HAL Id: pastel-00000759
https://pastel.archives-ouvertes.fr/pastel-00000759
Submitted on 31 Aug 2004HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Jérôme Minet
To cite this version:
Jérôme Minet. Synthèse et caractérisation de silicates de calcium hydratés hybrides. Chimie. Univer-sité Paris Sud - Paris XI, 2003. Français. �pastel-00000759�
N°D'ORDRE :
UNIVERSITÉ DE PARIS-SUD
U.F.R. SCIENTIFIQUE D'ORSAY
THÈSE
présentée pour obtenir le grade de
DOCTEUR ÈS SCIENCES
DE L'UNIVERSITE PARIS XI, ORSAY
Discipline : CHIMIE INORGANIQUE par :
M. MINET Jérôme
Sujet de la thèse :
SYNTHÈSE ET CARACTÉRISATION DE SILICATES
DE CALCIUM HYDRATÉS HYBRIDES
Soutenue le: 02/12/2003 devant la commission d'examen: M. Henri VAN DAMME, Rapporteur
M. Daniel ZAMBON, Rapporteur M. Philippe BOCH, Président M. Nicolas LEQUEUX, Directeur de thèse
M. Patrick JUDEINSTEIN M. Clément SANCHEZ Mme Angélique VICHOT, Invitée
Le guerrier ne pense pas en terme
de victoire ou de défaite,
ni même en terme de vie ou de mort,
il combat pour accomplir sa destinée…
Miyamoto Musashi, Gorin no sho. (Des cinq éléments, 1643).
Remerciements
Je souhaiterais en premier lieu remercier M. Henri Van Damme et M. Daniel Zambon de m'avoir fait l'honneur d'être mes rapporteurs, ainsi que tous les membres du jury: M. Sanchez, M. Boch, M. Judeinstein et Mme Vichot.
J'aimerais aussi remercier M. Philippe Boch d'avoir accepter que je finisse ma thèse dans son laboratoire, malgré des conditions d'arrivée assez… atypiques !
Mille(S) mercis à Roger Famery pour m'avoir fait participer activement à l'enseignement de la microscopie électronique à balayage durant l'année scolaire 2002-2003. Ce soutien m'a évité de manger des pissenlits (par la racine) toute l'année…
Je tiens particulièrement à remercier Nicolas Lequeux. Premièrement pour avoir confié à un métallurgiste un sujet traitant de l'hybridation de céramiques (ça n'a pas été facile tous les jours, mais comme l'a écrit Nietzsche "tout ce qui ne tue pas renforce"). Merci aussi d'avoir été à la fois mon directeur de thèse ainsi que mon responsable au quotidien. Merci enfin de m'avoir laissé une entière et pleine liberté sur la gestion de mon étude, sans pour autant en être désintéressé. (Finalement, on en a eu des résultats !)
Merci à l'infatigable Bruno Bresson pour son infaillible soutien sur cette formidable mais terrible machine qu'est la RMN, pour son consulting en ressources humaines et ses nombreuses séances de lecture. Merci aussi à Florence Babonneau pour ses conseils avisés (en particulier sur l'utilisation des tableurs !).
Le moment est venu de remercier l'ensemble des personnes qu'il m'a été donné de rencontrer et d'apprécier, sans lesquelles faire une thèse ne serait pas vraiment la même chose !
Au SESI: Alla la cosaque, Marie-Noëlle 100000V, Fred qui a un petit vélo dans la tête, Stéphane Berry-Berry.
Aux anciens du CMM: Jérôme et Gaëlle, Marie-Pierre, Geneviève, Arnaud le marathon-man, Florence, Isabelle ma colloc' d'ordi, Valerio mio palmo, et Sébastien qui hérite du reste (et du gros) du boulot ! Les thermites: Dame Fabienne, Charlie the excecutive woman, Philippe l'éternel boute-en-train, Sylvain le dandy-ludique (maintenant je peux le dire: merci pour le spectro !), Gwenn, Duñeska l'agent venu du…Vénézuéla, Laurentiu l'homme qui a lu l'intégrale de Balzac mérite qu'on le salut ici !, Samir en
Anne et ses photos d'Iran, Rémi le kung-fu boy, Julien l'homme de la route, Guy une fois, Jean, Caroline, Mister Manu, Cécile (merci pour ta manip et ses belles images!), Olivier les bons tuyaux… Bon courage à tous ceux qui vont entrer dans la carrière alors que leurs aînés n'y seront plus: Julie, Laetitia, Florence, Erwan, Benjamin, et qui sait Claire et Aurélie !
J'allais presque oublier mes poulains: Pierre-Yves, Martin et Blandine: bon courage pour la suite… Merci à tous pour les excellents moments que l'on a passé…
Merci aussi à mes amis qui ont supporté sans mot dire pendant ces longues années mes explications sur mon travail ainsi que sur la physique en général ! A ce titre vous méritez plus que des remerciements…
Remerciements particuliers à l'ami Stéphane qui m'a toujours chaleureusement hébergé chaque fois qu'il y avait grève de RER (c'est dire !) ou en cas de rush critique. Merci Steph ! Sans toi je crois bien que j'aurai mis au moins trois mois de plus pour finir.
Je tiens enfin à remercier mes parents qui m'ont laissé m'embarquer -intrigués- dans cette galère sans vraiment comprendre où tout ça me mènerait. Maintenant que j'ai fini, je ne serais toujours pas dire exactement où tout cela m'a mené, mais en tout cas j'ai réussi à revenir au port (ce qui n'est pas si mal).
Mes derniers mots vont à Géraldine, que je remercie de tout mon cœur de m'avoir supporté par monts et par vaux, sans un sous vaillant, jamais disponible et avec la science pour amante…
A Baptiste (et je l'espère aux autres) qui trouveront sûrement tout ceci bien fantasque si un jour ils lisent cette étude, j'aimerais leur faire part de ces mots:
…Si tu sais méditer, observer et connaître Sans jamais devenir sceptique ou destructeur ; Rêver, mais sans laisser ton rêve être ton maître,
Penser sans n'être qu'un penseur… Tu seras un homme, mon fils.
Sommaire
REMERCIEMENTS... 2
SOMMAIRE... 4
INTRODUCTION... 7
CHAPITRE 1: SILICATES HYDRATES LAMELLAIRES... 11
RESUME... 12
1.1. SILICATES HYDRATES LAMELLAIRES... 13
1.1.1. Introduction... 13
1.1.2. Classification des silicates ... 14
1.2. PHYLLOSILICATES... 15
1.2.1. Généralités ... 15
1.2.2. Structure générale ... 16
1.2.2.1. Structure de base de la couche de silicium... 16
1.2.2.2. Structures de base de la couche octaédrique ... 17
1.2.3. De l'unité structurale au phyllosilicate ... 18
1.2.4. Phyllosilicates 1:1 ... 19
1.2.5. Phyllosilicates 2:1 ... 20
1.2.6. Phyllosilicates 2:1:1... 23
1.3. SILICATES DE CALCIUM HYDRATES (C-S-H)... 25
1.3.1. Généralités ... 25
1.3.2. Synthèse des C-S-H ... 26
1.3.3. Microstructure des C-S-H ... 27
1.3.4. Structure des lamelles de C-S-H ... 29
1.3.4.1. Unités structurales de base ... 29
1.3.4.2. Feuillet de tobermorite: un feuillet de C-S-H idéal... 30
1.3.4.3. Trois modèles de feuillets pour les C-S-H réels... 31
1.3.4.4. Paramètres inconnus dans la structure des C-S-H... 33
1.4. C-S-H ET PHYLLOSILICATES 2:1... 35
CHAPITRE 2: PHYLLOSILICATES HYBRIDES... 37
RESUME... 38
2.1. DEFINITIONS... 39
2.1.1. Matériaux composites et matériaux hybrides... 39
2.1.2. Hybrides interstratifiés et dispersés... 40
2.2. METHODES DE SYNTHESE DE PHYLLOSILICATES HYBRIDES... 41
2.2.1. Synthèses des hybrides de classe I ... 41
2.2.1.1. Synthèse par intercalation ... 41
2.2.1.2. Synthèse par polymérisation in situ ... 43
2.2.2.1. Synthèses post creatione d'hybrides de classe II ... 46
2.2.2.2. Synthèses ab initio d'hybrides de classe II ... 47
2.2.3. Bilan sur les méthodes de synthèse ... 50
2.3. STRUCTURE DES FEUILLETS INORGANIQUES DANS LES PHYLLOSILICATES HYBRIDES. 51 2.3.1. Feuillets obtenus par méthode post creatione... 51
2.3.2. Feuillets obtenus par méthode ab initio... 51
CHAPITRE 3: SYNTHESE DE C-S-H HYBRIDES ... 56
RESUME... 57
3.1. SYNTHESE DE C-S-H HYBRIDES DE CLASSE I... 58
3.2. SYNTHESE DE C-S-H HYBRIDES DE CLASSE II ... 60
3.2.1. Positionnement de l'étude... 60
3.2.2. Principes de synthèse des C-S-H hybrides... 61
3.2.2.1. La méthode sol-gel ... 61
3.2.2.2. Synthèse de C-S-H par voie sol-gel ... 61
3.2.3. Modes opératoires... 62
3.2.3.1. Etapes de synthèses et mécanismes réactionnels ... 62
3.2.3.2. Composition des hybrides synthétisés... 65
3.2.3.3. Synthèse "aqueuse" de C-S-H hybrides ... 66
3.2.3.4. Synthèse "alcoolique" de C-S-H hybrides... 67
CHAPITRE 4: ORGANO-SILANES DE CALCIUM ... 68
RESUME... 69
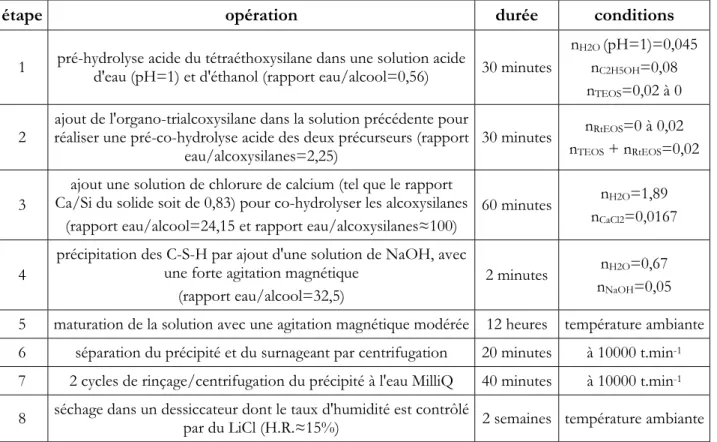
4.1. C-S-H DE REFERENCE... 70
4.1.1. Données expérimentales... 70
4.1.2. Analyses... 73
4.2. ORGANO-SILANES DE CALCIUM SYNTHETISES PAR VOIE "ALCOOLIQUE"... 74
4.2.1. Synthèses avec les radicaux aliphatiques... 74
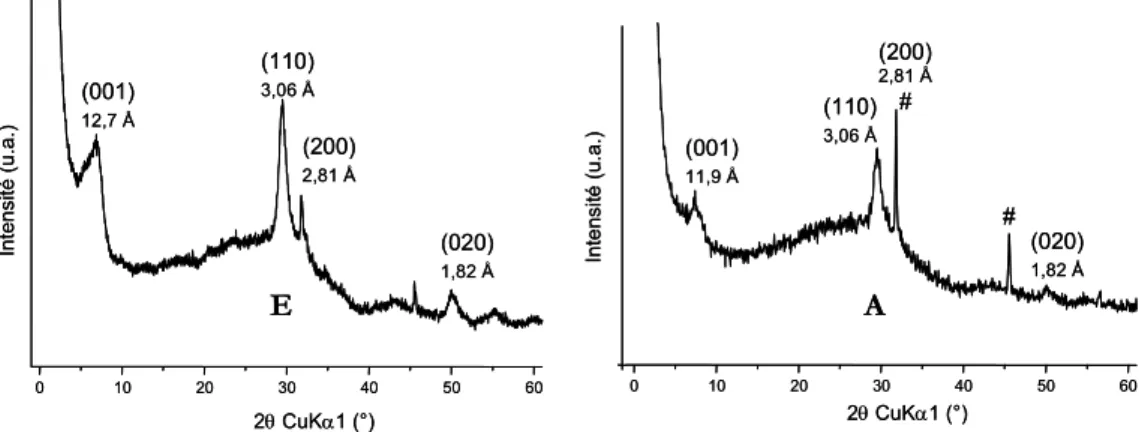
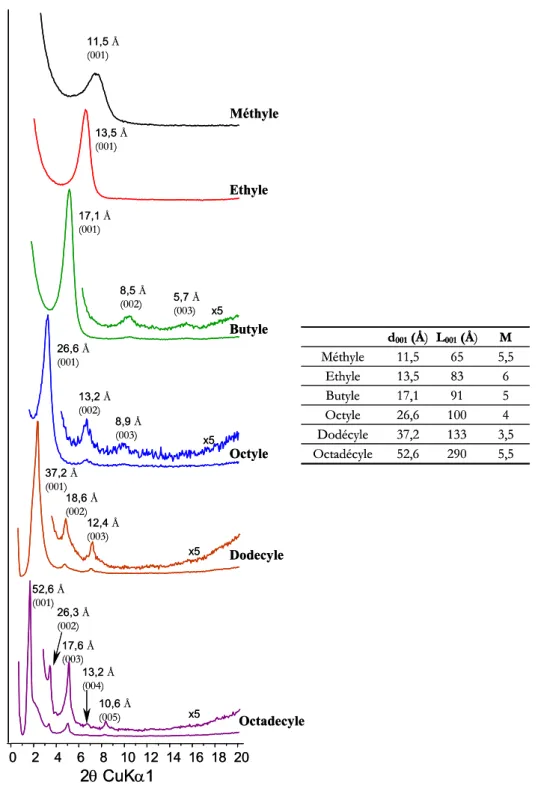
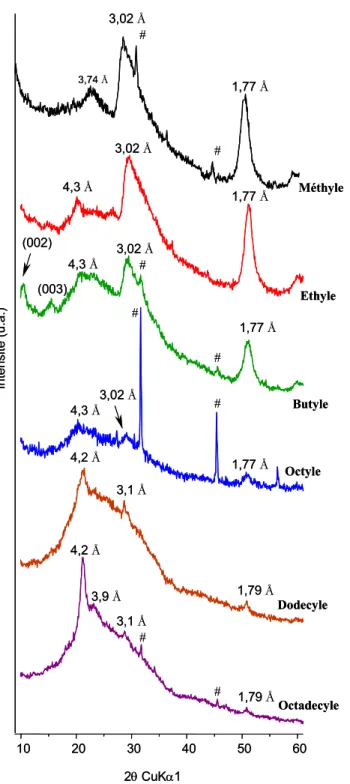
4.2.1.1. Données expérimentales... 74
4.2.1.2. Structure des organo-silanes de calcium obtenus... 84
4.2.2. Synthèse avec le radical fonctionnel Phényle ... 89
4.2.3. Conclusion sur les organo-silanes de calcium obtenus par voie alcoolique ... 91
4.3. ORGANO-SILANES DE CALCIUM SYNTHETISES PAR VOIE "AQUEUSE" ... 92
4.3.1. Synthèses avec les radicaux aliphatiques... 92
4.3.1.1. Données expérimentales... 92
4.3.1.2. Structure des organo-silanes de calcium formés ... 95
4.3.2. Synthèse avec le radical fonctionnel Vinyle... 97
4.3.3. Synthèse avec le radical Aminopropyle ... 99
4.3.4. Conclusion sur les organo-silanes de calcium obtenus par voie aqueuse... 101
4.4. DISCUSSION... 102
4.4.1. Comparaison entre les méthodes de synthèse par voie alcoolique et aqueuse 102 4.4.2. Co-assemblage ou réplication ?... 104
4.4.2.1. Radicaux organiques de faible taille ... 105
CHAPITRE 5: C-S-H HYBRIDES ... 111 RESUME... 112 5.1. INTRODUCTION... 113 5.2. RADICAUX ALIPHATIQUES... 115 5.2.1. Série M-C-S-H... 115 5.2.2. Série E-C-S-H... 119 5.2.3. Série B-C-S-H... 122 5.2.4. Série O-C-S-H ... 125 5.3. RADICAUX FONCTIONNELS... 128 5.3.1. Série V-C-S-H... 128 5.3.2. Série A-C-S-H... 131 5.3.3. Série P-C-S-H... 134 5.4. DISCUSSION... 137
5.4.1. Comparaison entre les différentes séries ... 137
5.4.2. Hybride homogène ou hétérogène ?... 139
CONCLUSION ET PERSPECTIVES ... 147
ANNEXES... 152
ANNEXE 1:CONDITIONS EXPERIMENTALES... 153
Réactifs ... 153
Préparation des échantillons ... 153
Analyses chimiques ... 153
Diffraction des Rayons X... 153
FTIR ... 153
RMN ... 154
ANNEXE 2:ANALYSES CHIMIQUES DES ORGANO-SILANES DE CALCIUM REALISES PAR VOIE "ALCOOLIQUE" ... 154
ANNEXE 3:DIFFRACTOGRAMMES X AUX PETITS ANGLES DES C-S-H HYBRIDES... 155
Introduction
Ignoti nulla cupido.
Les matériaux nanocomposites organique/inorganique sont l'enjeu d'un développement industriel majeur depuis les années 1950. Tirant bénéfice de l'association au niveau moléculaire de leurs deux composantes, ils sont aujourd'hui couramment employés dans des applications de surface comme: les peintures, les revêtements de surfaces (hydrophobe, anticorrosion, antirayure, etc.), les adhésifs, les verres de spécialité (antireflets, photochromiques, autonettoyants, etc.) [1-3].
Récemment, un intérêt s'est développé pour les approches de synthèse bio-minérales de matériaux hybrides organique/inorganique massiques.
La nature développe en effet une remarquable diversité d'organismes mettant à profit les effets d'une telle synergie: coquillages, carapaces, os, dents, etc. Ces matériaux présentent des propriétés mécaniques uniques du point de vue de leur ténacité, de leur résilience, de leur résistance à la flexion, etc., et ce pour des teneurs en phase organique extrêmement faibles.
La compréhension et la reproduction des stratégies d'association des phases organique et inorganique utilisées par la nature ouvrent donc de larges perspectives en ce qui concerne la réalisation de matériaux de synthèse aux propriétés atypiques.
Les matériaux développés à partir de ces recherches concernent à ce jour les domaines d'applications variés tels que la mécanique, l'optique, l'électronique, la catalyse, la synthèse de molécules énantiomères, le développement de membranes, la biologie etc. [4-8]
Cette démarche se retrouve également dans le domaine des matériaux cimentaires, dans lesquels des associations organique/inorganique sont couramment utilisées.
Le ciment est un mélange de calcaire, d'argile et de gypse porté à 1450°C dont les propriétés de liant hydraulique, d'ouvrabilité et de durabilité sont utilisées industriellement depuis le milieu du XIXème siècle. Cependant, les molécules organiques n'ont été que récemment incorporées dans le ciment dans l'objectif principal d'améliorer son ouvrabilité sur le terrain, en ajoutant des: retardateurs de prise, "superplastifiants", agents entraîneurs d'air…
En ce qui concerne l'amélioration des propriétés mécaniques du ciment par l'emploi de molécules organiques, la plus remarquable réalisation actuelle sont les ciments "MDF" (Macro Defect Free cement). Dans ces ciments, une association moléculaire du polymère d'alcool-polyvinylique (PVA) avec les phases d'aluminates de calcium augmente le module de flexion de 10 MPa à 150 MPa, ce qui font des MDF des ciments de très hautes performances.
Cependant, ces propriétés exceptionnelles des MDF ne sont pas dues à des interactions spécifiques entre la phase polymérique et les hydrates de ciment, mais sont plutôt la conséquence de la réduction globale de la porosité du matériau (la porosité étant le facteur limitant principal des charges à la rupture des matériaux fragiles) [9-11].
Quelle(s) stratégie(s) faut-il alors employer pour faire interagir des molécules organiques et les hydrates de ciment afin de développer de nouvelles propriétés pour les matériaux cimentaires?
Dans cette problématique, une des voies les plus prometteuses est de contrôler au niveau moléculaire la texture des hydrates de ciments par association avec des phases organiques.
Dans les ciments ordinaires, la phase majoritaire à l'origine des propriétés cohésives des matériaux sont les silicates de calcium hydratés (C-S-H). Les C-S-H sont des matériaux nanocristallins lamellaires formant des empilements de faible extension spatiale (de l'ordre d'une centaine d'Angströms).
Pour développer des interactions entre des molécules organiques et les C-S-H, des approches visant à reproduire des stratégies bio-minérales ont été récemment entreprises. Matsuyama et Young, en se basant sur des résultats d'intercalations de polymères entre les feuillets de minéraux argileux, ont les premiers employé des stratégies d'intercalation afin d'insérer différents polymères entre les feuillets de C-S-H. Ils ont ainsi pu observer que certains polymères comme l'alcool-polyvinylique (PVA) et le polydiallyldiméthylamonium (PDDMA) avaient tendance à s'intercaler entre les feuillets de C-S-H lors de la formation de ces derniers à partir d'une solution aqueuse [12-14].
Puis, Popova et al. [15] ainsi que Merlin et al. [16] se sont basés sur cette approche pour tenter de réaliser l'intercalation d'autres types de polymères entre les lamelles de C-S-H, voire de réitérer les expériences de Matsuyama et al. Cependant aucune de leurs expériences n'a confirmé les observations de la première équipe. Les résultats de Matsuyama et al. sont donc sujet à controverse.
Nous pouvons alors remarquer que -a contrario des argiles- les empilements de C-S-H ne sont pas gonflants. Dès lors, l'emploi de stratégies d'intercalation ne semble pas très favorable pour la réalisation d'hybrides polymères/C-S-H, puisque cette stratégie est basée directement sur la capacité des lamelles à accommoder des molécules organiques dans leur interfeuillet (c'est à
Pour réaliser des hybrides organique/C-S-H, nous avons donc choisi de nous orienter vers un nouveau type d'approche consistant à développer des interactions fortes entre des molécules organiques et les C-S-H.
Cette approche consiste plus précisément à greffer directement de façon covalente des molécules organiques sur les feuillets de C-S-H au cours de leur synthèse.
Pour y parvenir nous avons développé une méthode de synthèse de C-S-H hybrides par voie sol-gel qui consiste à faire précipiter le matériau à partir d'un mélange homogène de précurseurs en solution.
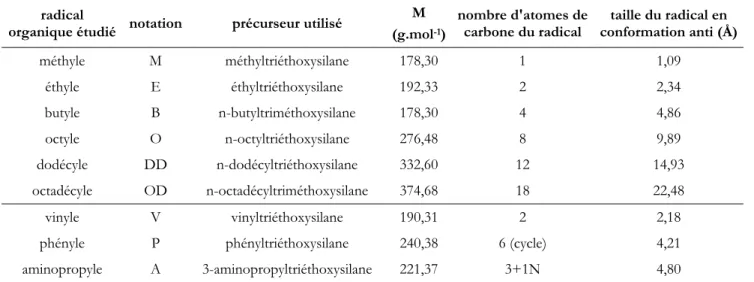
Dans notre étude, nous avons choisi d'étudier l'influence de la taille et de la teneur des radicaux organiques sur la formation des C-S-H hybrides.
Nous avons pour cela employé des radicaux aliphatiques allant du méthyle jusqu'à l'octadécyle. En outre, nous avons sondé l'influence de la nature du groupement organique utilisé sur la formation des C-S-H hybrides en utilisant les radicaux: vinyle, phényle et aminopropyle.
La caractérisation des matériaux hybrides obtenus s'est faite principalement par l'utilisation de la diffraction des rayons X, de la spectrométrie infrarouge et de la résonance magnétique nucléaire.
Ces méthodes d'investigation ont eu pour principal objectif de déterminer la nature des phases hybrides observées. Elles nous ont permis en particulier d'observer la formation de phases adoptant une structure de type C-S-H pour des teneurs et des tailles de radicaux organiques peu importantes. Puis la résonance magnétique nucléaire a été employée afin de démontrer qu'il existait bien une association structurale entre les atomes de silicium portant les radicaux organiques et les autres atomes de silicium. Nous avons ainsi pu montrer que les phases ayant une structure de type C-S-H étaient bien des C-S-H hybrides.
Chapitre 1:
Silicates hydratés lamellaires
De omni re scibili…
…et quibusquam aliis.
Pic de la Mirandole… … et Voltaire.
Résumé
Nous nous intéresserons dans cette étude à deux types de silicates hydratés lamellaires. Le premier est constitué par les phyllosilicates hydratés (smectites, vermiculites et pseudo-chlorites). Le deuxième est formé par les silicates de calcium hydratés (C-S-H).
Les deux types de matériaux adoptent un mode d'organisation multi-échelle. Ces minéraux sont en effet composés d'agglomérats, les agglomérats sont formés par un assemblage de particules, les particules sont constituées d'empilements de feuillets (les feuillets étant séparés par un interfeuillet d'eau), enfin, les feuillets sont constitués d'empilements de couches atomiques.
La différence entre ces deux types de matériaux réside principalement dans la nature et la structure de leurs couches atomiques:
- les feuillets de phyllosilicates hydratés sont constitués par une couche cationique en coordinence octaédrique encadrée par deux couches de silicium en coordinence tétraédrique décrivant un motif hexagonal. Leur structure est notée: Te(Q3) Oc Te(Q3).
- les silicates de calcium hydratés adoptent une structure formée par une double couche pseudo-octaédrique (heptaédrique) de calcium encadrée par deux couches de chaînes de silicium. Cette structure est notée: Te(Q1 Q2) OcOc Te(Q1 Q2).
1.1. Silicates hydratés lamellaires
1.1.1. Introduction
Nous savons aujourd'hui que le silicium est un constituant majeur de l'univers [17]. Présent dans les astres, les gaz interstellaires et les poussières cosmiques, il est le septième élément le plus abondant de l'univers (derrière l'hydrogène, l'hélium, l'oxygène, le carbone, l'azote et le néon). C'est donc l'élément métallique le plus abondant de l'univers.
Sur Terre, la répartition des éléments est encore plus remarquable puisque le silicium est l'élément le plus abondant (27,72 %) juste après l'oxygène (46,60 %) [18]. Sa présence s'y décline d'ailleurs avec une étonnante diversité, aussi bien dans le monde des minéraux que dans le monde des biomatériaux et est même indispensable à la biochimie du monde du vivant (plantes, bactéries)…
Dans le monde minéral, les minéraux silicatés -ou silicates- occupent donc une place privilégiée. Composés d'unités {SiO4} (voir chapitre 1.1.2), ils constituent plus de 90 p.100 de la
masse de la croûte terrestre [18], et forment le groupe de minéraux le plus vaste que nous connaissons sur Terre (voir Tableau 1. 1).
minéral % massique
feldspaths 60
pyroxènes et amphiboles 17
quartz 12 micas 4 Tableau 1. 1 : Répartition des silicates dans l'écorce terrestre.
Leur abondance, leur diversité et leur accessibilité sont autant de qualités qui ont suscité l'intérêt chez l'homme. L'homo habilis, le premier, les a utilisé pour en faire les premiers outils de l'histoire de l'humanité. Dès lors, leur utilisation a jalonné l'évolution de l'homme, qui n'a eu de cesse de se les approprier.
De nos jours, les silicates sont employés couramment dans de nombreux domaines de l'industrie, aussi bien de manière directe (matière première des céramiques, verres, matériaux de construction) que pour réaliser des produits plus élaborés (papiers, peintures, cosmétiques, plastiques). C'est à ce titre que de nombreuses recherches sont menées aujourd'hui, visant à développer des matériaux silicatés de haute technologie (catalyse, matériaux composites,
1.1.2. Classification
des
silicates
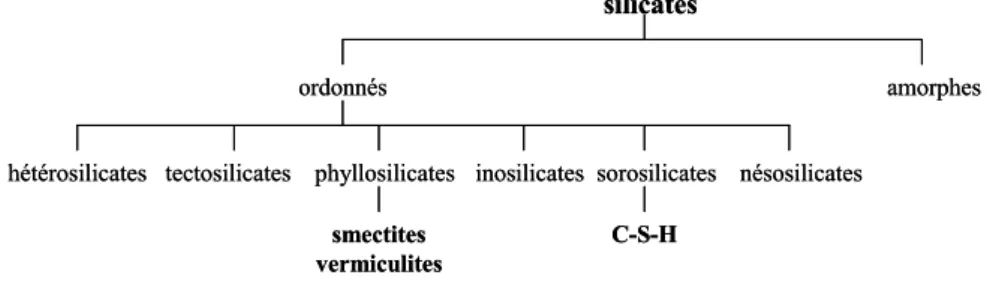
Les silicates forment une classe de matériaux extrêmement vaste, en composition, en structure, et en propriétés. Pour établir une classification de ces minéraux, il est d'usage d'employer des critères cristallochimiques. Comme les atomes de silicium sont presque toujours en coordinence tétraédrique et que les tétraèdres se connectent toujours entre eux par le sommet [19], il est alors commode de considérer l'arrangement de ces unités entre elles pour établir une classification des minéraux (plutôt que de les classer selon leur groupe d'espace). Dès lors, on retrouve les groupes de silicates bien connus [19], voir Figure 1. 1:
- les nésosilicates: silicates formés de tétraèdres de silicium indépendants des uns des autres (la forstérite par exemple)
- les sorosilicates: silicates formés de groupes finis de tétraèdres (thortveitite). Si les tétraèdres s'organisent en anneaux, ils forment le sous-groupe des cyclosilicates (béryl)
- les inosilicates: silicates formés de chaînes de tétraèdres (wollastonite). Dans lesquels on peut distinguer deux sous-groupes principaux: les pyroxènes (dont le motif de la chaîne silicatée est constitué de deux tétraèdres de silicium), et les amphiboles (constitués de chaînes unies)
- les phyllosilicates: (dits silicates lamellaires) dans lesquels les tétraèdres de silicium forment des plans (argiles, micas)
- les tectosilicates: silicates dont les tétraèdres s'organisent de façon tridimensionnelle (quartz, feldspaths, zéolites)
- les hétérosilicates: classe de silicates dans lesquels l'ordre entre les tétraèdres ne prédomine plus, et dans lesquels plusieurs modes d'arrangement entre les tétraèdres de silicium coexistent (clinozoïsite).
silicates
ordonnés amorphes
tectosilicates phyllosilicates inosilicatessorosilicates
hétérosilicates nésosilicates
silicates
ordonnés amorphes
tectosilicates phyllosilicates inosilicatessorosilicates
hétérosilicates nésosilicates
Figure 1. 1 : Classification des silicates.
Dans la suite de notre étude, nous nous intéresserons spécifiquement à deux types de silicates hydratés lamellaires: les silicates de calcium hydratés (notés C-S-H) et les phyllosilicates. Nous nous proposons de décrire dans un premier temps la structure des phyllosilicates, de façon à mettre en relief les spécificités structurelles des C-S-H par la suite.
1.2. Phyllosilicates
1.2.1. Généralités
Les phyllosilicates comptent plus de 200 variétés de minéraux différents [20, 21], parmi lesquelles on trouve les groupes très importants des micas et des argiles [18]. Les minéraux de ce groupe présentent un certain nombre de spécificités remarquables:
- La première est qu'il est difficile de décrire les phyllosilicates comme un groupe partageant une série de propriétés communes, puisqu'elles ne sont en général représentatives que de certains sous-groupes, et non de l'ensemble des minéraux. Cependant, ils présentent tous la propriété d'être facilement clivable selon leur plan basal (ce qui est lié à leur structure commune).
Malgré l'absence de propriétés communes, nous pouvons quand même rappeler celles du sous-groupe des argiles: plasticité, propriétés d'hydratation et d'absorption, durcissement en température (ces propriétés dépendent fortement de l'état d'hydratation des minéraux).
- Au niveau structural, ils présentent le même mode d'organisation multi-échelle. En effet, les phyllosilicates sont composés d'agglomérats, ces agglomérats sont composés de particules, chacune des particules est composée d'un empilement de lamelles (ou feuillets), une lamelle étant composée d'une couche planaire formée par un cation en coordinence octaédrique encadrée d'une ou deux couches planaires de silicium en coordinence tétraédrique (voir Figure 1. 2).
Les phyllosilicates peuvent se définir comme des silicates ayant une structure bidimensionnelle tripériodique, ordonnés jusqu'à un ordre mésoscopique (voir Figure 1. 2).
minéral: assemblage d'agglomérats 10-2m agglomérat: assemblage de particules 10-4m particule: empilement de feuillets 10-6m feuillet: empilement de couches atomiques 10-9m minéral: assemblage d'agglomérats 10-2m agglomérat: assemblage de particules 10-4m particule: empilement de feuillets 10-6m feuillet: empilement de couches atomiques 10-9m Figure 1. 2: Observation d'un phyllosilicate (montmorillonite) à différentes échelles
1.2.2. Structure
générale
En l'absence de propriétés macroscopiques communes, il a pendant longtemps été très difficile de classer et de définir les phyllosilicates comme tels. Cependant l'apport des techniques d'investigations structurales, telle que la diffraction des rayons X, a permis de résoudre complètement leur structure et de définir le groupe des phyllosilicates comme étant des matériaux possédant le même mode d'organisation des atomes de silicium.
1.2.2.1. Structure de base de la couche de silicium
Par définition (cf. chapitre 1.1.2), les atomes de silicium dans les phyllosilicates s'organisent selon un motif hexagonal planaire.
Dans cet agencement, chaque atome de silicium est tétra-coordonné (c'est à dire au centre d'un tétraèdre dont les quatre sommets sont occupés par des atomes d'oxygène). Pour être dans une configuration planaire régulière, chacun des tétraèdres partage les trois sommets de sa base avec trois tétraèdres voisins (l'oxygène apical -du sommet principal- étant non engagé), voir Figure 1. 3.
plan d'oxygène
plan d'oxygènes apicaux plan silicium
plan d'oxygène
plan d'oxygènes apicaux plan silicium
Figure 1. 3 : Agencement des atomes dans la couche tétraédrique.
Selon les notations en vigueur dans la cristallochimie des silicates [19], les tétraèdres de silicium sont en configuration Q3 (nombre de coordinence), L1 (liés par les sommets). La
mono-couche d'atomes est couramment qualifiée de tétraédrique, et notée Te.
Nous pouvons alors distinguer dans cette couche tétraédrique trois plans atomiques parallèles: un plan formé par les atomes d'oxygène de la base des tétraèdres, un plan atomique de silicium et un plan d'oxygènes apicaux (voir Figure 1. 3).
En outre, nous pouvons remarquer que les tétraèdres de silicium décrivent des motifs hexagonaux. La maille primitive de cet agencement peut être décrite avec un motif pyroxénique Si2O5 (voir Figure 1. 4). Ce mode d'arrangement structural est commun à tous les phyllosilicates
Figure 1. 4 : Motifs hexagonaux dans la couche tétraédrique de silicium des minéraux phylliteux,
vus selon un axe respectivement normal et parallèle à la maille primitive.
1.2.2.2. Structures de base de la couche octaédrique
La couche octaédrique est également planaire. Elle est formée par un cation métallique hexa-coordonné (le cation occupe le centre d'un octaèdre dont les six sommets sont des atomes d'oxygène). Dans cette configuration planaire régulière, chacun des oxygènes est partagé par trois octaèdres voisins, qui se trouvent ainsi liés par leurs arêtes (voir Figure 1. 5).
Comme précédemment, nous pouvons observer que cette couche est formée par l'empilement de trois plans atomiques parallèles: un plan cationique central encadré de deux plans d'atomes d'oxygène. La mono-couche est qualifiée d'octaédrique, et notée Oc.
Figure 1. 5 : Agencement des atomes dans la couche octaédrique.
En fonction de la nature divalente ou trivalente du cation, il est en outre possible de distinguer deux types de couche octaédrique:
- si le cation est divalent (ex Mg2+) tous les octaèdres sont occupés, la couche est dite
trioctaédrique. Si la mono-couche est isolée, la neutralité électrique est atteinte si tous les oxygènes sont hydroxylés (comme dans la Brucite Mg(OH)2).
- si le cation est trivalent (ex Al3+) seuls deux octaèdres sur trois sont occupés, la couche
est dite dioctaédrique. Là encore, si la mono-couche est isolée la neutralité électrique est atteinte si tous les oxygènes sont hydroxylés (cas de la Gibbsite Al(OH)3).
1.2.3.
De l'unité structurale au phyllosilicate
Il convient maintenant de passer de la description d'unités structurales de base à celle des phyllosilicates. Cette étape est complexe, car elle consiste à passer de la description de plans atomiques infinis et parfaits, à la description précise de minéraux lamellaires réels.
Pour y parvenir, il faut considérer tout d'abord les paramètres caractérisant la structure intrinsèque d'une lamelle individuelle:
- le mode d'association des couches tétraédrique et octaédrique entre elles, - le caractère tri ou dioctaédrique du plan octaédrique,
- le caractère continu ou discontinu du plan octaédrique,
- les défauts structuraux, la taille finie des feuillets, ainsi que leur forme.
Mais il faut aussi considérer les paramètres décrivant le mode d'empilement des lamelles dans une particule de taille finie:
- le nombre de lamelles d'un empilement (pouvant varier d'un dizaine à une centaine), - la nature, et l'épaisseur de l'espace interfoliaire,
- le caractère mono ou polyphylliteux d'un empilement. En cas d'interstratification, il faut déterminer le mode de succession entre les feuillets de natures différentes (alternance régulière, distribution aléatoire, etc.),
- la polytypie, c'est à dire caractériser la façon dont s'oriente un feuillet par rapport à ses voisins (empilement ordonné, semi-ordonné, à désordre translationnel, à désordre turbostratique (i.e. translationnel et rotationnel) [22]).
Tous ces paramètres sont indispensables pour rendre compte de la grande variété du groupe des phyllosilicates. Cependant, notre objectif ici n'est pas de le décrire exhaustivement, mais est plutôt de donner une description de structures moyennes, susceptibles de représenter l'ensemble des minéraux de ce groupe. C'est pourquoi nous nous restreindrons à la description des phyllosilicates ayant les caractères suivants (voir Figure 1. 6):
- couche octaédrique continue, - monophylliteux (non interstratifiés),
- sans rendre compte ni des défauts structuraux, ni de la taille et de la forme des feuillets, ni de leur polytypie.
Les minéraux modèles que nous allons décrire forment trois sous-groupes distincts: les phyllosilicates "1:1" , "2:1" et "2:1:1" et peuvent servir de base à la description des minéraux appartenant aux sous-groupes non développés de l'arborescence de la Figure 1. 6 [23].
phyllosilicates couche Oc continue phyllosilicates 1:1 ou Te Oc monophyllites couche Oc discontinue (palygorskite, sépiotite…) polyphyllites (interstratifiés) phyllosilicates 2:1 ou Te Oc Te phyllosilicates 2:1:1 ou Te Oc Te Oc phyllosilicates couche Oc continue phyllosilicates 1:1 ou Te Oc monophyllites couche Oc discontinue (palygorskite, sépiotite…) polyphyllites (interstratifiés) phyllosilicates 2:1 ou Te Oc Te phyllosilicates 2:1:1 ou Te Oc Te Oc
Figure 1. 6 : Les différents sous-groupes de phyllosilicates.
1.2.4. Phyllosilicates
1:1
Un premier mode d'agencement consiste à associer une seule couche tétraédrique à la couche octaédrique. C'est le sous-groupe des phyllosilicates 1:1, ou Te Oc.
Dans ce mode d'organisation deux oxygènes sur trois d'un plan d'oxygène de la couche octaédrique sont les oxygènes apicaux de la couche tétraédrique. Le tiers des atomes d'oxygène restant, ainsi que l'autre couche d'atomes d'oxygène de la couche octaédrique sont hydroxylés (voir Figure 1. 7). cavité hexagonale a b cavité hexagonale a b
Figure 1. 7 : Structure et maille primitive des phyllosilicates 1:1.
Si l'on observe leur structure selon l'axe cr, normal au feuillet, on peut mettre en évidence des cavités hexagonales à l'intérieur desquelles se trouve un groupement OH appartenant à la couche octaédrique (voir Figure 1. 7). On peut, en outre, définir une maille orthorhombique primitive représentative de la globalité de la structure. Pour déterminer la composition de la maille, il faut toutefois considérer la nature du cation de la couche octaédrique.
Ceci nous amène à définir deux familles de phyllosilicates 1:1: celle de nature dioctaédrique de type kaolinite, et celle de nature trioctaédrique de type lizardite (voir Tableau 1. 2):
Dioctaédrique Trioctaédrique charge:
(e-/maille)
kaolinite Si4 Al4 O10 (OH)8
lizardite
Si4 Mg6 O10 (OH)8 0
Tableau 1. 2 : Classification des phyllosilicates 1:1.
De nombreux minéraux appartenant à ces familles présentent des substitutions atomiques dans leur couche Te et/ou Oc (minéraux de type antigorite ou de type berthiérine). Néanmoins, ces substitutions ne modifient pas la neutralité électrique des feuillets. De sorte qu'il n'existe pas dans la nature de phyllosilicates 1:1 ayant de feuillet chargé.
Dans une particule de phyllosilicates 1:1, l'empilement des feuillets se fait de telle sorte que la couche Oc d'un feuillet soit face à la couche Te du feuillet voisin. Ce mode d'organisation engendre des interactions de Van der Waals importantes entre les feuillets de l'empilement (plans OH face aux plans O). En conséquence, l'espace interfoliaire (aussi appelé interfeuillet), qui constitue dans les phyllosilicates l'espace entre deux feuillets consécutifs (et qui est généralement occupé par des cations compensateurs et/ou de l'eau), est inexistant dans les phyllosilicates 1:1. Ces minéraux ne sont donc ni gonflants, ni exfoliables.
Ayant la structure la plus simple des phyllosilicates (Te Oc), et n'ayant pas d'interfeuillet, les phyllosilicates 1:1 ont donc la plus faible distance basale de tout le groupe minéral. C'est pourquoi certains auteurs les dénomment aussi les "phyllosilicates à 7 Å".
1.2.5. Phyllosilicates
2:1
Un deuxième mode d'agencement des unités structurales de base consiste à associer une couche tétraédrique de chaque côté de la couche octaédrique. C'est le sous-groupe des phyllosilicates 2:1, ou Te Oc Te.
Dans ce mode d'organisation deux oxygènes sur trois des plans d'oxygènes de la couche octaédrique sont les oxygènes apicaux des couches tétraédriques. Le tiers des oxygènes non engagés sont hydroxylés et se trouvent au centre des cavités hexagonales (voir Figure 1. 8).
Te Te Oc Interfeuillet distance basale: d001 xM+ nH 2O Te Te Oc Interfeuillet distance basale: d001 xM+ nH 2O
Figure 1. 8 : Structure des phyllosilicates 2:1.
On peut, comme précédemment, définir une maille orthorhombique primitive de même base (ar,br) que celle des phyllosilicates 1:1 (voir Figure 1. 7). Chacune des mailles contient quatre cavités hexagonales. Notons de plus, que les cavités hexagonales supérieures et inférieures d'un même feuillet ne sont pas d'aplomb, mais décalées de ar/3 .
Là encore, il est nécessaire de définir deux familles de phyllosilicates 2:1, en fonction de la nature du cation de la couche octaédrique: les phyllosilicates 2:1 dioctaédriques de la famille de la pyrophyllite, et ceux de nature trioctaédriques de la famille du talc (voir Tableau 1. 3):
Dans tous les phyllosilicates 2:1 autre que le talc et la pyrophyllite, il existe des substitutions isomorphes des cations du feuillet par des cations de charge différente.
Ces substitutions ont pour conséquence de rompre la neutralité électrique du feuillet. L'édifice cristallin devant rester neutre dans son ensemble, la compensation de ces charges négatives se fait par la présence dans l'espace interfoliaire de cations compensateurs (hydratés ou non). La quantité et la localisation des substitutions isomorphes dans les couches tétraédriques et/ou octaédrique conduit à distinguer plusieurs sous-groupes de minéraux, que l'on peut classer en fonction de leur charge par maille (voir Tableau 1. 3).
Par exemple, nous pouvons voir dans ce tableau que les smectites constituent le sous-groupe de phyllosilicates 2:1 dont la charge par maille est la plus faible. Ce sous-sous-groupe englobe une série de minéraux dont la charge (en électrons par maille) varie continûment en fonction de la localisation des substitutions: depuis 0,4 pour la montmorillonite et l'hectorite (dont la charge n'est due qu'à des substitutions dans la couche octaédrique) jusqu'à 1,2 pour la beidellite et la saponite (dont la charge n'est due qu'à des substitutions dans la couche tétraédrique).
Dioctaédrique Trioctaédrique charge: (e-/maille) pyrophyllite Si8 Al4 O20 (OH)4 talc Si8 Mg6 O20 (OH)4 0 smectites montmorillonite
Si8 (Al4-yMgy) O20 (OH)4 , M+y.nH2O
hectorite Si8 (Mg6-yLiy) O20 (OH)4 , M+y.nH2O beidellite (Si8-xAlx) Al4 O20 (OH)4 , M+x.nH2O saponite (Si8-xAlx) Mg6 O20 (OH)4 , M+x.nH2O 0,4 à 1,2 vermiculites
(Si8-xAlx) (Al4-yMg2+y) O20 (OH)4 , M+x+y.nH2O (Si8-xAlx) (Mg6-yN3+y) O20 (OH)4 , M+x-y.nH2O 1,2 à 1,8
micas muscovite (Si6Al2) Al4 O20 (OH)4 , K2 phlogopite (Si6Al2) Mg6 O20 (OH)4 , K2 2 margarite (Si4Al4) Al4 O20 (OH)4 , Ca2 clintonite (Si4Al4) Mg6 O20 (OH)4 , Ca2 4
Tableau 1. 3 : Classification des phyllosilicates 2:1.
Le seul mode d'organisation possible d'un empilement de feuillets de phyllosilicates 2:1, consiste en ce que les couches Te de feuillets consécutifs soient en vis à vis.
Cet agencement engendre des interactions de Van der Waals faibles entre les feuillets de l'empilement (plans O face aux plans O).
Cependant, la présence en quantité plus ou loin importante de cations compensateurs et/ou d'eau entre les feuillets influe sur ces interactions:
- La présence de cations compensateurs dans l'espace interfoliaire ajoute un caractère ionique aux interactions entre feuillets. Ce caractère est d'autant plus fort que la charge par maille est grande. Le cas extrême étant représenté par le sous-groupe des micas dit "cassants" (margarite, clintonite) dans lequel le caractère ionique prévaut, et donne aux minéraux des propriétés mécaniques de type solide ionique (fragilité).
- A l'inverse, la présence de l'eau dans l'espace interfoliaire tend à écranter les interactions Van der Waals existant entre les feuillets, ainsi que les interactions ioniques due à la présence de cations compensateurs. Les feuillets sont donc moins liés les uns aux autres, ce qui leur confère la capacité d'être "gonflants", c'est à dire de pouvoir adapter leur mode d'organisation en fonction du degré d'hydratation (smectites, vermiculites).
Cette propriété dépend toutefois grandement de la nature des cations compensateurs [24]. Dans les smectites, par exemple, si les cations compensateurs sont Na+ ou Li+ [24, 25], la quantité d'eau adsorbée entre les feuillets est telle que la distance
basale peut atteindre plusieurs centaines d'Angströms, et les feuillets peuvent même aller jusqu'à s'exfolier complètement dans l'eau [24, 25]. L'évolution de la distance basale caractéristique d'un empilement est alors un excellent indicateur du degré d'hydratation du minéral étudié [25, 26].
Malgré une structure Te Oc Te commune, les phyllosilicates 2:1 présentent de grandes hétérogénéités en ce qui concerne la nature et l'épaisseur de leur espace interfoliaire. Cependant, certains auteurs les qualifient de "phyllosilicates à 10 Å", en rapport avec l'épaisseur du seul feuillet Te Oc Te.
1.2.6. Phyllosilicates
2:1:1
La description du groupe des phyllosilicates s'achève par celle d'un dernier sous-groupe: celui des phyllosilicates 2:1:1. Ces minéraux sont constitués d'une alternance régulière de feuillets Te Oc Te (de type phyllosilicate 2:1) et de feuillets octaédriques (de type brucitique) (voir Figure 1. 9). Ils sont donc en conformation Te Oc Te Oc.
Te Te Oc Oc Te Te Oc Oc
Figure 1. 9 : Structure des phyllosilicates 2:1:1.
Le feuillet Te Oc Te est de type mica (voir chapitre 1.2.5), c'est à dire avec des substitutions de silicium de ses couches tétraédriques par des atomes trivalents (Al3+). Dès lors,
les défauts de charge du feuillet peuvent être compensés de deux manières distinctes, à l'origine de deux familles de phyllosilicates 2:1:1:
- groupe des chlorites (ou chlorites vraies), dans lequel les défauts de charges sont compensés directement par le feuillet Oc. S'il est de nature trioctaédrique, la génération d'un excès de charge positive se fait par substitution de cations divalents par des cations trivalents. S'il est dioctaédrique la compensation se fait par la présence d'un excès de cations trivalents dans le feuillet (cf. chapitre 1.2.2.2.).
Ce mode d'organisation engendre de fortes interactions entre les feuillets: à la fois de type Van der Waals (plans OH du feuillet Oc face aux plans O des couches Te), ainsi que de type ionique (voir Figure 1. 9). Les chlorites ne sont donc pas gonflantes et présentent une distance basale de l'ordre de 14 Å, dont certains auteurs se servent pour qualifier l'ensemble des phyllosilicate 2:1:1 de "phyllosilicates à 14 Å".
- groupe des pseudo-chlorites (ou chlorites gonflantes), dans lequel la compensation de charge se fait, comme dans les smectites, par la présence de cations compensateurs hydratés (le feuillet Oc restant électriquement neutre).
A contrario des chlorites vraies, les feuillets de pseudo-chlorite ne sont donc liés entre eux que par des interactions de Van der Waals. En outre, la présence d'eau entre les feuillets Oc et Te Oc Te écrante ces intractions, conférant à ces minéraux la propriété d'être gonflantes (comme les smectites).
Pour établir une classification des phyllosilicate 2:1:1, il aurait fallu, si nous avions suivi les divisions utilisées pour les autres sous-groupes, considérer les caractères di- ou trioctaédrique de chacun des feuillets constitutifs. Cependant, la très grande majorité de ces minéraux ont leurs deux feuillets constitutifs de nature trioctaédrique [21]. Nous nous contenterons de leur description pour établir une classification de ce sous-groupe (voir Tableau 1. 4), en rappelant toutefois que les autres espèces minérales existent mais sont peu représentées dans la nature.
Phyllosilicates 2:1:1 charge:
(e-/maille)
chlorites
(Si8-xAlx) Mg6 O20 (OH)4 , (Mg6-xAlx)(OH)12
0,4 à 2
pseudo-chlorites
(Si8-xAlx) Mg6 O20 (OH)4 , M+x.nH2O , Mg6(OH)12
0,4 à 1,2
Tableau 1. 4 : Classification des phyllosilicates 2:1:1 (feuillets Te Oc Te et Oc trioctaédriques).
1.3. Silicates de calcium hydratés (C-S-H)
1.3.1. Généralités
A contrario des phyllosilicates, les silicates de calcium hydratés (C-S-H) n'existent pas à l'état naturel, mais sont formés lors de l'hydratation du ciment.
Les ciments ont été utilisés dans l'Antiquité pour l'édification d'ouvrages monumentaux, leur conférant une durabilité exceptionnelle [27] (mélangés avec du sable pour faire du mortier, ou avec du gravier pour donner du béton). Mais ce n'est que tardivement que les ciments réapparaissent dans l'époque moderne. En effet, ce n'est qu'en 1756 que Smeaton redécouvre les propriétés hydrauliques de mélanges de calcaire et d'argile. Mais la production industrielle de ciment ne débute que sous l'impulsion d'Apsdin vers le milieu du XIXème siècle [28], offrant ainsi
à l'Angleterre en début d'industrialisation, un matériau de construction économique permettant de s'affranchir de la taille de la pierre. Il baptisa son ciment "Portland" en raison de sa ressemblance avec la pierre de la presqu'île de Portland (au Sud de l'Angleterre), appellation qui devait être conservée jusqu'à nos jours [29].
De nos jours, on utilise couramment pour la réalisation d'ouvrages du Ciment Portland Artificiel (dit CPA). Ce ciment est obtenu en ajoutant du gypse finement broyée à un clinker composé d'un mélange de 80% de calcaire et 20% d'argile porté à 1450°C.
Le CPA est composé de phases anhydres dont les teneurs massiques moyennes sont [30]:
silicate tricalcique Ca3SiO5 50 à 70%
silicate dicalcique Ca2SiO4 15 à 30%
aluminate tricalcique Ca3Al2O6 5 à 10%
alumino-ferrite tétracalcique Ca4Al2Fe2O10 5 à 10%
Tableau 1. 5 : Composition moyenne des ciments Portland.
Le CPA est un liant hydraulique, c'est à dire qu'il acquiert ses propriétés de cohésion et de durcissement par ajout d'eau. Ces propriétés sont dues principalement aux silicates de calcium hydratés, résultants de l’hydratation des principaux composants anhydres: le silicate tricalcique (Ca3SiO5) et le silicate dicalcique (Ca2SiO4).
1.3.2.
Synthèse des C-S-H
Il existe différentes méthodes de synthèse des C-S-H. La première étant bien sûr d'obtenir des C-S-H par hydratation des silicates de calcium présents dans le CPA. Cependant, la composition des C-S-H est très variable dans un ciment [30]. En effet, leur composition dépend à la fois des conditions d'hydratation du ciment (rapport eau/ciment) ainsi que du mode d'arrangement local des phases anhydres au moment de l'hydratation. De sorte que les C-S-H obtenus par hydratation du ciment ont une composition variable, pouvant s'écrire: (CaO)x(SiO2)y(H2O)z avec: 0,6<x/y<2 et 1<z<4 (noté CxSyHz en abrégé). Leur composition
moyenne est cependant proche de (CaO)1,7(SiO2)(H2O)4 [30], et peuvent également inclure
quelques impuretés comme des aluminates et des sulfates.
Un autre mode d'obtention de C-S-H est la méthode de synthèse par réaction dite pouzzolanique (nom donné par analogie avec la méthode de fabrication des ciments des Romains, réalisés à partir de chaux et de cendres volcaniques (riches en silice) de la région de Pouzzole). Cette méthode consiste à laisser mûrir une solution de chaux (CaO) et de silice vitreuse (SiO2), à partir de laquelle les C-S-H vont se former par mécanisme de
dissolution/précipitation. Les C-S-H obtenus, voient leur composition varier en fonction de la concentration d'équilibre des ions Ca2+ en solution. L'évolution du rapport x/y -ou Ca/Si- du
solide a été étudiée par de nombreux auteurs [30-34]. Leurs études montrent clairement l’existence de trois domaines distincts comme le montre la Figure 1. 10:
0 1 2 3 0 5 10 15 20 25 Ca/Si molaire Greenberg Fujii Taylor [Ca2+] en mmol.l-1
Figure 1. 10 : Rapport Ca/Si dans les C-S-H en fonction de la concentration d'équilibre en Ca2+
dans la solution.
- [Ca2+]<2.10-3M: domaine de coexistence du gel de silice et des C-S-H de Ca/Si=0,66
- 2.10-3M<[Ca2+]<22.10-3M: domaine d’existence des C-S-H de rapport 0,66<Ca/Si<1,7
- [Ca2+]>22.10-3M: domaine de coexistence des CSH de Ca/Si=1,7 et de la portlandite (Ca(OH) 2)
θ βcosλ =
L
Une autre méthode de synthèse des C-S-H est la précipitation. Elle consiste à faire précipiter le solide à partir de sels dissous servant à la fois de source de calcium et de silicium (chlorure de calcium, nitrate de calcium, métasilicate de sodium, etc.). En augmentant le pH au delà de 10, l'espèce [SiO(OH)3] - devient majoritaire en solution [35] et provoque la condensation
des C-S-H, selon la réaction: xCa2+ + ySiO(OH)3- + 2(x -y)OH
+ zH2O → CxSyHz.
Cette méthode permet, comme celle précédente, de contrôler la composition des C-S-H (rapport Ca/Si) à partir de la teneur des ions calcium en solution (voir Figure 1. 10).
1.3.3.
Microstructure des C-S-H
Pendant longtemps il a été difficile d'appréhender la structure des C-S-H et d'en donner une description cohérente.
En effet, en plus de présenter des variations très importantes de composition, les C-S-H présentent un diffractogramme des rayons X atypique, caractéristique d'un mode d'organisation intermédiaire entre celui d'un cristal tridimensionnel et d'un solide amorphe (voir Figure 1. 11).
En particulier, si l'on détermine la taille du domaine de cohérence des C-S-H en utilisant la loi de Scherrer [36], on peut mettre en évidence une organisation du matériau sur une centaine d'Angströms (avec: β largeur à mi-hauteur et θ position de la raie considérée).
0 1 0 2 0 3 0 4 0 5 0 6 0 in ten sité 2 t h e t a C u 5 , 4 5 Å 2 , 8 1 Å 2 , 1 1 Å 1 2 , 8 5 Å 1 , 8 3 Å 1 , 6 0 Å 3 , 0 6 Å 0 1 0 2 0 3 0 4 0 5 0 6 0 in ten sité 2 t h e t a C u 5 , 4 5 Å 2 , 8 1 Å 2 , 1 1 Å 1 2 , 8 5 Å 1 , 8 3 Å 1 , 6 0 Å 3 , 0 6 Å
Figure 1. 11 : Diffractogramme X d'un C-S-H de Ca/Si=0,83 obtenu par réaction
pouzzolanique.
La taille extrêmement restreinte du domaine de cohérence des C-S-H est un obstacle à une résolution structurale directe. De sorte que la diffraction des rayons X s'est montrée insuffisante pour résoudre leur structure (a contrario des phyllosilicates qui ont des domaines de cohérence bien supérieurs [20]). Elle rend en outre l'établissement de modèles structuraux difficile.
Malgré cela, de nombreux modèles tentant de décrire leur structure se sont succédés et complémentés, parmi lesquels nous pouvons citer ceux de: Taylor 1950 [37], Powers 1964 [38], Feldman 1968 [39], Ramachandran 1981 [40], Fujii 1983 [41], Taylor 1986 [42], Richardson 1992 [43], Cong et Kirkpatrick 1996 [44].
L'évolution des ces modèles, reflètent bien la gageure que représente la description d'un solide de composition variable, ordonné localement, et pouvant accommoder de nombreux défauts structuraux. Cependant, l'intégration progressive de données expérimentales acquises avec des méthodes de caractérisation de plus en plus performantes (MET [16, 45], FTIR [46], RAMAN [47], RMN du 29Si [44, 48-52], RMN de 17O [50, 53], EXAFS [54-56], SANS [57],
relaxation RMN [58-60]) permet aujourd'hui de présenter une structure moyenne cohérente, susceptible de représenter l'ensemble des conformations adoptées par les C-S-H.
Les C-S-H ont un mode d'organisation structurel multi-échelle (voir Figure 1. 12) [16, 44-60]: - Au niveau macroscopique, les C-S-H sont formés d'agglomérats.
- Les agglomérats sont composés de particules.
Comme les particules ont une faible extension planaire comparativement à leur hauteur, elles peuvent être assimilées à des "briques". Un agglomérat peut alors être assimilé à un empilement désordonné de briques.
- Une particule est composée d'un empilement d'un nombre restreint de lamelles (une dizaine de feuillets dans les conditions de synthèse les plus favorables).
- Chaque lamelle est composée de deux couches de chaînes de silicium en coordinence tétraédrique encadrant une double couche de calcium en coordinence pseudo-octaédrique, notée Te OcOc Te (voir chapitre 1.3.4).
particules: empilement de feuillets 10-8m feuillet: empilement de couches atomiques 10-9m agglomérat: assemblage de particules 10-6m minéral: assemblage agglomérats 10-2m particules: empilement de feuillets 10-8m feuillet: empilement de couches atomiques 10-9m agglomérat: assemblage de particules 10-6m minéral: assemblage agglomérats 10-2m
1.3.4.
Structure des lamelles de C-S-H
1.3.4.1. Unités structurales de base
Un modèle d'arrangement structural pour les couches de silicium est la wollastonite β-CaSiO3[19, 61]. Dans cet inosilicate, chacun des tétraèdres est lié par les sommets à deux voisins,
décrivant ainsi des rangées de chaînes infinies de tétraèdres parallèles entre elles (configuration de type Q2 , L1 [19]).
Les chaînes ont un motif de trois unités (dit "dreierketten"), dans lequel on peut distinguer deux types de silicium distincts (voir Figure 1. 13): ceux constituant un dimère (Q2), et celui
reliant deux dimères entre eux, dit "pontant" (noté Q2 P). Q2 P Q2 Q2 P Q2
Figure 1. 13 : Modèle de chaîne de tétraèdres de silicium dans les C-S-H.
Motif dreierketten représenté.
Les atomes de calcium adoptent quant à eux le type de conformation structurel commun à la tobermorite et la jennite [62, 63].
Dans ces minéraux, les couches d'atomes de calcium sont en coordinence pseudo-octaédrique (heptaédrique). Les octaèdres sont non réguliers et forment une structure planaire en deux niveaux. Les octaèdres de niveaux différents sont liés entre eux par les sommets, tandis que ceux d'un même niveaux sont liés entre eux par les arêtes (voir Figure 1. 14). La double couche est infinie selon son plan, c'est à dire sur la Figure 1. 14 selon les axes de la figure [100] et normal au plan de la figure [010].
Figure 1. 14 : Modèle d'agencement en double couche des pseudo-octaèdres de calcium dans les
1.3.4.2. Feuillet de tobermorite: un feuillet de C-S-H idéal
Les feuillets de C-S-H ont pour modèle ceux de tobermorite 11 et 14 Å [62, 64], de composition Ca4[Si6O16(OH)2].nH2O (avec 2≤n≤5).
d001 c c b a d001 c c b a
Figure 1. 15 : Feuillet et maille idéaux de C-S-H, vue selon l'axe [010] à gauche, et [100] à droite.
De tels feuillets (de rapport Ca/Si=0,66) sont composés d'une double couche d'octaèdres de calcium sur laquelle sont liées des rangées infinies de tétraèdres de silicium (voir Figure 1. 15).
Dans ce mode d'organisation tous les octaèdres de calcium sont liés à des polyèdres voisins (de calcium, ou de silicium). Les tétraèdres de silicium sont liées aux octaèdres de calcium de façon différente en fonction de leur nature: les tétraèdres du dimère du dreierketten engagent leurs deux liaisons restantes avec des octaèdres voisins, tandis que les tétraèdres pontants n'engagent qu'une seule liaison avec un octaèdre, la dernière liaison étant hydroxylée pour maintenir la neutralité électrique de l'édifice cristallin. Une maille orthorhombique primitive rendant compte de cette structure idéale est représentée sur la Figure 1. 15.
Cette maille peut aussi être décrite simplement par le schéma représenté Figure 1. 16. Ce schéma, communément utilisé, met en relief la structure Te Oc Te des C-S-H, et permet en outre de dénombrer facilement les atomes de la maille.
Remarquons que la distance basale d001 peut varier de 11 ou 14 Å en fonction de
l'hydratation de l'espace interfoliaire (qui dépend du mode de synthèse de la tobermorite).
Plan CaO Te Oc Te c b interfeuillet (eau) 4 Ca , 10 O 2 Si , 3 O 1 Si , 1 OH n H2O 2 Si , 3 O 1 Si , 1 OH feuillet tobermoritique, feuillet modèle de C-S-H: Ca4[Si6O16(OH)2].nH2O Plan CaO Te Oc Te c b interfeuillet (eau) 4 Ca , 10 O 2 Si , 3 O 1 Si , 1 OH n H2O 2 Si , 3 O 1 Si , 1 OH feuillet tobermoritique, feuillet modèle de C-S-H: Ca4[Si6O16(OH)2].nH2O
1.3.4.3. Trois modèles de feuillets pour les C-S-H réels
Il est à présent possible de proposer des modèles de feuillets de C-S-H en se basant sur la description du feuillet de tobermorite. Néanmoins, pour rendre compte de la structure pouvant être adoptée par un matériau de composition très variable (rapports Ca/Si allant de 0,66 à 1,7), il est nécessaire d'utiliser des sous-modèles.
Pour décrire complètement les C-S-H, nous utiliserons donc trois sous-modèles de rapports Ca/Si respectifs de: 0,66 ; 1,0 ; et 1,5 (chacun des sous-modèles pouvant être amené à évoluer de façon à mieux décrire la structure du matériau).
Modèle pour les C-S-H de faible Ca/Si (0,66≤Ca/Si<1):
Le modèle de feuillet pour les C-S-H de faible Ca/Si est conforme à celui de tobermorite, et a pour composition moyenne: Ca4[Si6O16(OH)2].nH2O.
Les chaînes de silicium n'y sont cependant pas infinies comme dans le modèle tobermoritique, mais leur longueur diminuent progressivement avec l'augmentation du rapport Ca/Si (typiquement plusieurs dizaines de tétraèdres pour Ca/Si=0,66).
La continuité des chaînes s'interrompt par lacunes de tétraèdres pontants Q2
P , ce qui fait
apparaître dans la structure (pour chaque lacune) deux tétraèdres de bout de chaînes, ainsi qu'une liaison CaOH. Les tétraèdres de bout de chaînes ne sont plus liés qu'à un seul tétraèdre de silicium, ils sont alors notés Q1 [19] (leur liaison pendante étant hydroxylée).
Pour les faibles rapports Ca/Si, le pH de l'eau de l'espace interfoliaire est élevé (pH≈10 [33]), mais insuffisamment basique pour déprotoner la liaison hydroxylée des sites SiOH.
Toutefois, à partir de rapports Ca/Si de 0,8 le pH augmente au delà de 11,5 [33], ce qui a pour conséquence de déprotoner une partie des liaisons hydroxylées des tétraèdres pontants et des tétraèdres de bout de chaînes. Pour maintenir la neutralité électrique de l'édifice cristallin, la charge négative du feuillet est compensée par la présence d'ions calcium dans l'espace interfoliaire (ce qui augmente légèrement le rapport Ca/Si).
Ci-dessus, un exemple de compensation de charge par ajout d'un cation Ca2+ dans une
maille de C-S-H dans laquelle les deux tétraèdres pontants sont déprotonés. Cette maille, a une
4 Ca , 10 O 2 Si , 3 O 1 Si , 1 OH n H2O 2 Si , 3 O 1 Si , 1 OH
C-S-H Ca/Si=0,66: Ca4[Si6O16(OH)2].nH2O
4 Ca , 10 O 2 Si , 3 O 1 Si , 1 OH n H2O 2 Si , 3 O 1 Si , 1 OH
C-S-H Ca/Si=0,66: Ca4[Si6O16(OH)2].nH2O
4 Ca , 10 O 2 Si , 3 O 1 Si , 1 O -n H2O , 1Ca2+ 2 Si , 3 O 1 Si , 1 O -C-S-H Ca/Si=0,83: Ca5[Si6O18].nH2O 4 Ca , 10 O 2 Si , 3 O 1 Si , 1 O -n H2O , 1Ca2+ 2 Si , 3 O 1 Si , 1 O -C-S-H Ca/Si=0,83: Ca5[Si6O18].nH2O
Modèle pour les C-S-H de Ca/Si intermédiaire (1≤Ca/Si<1,5):
Le modèle de feuillet des C-S-H de rapports Ca/Si intermédiaires peut être considéré comme un feuillet de type tobermoritique, dans lequel environ la moitié des tétraèdres pontants sont manquants (soit un par maille). Ce qui a pour incidence de réduire la longueur moyenne les chaînes de silicium à environ cinq unités.
Les liaisons pendantes des tétraèdres de fin de chaînes sont majoritairement déprotonées, l'excès de charge négative étant compensé par la présence d'ions calcium dans l'espace interfoliaire. Le schéma ci-dessus représente un C-S-H de Ca/Si=1 de composition moyenne Ca5[Si5O16(OH)2].nH2O.
Modèle pour les C-S-H de fort Ca/Si (Ca/Si≥1,5):
Les feuillets de C-S-H de forts rapports Ca/Si ne contiennent plus de tétraèdres pontants. Les "chaînes" de silicium sont réduites à un ensemble discontinu de dimères, dont la charge est compensée par la présence d'un ion calcium par dimère (soit deux par maille). Le schéma ci-dessus représente un C-S-H de rapport Ca/Si=1,5 et de composition moyenne Ca6[Si4O16(OH)2].nH2O.
Toutefois, il est aussi possible d'observer des C-S-H de rapports Ca/Si supérieurs à 1,5. La structure de ces feuillets de C-S-H est similaire à celle des feuillets de Ca/Si=1,5 mais présente en outre des lacunes de dimères entiers (ce qui augmente le rapport Ca/Si du feuillet). Ces feuillets ont donc localement une structure de type jennite [30, 63].
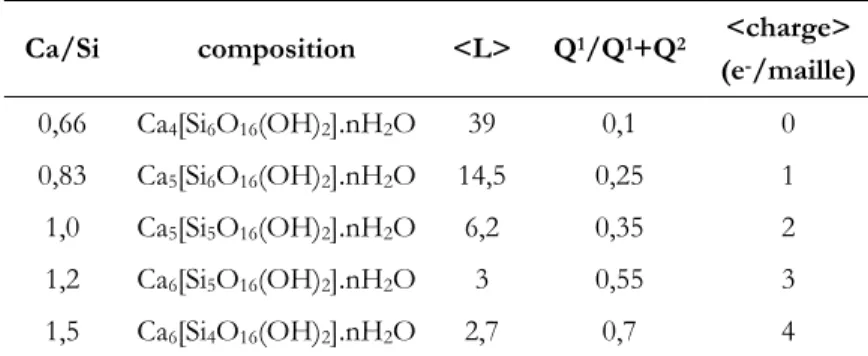
Nous nous proposons d'illustrer ces modèles par les travaux de Cong et Kirkpatrick, qui ont mené de larges investigations sur un ensemble de C-S-H obtenus par différentes méthodes de synthèses [44, 47, 49, 50, 53, 54]. Nous nous intéresserons en particulier aux résultats qu'ils ont obtenus pour des C-S-H de différents rapports Ca/Si, synthétisés par réaction pouzzolanique pendant six mois à 40°C [44].
Le Tableau 1. 6 reporte l'évolution de la longueur moyenne des chaînes de silicium <L> (avec L=2(1+Q2/Q1)), de la proportion de sites Q1 dans les chaînes de silicium (rapport
Q1/Q1+Q2), ainsi que la charge moyenne du feuillet, en fonction du rapport Ca/Si des feuillets
de C-S-H . 4 Ca , 10 O 2 Si , 2 O-, 1 O , 1 OH n H2O , 1 Ca2+ 2 Si , 3 O 1 Si , 1 OH
C-S-H Ca/Si=1,0: Ca5[Si5O16 (OH)2].nH2O
4 Ca , 10 O
2 Si , 2 O-, 1 O , 1 OH
n H2O , 1 Ca2+
2 Si , 3 O 1 Si , 1 OH
C-S-H Ca/Si=1,0: Ca5[Si5O16 (OH)2].nH2O
4 Ca , 10 O
n H2O, 1Ca2+
2 Si , 2 O-, 1 O , 1 OH
2 Si , 2 O-, 1 O , 1 OH
1Ca2+
C-S-H Ca/Si=1,5: Ca6[Si4O16 (OH)2].nH2O
4 Ca , 10 O
n H2O, 1Ca2+
2 Si , 2 O-, 1 O , 1 OH
2 Si , 2 O-, 1 O , 1 OH
1Ca2+
Notons que la composition moyenne de la maille des feuillets de C-S-H, ainsi que leur charge moyenne ne proviennent pas de données expérimentales, mais sont proposées à partir des modèles structuraux de feuillets venant d'être présentés.
On peut en outre remarquer que les auteurs n'ont pas reporté dans leur étude l'évolution de la distance basale d001 des C-S-H en fonction de leur rapport Ca/Si. Ils constatent même, que
quelque soit l'échantillon, l'évolution de la distance basale est indépendante du rapport Ca/Si du matériau, et se situe toujours entre 10 et 14 Å. Ils suggèrent ainsi que la distance basale, donc le degré d'hydratation de l'espace interfoliaire, est plus sensible au mode de synthèse du matériau qu'à la structure du feuillet lui même.
Ca/Si composition <L> Q1/Q1+Q2 <charge>
(e-/maille) 0,66 Ca4[Si6O16(OH)2].nH2O 39 0,1 0 0,83 Ca5[Si6O16(OH)2].nH2O 14,5 0,25 1 1,0 Ca5[Si5O16(OH)2].nH2O 6,2 0,35 2 1,2 Ca6[Si5O16(OH)2].nH2O 3 0,55 3 1,5 Ca6[Si4O16(OH)2].nH2O 2,7 0,7 4
Tableau 1. 6 : Evolution de la structure des chaînes de silicium des C-S-H en fonction de leur
rapport Ca/Si, d'après Cong et Kirkpatrick [44].
1.3.4.4. Paramètres inconnus dans la structure des C-S-H
Il est nécessaire de rappeler ici que la structure réelle des C-S-H n'a pas été résolue. Les modèles structuraux présentés précédemment sont certes cohérents avec les données expérimentales acquises sur le matériau et rendent compte de sa composition variable, mais ils restent incomplets.
En effet, des incertitudes sur l'organisation structurales des C-S-H demeurent sur trois domaines: - la structure du feuillet
- la nature de l'espace interfoliaire
- le mode d'empilement des feuillets au sein d'une particule
• La structure du feuillet est le domaine sur lequel on bénéficie du plus grand nombre de données expérimentales, et est donc relativement bien appréhendé. Cependant la proportion réelle de sites SiOH déprotonés en fonction du pH n'est pas clairement établi. Cette proportion influe à la fois sur la charge portée par le feuillet et sur le nombre moyen d'atomes de calcium compensateur dans l'interfeuillet.
En outre, aucun défaut structurel (autre que les lacunes de tétraèdres pontants), n'a été considéré à ce jour, que ce soit pour les chaînes de silicium ou pour la double couche de calcium.
En particulier, on peut tout à fait considérer que certains tétraèdres pontants des C-S-H peuvent effectuer une rotation autour de leur axe ("tilt"). Cette rotation fait apparaître deux sites SiOH et un site CaOH (voir Figure 1. 17). En fonction des conditions de pH de l'eau interfoliaire [33], tout ou partie des sites SiOH peuvent alors être déprotonés, modifiant ainsi la répartition des charges du feuillet.
Figure 1. 17 : Rotation d'un tétraèdre pontant dans un chaîne de C-S-H.
Les sites hydroxylés sont représentés par une sphère.
• La nature de l'espace interfoliaire est relativement mal appréhendée car il est très difficile de contrôler précisément son degré d'hydratation. En effet, comme il l'a été présenté au chapitre 1.3.3, les agglomérats forment un agencement désordonné et non compact de particules. Cet agencement présente différents niveaux de porosité saturés d'eau (la surface spécifique des C-S-H étant de l'ordre 500 m2.g-1). Dès lors, il est très difficile de déterminer le degré d'hydratation de
l'espace interfoliaire seul.
De plus, des incertitudes sur la position et nature des cations compensateurs Ca2+
demeurent. En particulier, des études tendent à monter que, contrairement aux cations compensateurs des phyllosilicates 2:1 hydratés, ils ne sont que partiellement échangeables [65-67]. Ce qui soulève des interrogations quant à leur caractère labile dans la structure.
• Enfin, si le nombre moyen de feuillet constituant un empilement peut être déterminé (typiquement de 3 à 10 feuillets), les modes d'orientation des feuillets les uns par rapport aux autres au sein d'un empilement (polytypie) n'ont jamais été étudiés.
En l'absence d'études, nous considérerons que les C-S-H adoptent les configurations orientationnelles les plus désordonnées (c'est à dire forment des empilements turbostratiques).
1.4. C-S-H et phyllosilicates 2:1
Nous pouvons maintenant établir une comparaison entre les C-S-H et les phyllosilicates. En particulier, nous comparerons précisément les C-S-H avec les phyllosilicates 2:1 hydratés (smectites et vermiculites).
Ces matériaux partagent en premier lieu le fait d'être des silicates hydratés lamellaires. Ils partagent de plus le même mode d'organisation structurale multi-échelle, et sont formés d'empilements de feuillets.
Leurs feuillets sont d'épaisseur comparable (9,4 Å pour les C-S-H [64], et 9,6 Å pour les phyllosilicates 2:1 [20, 21]). Ils sont composés d'un agencement de couches de type Te Oc Te, et sont chargés négativement. En outre, leurs interfeuillets sont composés d'eau et de cations compensateurs.
Du fait de leur mode commun d'organisation, ces deux matériaux sont souvent associés. Néanmoins ces ressemblances cachent des différences structurales importantes, qui engendrent des propriétés macroscopiques très différentes.
En premier lieu, la taille des domaines de cohérence ne sont pas comparables: quelques dizaines de nanomètres pour les C-S-H, à quelques micromètres pour les phyllosilicates 2:1.
De même, les empilements de feuillets de C-S-H sont constitués d'au plus une dizaine d'unités, tandis qu'ils peuvent atteindre des centaines d'unités pour les phyllosilicates.
En ce qui concerne la structure individuelle des feuillets, si les deux matériaux partagent une organisation de type Te Oc Te, il faut néanmoins préciser qu'elle est de type Te(Q1 Q2) OcOc
Te(Q1 Q2) pour les C-S-H et de type Te(Q3) Oc Te(Q3) pour les phyllosilicates. Ce qui a pour
conséquence que les C-S-H sont des sorosilicates et non des phyllosilicates (voir Figure 1. 18). Enfin, l'origine des charges des feuillets des matériaux est de nature différentes. Dans les phyllosilicates les charges sont créées par des substitutions isomorphes dans les feuillets. Ces défauts de charge peuvent être considérées comme délocalisés et répartis de façon homogène dans les feuillets. Dans les C-S-H les charges sont créées par la déprotonation des sites SiOH des chaînes de silicium. Ce sont des excès de charge qui sont localisées spécifiquement sur des sites de surface des feuillets.
![Figure 1. 15 : Feuillet et maille idéaux de C-S-H, vue selon l'axe [010] à gauche, et [100] à droite](https://thumb-eu.123doks.com/thumbv2/123doknet/2824613.67853/32.892.237.656.219.452/figure-feuillet-maille-idéaux-vue-axe-gauche-droite.webp)