Cozzolino, 2011; Mattiazzi et al., 2002; Tafuri et al., 2015; Tan et al., 2014). Son rôle principal dans le traitement des ROS sur le plan cellulaire permet aussi de penser qu’une mutation pourrait avoir un impact important dans le stress oxydatif et associé au réticulum endoplasmique (Chen et al., 2015; Labarre
et al., 2013; Nishitoh et al., 2008; Urushitani et al., 2008). Malgré toutes les théories avancées et scientifiquement
validées, il est encore difficile aujourd’hui de déterminer le phénomène principal ou unique qui caractérise la toxicité de la SOD1 mal repliée et des mutations du gène SOD1. Les pistes de solution principales proviennent du modèle murin de SOD1, longtemps utilisé afin d’étudier les phénomènes physiopathologiques de la SLA et de l’association de la protéine au développement de la maladie. Les modèles murins ont entre autres permis de déterminer que la grande majorité des mutations associées à la SLAF dans ce gène n’inhibent pas l’activité enzymatique de la protéine (Borchelt et al., 1994; Sau et al., 2007). Contrairement à différents modèles murins portant une mutation du gène SOD1 et qui présentent un phénotype moteur semblable à la SLA chez l’humain (Bruijn et al., 1997; Gurney et al., 1994a; Julien and Kriz, 2006; Wong et al., 1995), le knock-out (K.O.) du gène chez la souris ne mène pas au développement de ces mêmes problèmes moteurs (Reaume et al., 1996). Il a par contre été démontré qu’une accumulation importante de SOD1 mal repliée est présente et détectable dès le jour 7 de vie dans les neurones moteurs facilement fatigables (« fast fatigable » (FF)) chez les souris SOD1-G93A (Saxena et al., Neuron 2013). Originalement, il a été avancé comme théorie que le développement de la SLA associée à la SOD1 était dû à une perte de fonction dismutase de la protéine. Cette théorie a par contre rapidement été démentie puisque des souris n’exprimant pas du tout le gène SOD1 n’ont pas développé la maladie après 6 mois de vie (Reaume et al., 1996). Cette étude est aussi appuyée par le fait que ces souris, bien que présentant certaines axonopathies, ne présentent aucune perte neuronale (Shefner et al., 1999). Différentes études ont en effet pu démontrer que la SOD1 de type sauvage n’est pas impliquée dans le développement de la maladie puisque son expression chez la souris ne semble pas causer de perte de neurones moteurs (Shefner et al., 1999). À contresens, l’expression de la SOD1 de type sauvage à des niveaux semblables à la SOD1G93A mène au développement de phénotypes neuropathologique et moteur semblables à la SLA (K. S. Graffmo et al., 2013).
9
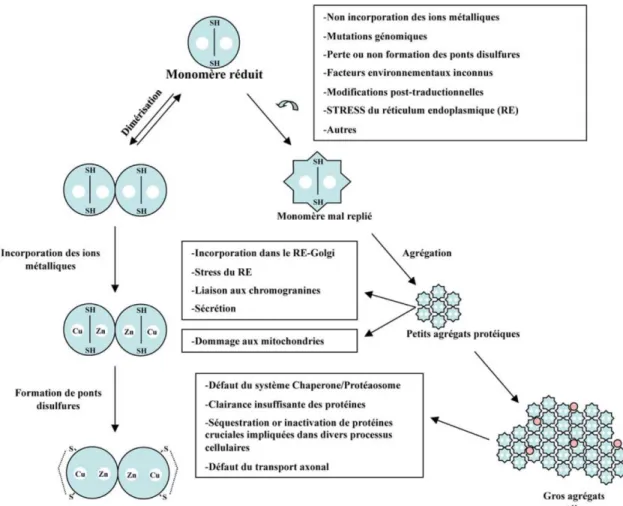
Le mauvais repliement de la protéine SOD1 peut être observé en présence d’une des mutations associées à la SLAF. Ce phénomène, bien qu’aléatoire, peut être expliqué par différents facteurs. Ses conséquences sont présentées en figure 1.
Figure 1 : Toxicité associée au mauvais repliement de la protéine SOD1
Dans des conditions normales, le monomère réduit (non métallé) de la SOD1 se dimérise avec lui-même avant que les ions métalliques du cuivre (Cu) et du zinc (Zn) puissent s’incorporer au dimère. La formation de ponts disulfures intramoléculaires permet ensuite la stabilisation de l’enzyme. Le mauvais repliement de la protéine SOD1 par divers facteurs qui favorisent la conversion des monomères réduits vers sa forme mal repliée mène à la formation de petits et de gros agrégats protéiques avec des propriétés toxiques. Figure tirée et adaptée
de Labarre, Paré et al., 2013.
De façon intéressante, il a dernièrement été démontré que la protéine mal repliée a la capacité de voyager d’une cellule à l’autre grâce à divers mécanismes, dont les vésicules extracellulaires, qui seront abordés
10
plus longuement dans une autre section (Hanspal et al., 2017; Silverman et al., 2016). De plus, certaines espèces de SOD1 mal repliée présenteraient de réelles caractéristiques prioniques, dont l’accélération du développement de la SLA chez des souris transgéniques injectées avec d’infimes quantités de celle-ci (Ekhtiari Bidhendi et al., 2016; Jonsson et al., 2004). Ces différentes observations permettent de penser que la forme mal repliée de la SOD1 pourrait être à la base de la toxicité qui lui est associée, sans pour autant permettre d’identifier le phénomène biologique affecté par les mutations associées à la SLAF, qui reste à être élucidé.
La SLA est une maladie neurodégénérative dont une des principales caractéristiques est qu’elle atteint de façon importante les neurones moteurs du système nerveux central. Lorsque l’on parle de la SLA, les principaux neurones moteurs atteints et qui présentent une dégénération sont les alphas (Conradi and Ronnevi, 1993; Frey et al., 2000). Les neurones moteurs sont communément divisés en trois classes principales : les neurones moteurs alpha (), bêta () et gamma () (Manuel and Zytnicki, 2011).
Depuis quelques années déjà, l’aspect propre aux neurones moteurs de la SLA a été largement étudié et plusieurs autres types cellulaires ont été associés à la SLAF (SOD1) ainsi qu’à la pathologie de la maladie. Lorsque l’on parle du système nerveux central, de plus en plus d’études ont démontré que les astrocytes, les microglies, les cellules gliales et même les cellules de Schwann pourraient avoir une implication plus ou moins importante dans le développement de la maladie (pour revue, voir Ilieva et al., 2009). Une dégénérescence axonale des neurones moteurs ne portant pas de mutation associée à la SLAF peut être observée chez la souris dont les cellules gliales sont porteuses d’une mutation dans le gène SOD1 (M. Clement et al., 2003). Des milieux conditionnés dérivés d’astrocytes et de neurones moteurs vont de plus permettre d’obtenir le même type de phénomène in vitro (Haidet-phillips et al., 2012; Paolo Di Giorgio et al., 2013; Papadeas et al., 2011; Rajan et
al., 2011). La figure 2 présente les différents types cellulaires associés au développement de la SLA ou à sa
11
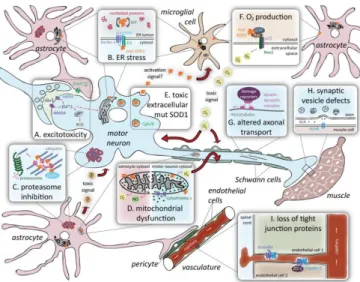
Figure 2 : Contribution de plusieurs types cellulaires dans le développement de la SLA
Bien que les neurones moteurs semblent démontrer un phénotype associé à la maladie ou à une dégénérescence, d’autres types cellulaires associés au système nerveux et même hors de celui-ci doivent être pris en considération afin de bien comprendre la pathologie de cette maladie neurodégénérative, dont les cellules musculaires, vasculaires, les astrocytes et les cellules microgliales. La pathologie est associée à différents processus biologiques distincts, dont excitotoxicité, le stress du réticulum endoplasmique, l’inhibition du protéasome, un dysfontionnement des mictochondries, la SOD1 extracullaire et sa toxicité, la production d’O2, des altérations du transport axonal, des vésicules synaptiques abérrantes ainsi que la perte de protéines de jonctions serrées. Figure tirée de Ilieva et al., 2009.
Transactive response DNA binding protein 43 kDa (TARDBP/TDP-43)
La protéine TDP-43 est encodée par le gène TARDBP et son rôle premier dans la cellule est de lier l’ADN et l’ARN (« DNA-RNA binding protein ») (Ou et al., 1995). Exprimée de façon ubiquitaire, TDP-43 se localise prioritairement dans le noyau des cellules, mais a la capacité de se déplacer du noyau vers le cytoplasme (Winton et al., 2008). Découverte en 1995, cette protéine a été démontrée comme étant un agent de modulation de l’expression génique du VIH-1 (Ou et al., 1995). Lorsque présente dans le noyau des cellules, la protéine joue un rôle important dans l’épissage de l’ARN et la synthèse de micro-ARN (Gregory et al., 2004; Xu, 2012). De plus, TDP-43 joue un rôle important dans le transport de granules d’ARN dans les neurones (Alami et al., 2014). Les mutations de TARDBP représentent environ 4 % de tous les cas de SLAF. Tout comme pour le gène
SOD1, la grande majorité des 14 mutations de TARDBP associées à la SLAF sont des mutations non-sens et
12
comme rôle principal la liaison aux protéines en raison de sa forte concentration en glycines (Mackenzie and Rademakers, 2008; Williams et al., 1988; Yang et al., 2010). Des inclusions et des accumulations cytoplasmiques de la protéine au cytoplasme des neurones moteurs sont actuellement considérées comme un élément de diagnostic neuropathologique de la SLA, et ce, tant chez les patients atteints de SLAS que ceux atteints de SLAF, avec ou sans mutations dans le gène TARDBP (Neumann et al., 2006). De façon exceptionnelle, cette délocalisation n’est observée que chez un très petit nombre de patients présentant une mutation du gène SOD1 (Jeon et al., 2018; Mackenzie et al., 2007; Shan et al., 2009). C’est cette caractéristique de la protéine TDP-43, analysée sur des tissus post-mortem de patients atteints de la SLA, qui fait de TDP-43 le seul biomarqueur de la maladie. Malheureusement, sa détection à la suite du décès des patients n’en fait pas un outil de diagnostic prédictif ou de suivi lors des études cliniques d’intérêt. L’agrégation de cette protéine se voudrait transitoire entre sa forme régulière et sa forme agrégée. Ce phénomène pourrait être associé à l’évolution de la maladie puisque reconnu comme étant plus sévère en situation de stress cellulaire et en présence de granules de stress (Boeynaems et al., 2018; Boeynaems and Gitler, 2018; Johnson et al., 2009; Wang et al., 2018).
Physiopathologie de TDP-43 dans la SLAF
La principale association de TDP-43 et de la SLA se veut être sa délocalisation du noyau au cytoplasme et la formation d’agrégats cytoplasmiques ubiquitinés, détectables chez tout près de 97 % des patients atteints de SLA à la suite de l’autopsie (Mackenzie et al., 2007; Maekawa et al., 2009; Neumann et al., 2006). Les agrégats cytoplasmiques de TDP-43 sont formés de façon générale de la protéine complète, de différentes formes tronquées de celle-ci ainsi que des formes ubiquitinées et phosphorylées (Neumann et al., 2006). Il est encore impossible de déterminer avec certitude si ces agrégats cytoplasmiques sont une cause associée à la pathologie de la SLA ou une conséquence. Bien que les agrégats soient considérés comme une des caractéristiques principales de la maladie sur le plan neuropathologique, leur rôle reste à être élucidé. La formation d’agrégats de TDP-43 au cytoplasme des cellules est toxique, sans pour autant être associée à la protéine tronquée ou native (Barmada et al., 2010; Igaz et al., 2011).
La surexpression de la protéine de type sauvage TDP-43 mène à l’agrégation de celle-ci au cytoplasme des cellules chez la souris, la mouche Drosophila melanogaster et le nématode Caenorhabditis elegans (Ash et
al., 2010; Li et al., 2010; Wils et al., 2010). Par contre, ces études n’ont pas pu démontrer hors de tout doute
que cette surexpression mène au développement de déficits moteurs semblables à ce qui est observé chez les patients sur le plan clinique, puisque les souris présentant le gène humain muté présentent un phénotype
13
semblable à la démence fronto-temporale et non à la SLA, alors que chez le nématode et la mouche, un phénotype moteur est présent (Igaz et al., 2011; Janssens et al., 2013; Li et al., 2010; Vaccaro et al., 2012a). À l’inverse, des délétions du gène TARDBP, qu’elles soient partielles ou complètes, vont mener au développement de phénotypes moteurs semblables à ce qui est observé chez les patients atteints de SLA (Iguchi et al., 2013a; Kabashi et al., 2010; Wu et al., 2012). Différentes modifications génétiques, comme des délétions ou des mutations au gène, lorsque appliquées à des cellules gliales ou à des cellules musculaires, mèneront aussi au développement d’un phénotype moteur caractéristique de la SLA (Diaper et al., 2013a; Yang et al., 2014). Une dérégulation de la protéine TDP-43 pourrait de plus mener à une pathologie associée au Nuclear Factor B et à sa sous-unité p65, qui seraient des partenaires de liaison (Swarup et al., 2011b). Le modèle pathologique général de la protéine TDP-43 dans la SLA est présenté à la figure 3. La protéine TDP-43 se veut un biomarqueur important de la SLA post-mortem. Par contre, cette protéine est difficilement analysable à titre de biomarqueur de diagnostic ou prédictif avec les connaissances actuelles sur la toxicité de sa délocalisation et des agrégats qui lui sont associés (Valle and Carrì, 2016). Une surexpression et une sous-expression de TDP-43 vont mener à une perte de neurones moteurs, ce qui met sur la table deux théories différentes associées à sa pathologie : un gain ainsi qu’une perte de fonction de la protéine. La présence d’agrégats cytoplasmiques de la protéine pourrait mener à un gain de fonction toxique, par exemple en séquestrant des protéines et des ARN importants à la survie cellulaire. La toxicité de TDP-43 semble être directement et étroitement associée à son niveau d’expression (Avendano-Vazques et al., 2012). Différentes études, réalisées chez la mouche et le nématode C. elegans, ont pu démontrer une toxicité de la protéine tant au cytoplasme qu’au noyau en présence de mutations affectant sa localisation nucléaire. D’autres études ont aussi pu démontrer une toxicité lors de la surexpression de la protéine ainsi qu’un lien direct avec son domaine liant l’ARN chez la mouche et le nématode (Miguel et al., 2011; Ritson et al., 2010). Ainsi, la théorie du gain de fonction semble être étroitement liée à la fonction intrinsèque de la protéine, sa capacité à lier l’ARN et sa localisation, et non à une fonction toxique nouvelle (Ash et al., 2010; Ihara et al., 2013; Voigt et al., 2010). La théorie de perte de fonction de TDP-43 a aussi été étudiée par plusieurs équipes de recherche. Chez le nématode C. elegans, tdp-1, l’orthologue de TDP-43, lorsque sous-exprimé, mène à une durée de vie plus longue, puisque ce gène n’est pas nécessaire à sa survie comme chez l’humain. Cette modification génétique mène par contre à d’importants problèmes de motilité et de fertilité (Vaccaro et al., 2012b; T. Zhang et al., 2012). Chez la mouche, les modèles de perte de fonction de TDP-43 (orthologue dTDP-43) mènent à une perte axonale importante, à une modification des boutons synaptiques ainsi qu’à une durée de vie diminuée (Li et al., 2010; Lin et al., 2011; J. W. Wang et al., 2011). Toujours dans ce même modèle d’étude, certaines pathologies de la SLA, dont une transmission synaptique déficiente, sont observées en présence d’une perte ou d’un gain de fonction de dTDP-43 ou d’une surexpression ou sous-expression de dTDP-43 (Vanden Broeck et al., 2013; Diaper et al., 2013b). Chez le poisson zèbre, deux orthologues de TDP-43 existent : tardbp et tardbpl. L’absence d’expression transitoire de tardbp mène à une
14
désorganisation des axones des neurones moteurs alors qu’une présence de mutations dans tardpl et tardp mène à une paralysie ainsi qu’à une dégénérescence musculaire (Bose et al., 2019; Hewamaddumal et al., 2013; Kabashi et al., 2009; Lissouba et al., 2018; Schmid et al., 2013). Chez la souris, une perte d’expression de TDP-43 mène à la mort de l’embryon (Hewamaddumal et al., 2013). Une perte d’expression de ce gène chez l’embryon mène à une atrophie musculaire et axonale (Iguchi et al., 2013b). Tant une perte de fonction qu’un gain de fonction ont été associés à TDP-43 dans un modèle cellulaire NSC-34 (Cascella et al., 2016).
Figure 3 : Modèle simplifié de la protéinopathie de TDP-43
En condition non pathologique (physiologique), la protéine TDP-43 se trouve principalement dans le noyau des neurones moteurs. En condition pathologique, comme pour 97 % des cas de SLA, la protéine se déplace du noyau vers le cytoplasme et forme des agrégats cytoplasmiques ubiquitinés. Figure tirée et adaptée de Buratti, 2008.
C9ORF72
Tout près de 50 % des cas de SLAF sont actuellement expliqués par la répétition d’un hexanucléotide GGGGCC dans le premier intron du gène C9ORF72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Présente entre 10 et 15 fois chez un individu sain et jamais plus de 23 fois, cette répétition peut être présente