HAL Id: hal-01593328
https://hal.archives-ouvertes.fr/hal-01593328
Submitted on 8 Nov 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Engineering of nano-crystalline drug suspensions:
Employing a physico-chemistry based stabilizer selection
methodology or approach
Jean-Rene Authelin, Mostafa Nakach, Jean-René Authelin, Tharwat Tadros,
Laurence Galet, Alain Chamayou
To cite this version:
Jean-Rene Authelin, Mostafa Nakach, Jean-René Authelin, Tharwat Tadros, Laurence Galet, et al..
Engineering of nano-crystalline drug suspensions: Employing a physico-chemistry based stabilizer
selection methodology or approach. International Journal of Pharmaceutics, Elsevier, 2014, 476 (1-2),
p. 277-288. �10.1016/j.ijpharm.2014.09.048�. �hal-01593328�
Engineering
of
nano-crystalline
drug
suspensions:
Employing
a
physico-chemistry
based
stabilizer
selection
methodology
or
approach
Mostafa
Nakach
a,*
,
Jean-René
Authelin
a,
Tharwat
Tadros
b,
Laurence
Galet
c,
Alain
Chamayou
caSanofiR&D,13,quaiJulesGuesde,VitrysurSeine94403,France b89NashGroveLane,Wokingham,BerkshireRG404HE,UK cEcoledesMinesd'Albi,CampusJarlard,RoutedeTeillet83013,France
Keywords: Topdownprocess Nano-crystallinesuspension Beadsmilling Stabilizer Wettingagent Dispersantagent ABSTRACT
Thispaperdescribesasystematicapproachtoselectoptimumstabilizerforthepreparationof nano-crystallinesuspensions ofanactivepharmaceutical ingredient(API). Thestabilizercanbe eithera dispersantoracombinationofdispersantandwettingagent.Theproposedscreeningmethodisaquick and efficientway to investigatealargenumberof stabilizersbased ontheprinciplesof physical-chemistryandemploysastepwiseapproach.Themethodologyhasbeendividedintwomainparts;the firstpartbeingfocusedonthequalitativescreeningwiththeobjectiveofselectingthebestcandidate(s) forfurtherinvestigation,thesecondparthasbeenfocusedonquantitativescreeningwiththeobjectiveto optimizetheratioandamountofwettinganddispersingagents,basedonwettability,surfacecharges measurement,adsorptionevaluation,process-abilityevaluationandstoragestability.
The results showed clearly that SDS/PVP 40/60% (w/w) (sodium dodecyl sulfate/poly(vinyl pyrrolidone))atatotalconcentrationof1.2%wastheoptimumstabilizercomposition,atwhichthe resultingnanosuspensionswerestableformorethan50daysatroomtemperature.
1.Introduction
Atpresent,thesmallmolecularentitiesproducedbythecurrent pharmaceuticaldiscoveryareshowinganincreasingtrendtoward pooraqueoussolubility(Lee,2002),(Sharmaetal.,2009),(Savjani etal.,2012).Such lowwatersolubilityisa challengetoachieve adequatebio-availability(Kipp,2004)afteroraladministration.It also limits the types of formulations suitable for parenteral administration (Wong et al., 2008). In the recent years, nano-crystallinesuspensionshavebeenappliedforthedeliveryofhighly water-insolubleactivepharmaceuticalingredients(APIs)(Shegokar
andMüller,2010),(Kawabataetal.,2011),(Singhetal.,2011),(Wang
etal.,2012).ByreducingtheparticlesizeoftheAPI,therateofthe dissolution whichisdirectlyproportionaltothespecificsurfacearea, aswellasthesolubilityoftheAPIcanbesignificantlyenhanced
(Kesisoglouetal.,2007;NoyesandWhitney,1897).IndeedOstwal–
Freundlichequation(Bormetal.,2006)showsthatthesolubility increasesexponentiallywithdecreaseofparticleradius,r. Signifi-cantincreaseinsolubilityistypicallyobservedwhenrislessthan 200nm.Inaddition,incontrarytotheformulationsmadefrompoor tolerablesolvents,suchasPolysorbate,theinjectabledosecanbe increased using nano-crystalline suspensions. Indeed they are essentiallymadeofpuredrugandtypicallyusearelativelysmall amountofexcipients(Bazile,2011)
Topdownprocess(particles sizereduction bymilling) is an efficientwaytopreparenano-crystallinesuspensions.Thismethod is the most commonly used one thanks to the possibility of controllingparticlessizebyaproperchoiceofwetting/dispersing agent, as well as by controlling the milling conditions (Leena
Peltonen, 2010).Thegeneratednano-crystalline particlesin the
dispersionmustbestabilizedagainstflocculation(Holthoffetal.,
1996; Lauten and Nystrom,2001)and crystal growth(Ostwald
ripening (Ostwald, 1901)), thus the selection of appropriate stabilizercompositionisacrucialstepinachievingstable nano-crystallinesuspensions.
The present paper proposes a physico-chemistry based method for the selection of suitable wetting/dispersing agent
* Correspondingauthor.Tel.:+33158932196;fax:+33158933210. E-mailaddresses:mostafa.nakach@sanofi.com(M.Nakach),
Jean-Rene.Authelin@sanofi.com(J.-R.Authelin),tharwat@tadros.fsnet.co.uk
(T.Tadros),laurence.Galet@mines-albi.fr(L.Galet),alain.chamayou@mines-albi.fr
formanufacturingnano-crystallinesuspensionsusingtop-down process.
2. Scientificbackgroundofformulationstrategy
AccordingtoTadros(2005),inaAPIpowderofanymaterialthe aggregates and agglomerates are held together by very strong attractive forces. When these aggregates and agglomerates are dispersedinaliquidmedium,theattractiveforcesarereducedbut stillsufficienttokeeptheparticlesstronglyattachedtogether.To separate theparticlein suchaggregatesoragglomeratesandto disintegratethem,acombinationofmillingprocessand supple-mentingwetting/dispersantagentsisrequiredtoovercomesuch attractiveforces.
Dependingonboththewetting/dispersantagentusedandon thedrugmaterialproperties,theparticlesizeswilldecreasedown toanequilibriumvalueduringmilling(watersolubility,density, molecularweight).Differentdrugswillrequiredifferentwetting/ dispersant agents. Approach for selecting the most suitable wetting/dispersantagentforspecificAPI,isthereforedesired.
From our point of view, an ideal wetting/dispersant agent shouldsatisfyseveralcriteria:
(i)Itshouldachieveamaximumreductioninthesurfaceenergy of the powder. In fact, Rehbinder and his collaborators investigatedtherole of surfactantsin thegrinding process. Theyfoundthat, asa resultofsurfactant adsorptionatthe solid/liquidinterface,thesurfaceenergyat theboundaryis reducedandthisfacilitatestheprocessofparticles deforma-tion or disintegration. The adsorption of surfactants at the solid/liquidinterfaceincracksfacilitatesthepropagationof this phenomena. This mechanism is referred to as the Rehbindereffect(Monteiroetal.,2013).Furthermore,foran efficientmilling,themigrationofwettingagentshouldbeas fastasthepropagationofcracks(Tadros,2005).
(ii)ThesuspensionshouldbestabletoavoidOstwaldripeningand flocculation or aggregation during the storage. Typically colloidalstability canbeobtainedeither byelectrostatic or steric stabilizationor a combination of both: “electrosteric stabilization”.Electrostaticstabilizationisbroughtby adsorp-tionofchargedspecies,likeionicsurfactantorphospholipids. Theefficiencyofelectrostaticrepulsioncanbeassessedfrom theknowledgeoftheionicconcentrationandionvalency,as well as by measuring the zeta potential of the particles
(Hunter,1988).Itiswellknownthattheelectrostaticrepulsion
increases with a decrease of electrolyte concentration, a decreaseofionvalencyandanincreaseofzetapotential(Adler etal.,2000).Thezetapotentialmeasurementsallow estimat-ing of colloidal suspension stability (Cosgrove, 2010). The colloidalsystemisstablewhenadominantroleisplayedby theforcescausingthemutualrepulsionoftheparticles.Higher is the absolute value of the zeta potential, greater is the probabilitythatthestudiedsuspensionwillbestable.Asmall valueof thezeta potential (from +5 to!5mV) indicates a tendency for the system destabilization (Iwona Ostolska, 2014).Reportedtypicalabsolutezetapotentialvalueforstable suspensionisbetween20and30mV,althoughvalueashigher as100mVcanbeobtained(Dery,2012).Non-ionicdispersants reduce flocculation through steric repulsion (Adler et al., 2000).Theseagents,mostlypolymers, formadsorbedlayers withthickness(
D
)whichisstronglyhydratedinwater.When twoparticleseachhavinganadsorbedlayerofthickness(D
) approacheachotheratasurface-to-surfacedistancehthatis smallerthan2D
,strongrepulsionoccursas aresultof two phenomena:(i)unfavorablemixingofthestabilizingchains when these are in a good solvent. (ii) Reduction of theconfigurational entropy on considerable overlap of the stabilizingchains(Fisher,1958;Sato,1980).
(iii)Minimizing Ostwald ripening: firstly, in order to limit the materialtransport,theAPIsolubilityshouldbemaintainedas lowaspossible.Indeed,duetoOstwaldripening,theaverage particle size may increase over time. The driving force for Ostwaldripeningisthehighersolubilityofsmallerparticles thanthelargerones(Hiemenz,1997).Thisresultsinashiftof theparticlesizedistributiontolargervaluesduringthestorage of nanosuspension, especially at higher temperatures. Sec-ondly,theadsorptionof polymeratthesurface ofparticles may also efficiently inhibit the crystal growth (Simonelli
etal.,1970).
(iv)Theformulationshouldbeeasytohandleandtoprocessina beadmill,itshouldinparticularnotbetooviscous.Indeed, rheological parameters arecritical duringmilling (Gordana
Matijasic and Glasnovic, 2008).High viscositymay require
longerprocessingtime(Leeetal.,2005).
3. Experimental 3.1.Materials
Amodelhydrophobicand nonionisablehighlyinsolubleAPI was obtained from Sanofi and the API was micronized by jet millingbeforeuse.Thephysico-chemicalpropertiesoftheAPIare providedinTable1.
Thedispersant/wettingagentsusedfortheinvestigationand theirintendeduseinsuspensionstabilizationarelistedinTable2
(SeeAppendixA).Severalchemicalcategorieswereused(cellulose
derivatives, povidones, phospholipides, poloxamers, polyethyl-eneglycoland derivatives,fattyacidsand fattyacidesters,SDS, sodiumployacrylate).
3.2.Methods
3.2.1.Preparationofnanosuspensions
Forthescreeningofdispersant/wettingagentsusinglowshear milling,a suspensioncontaining20%(w/w)ofAPI,3%(w/w)of dispersant/wettingagents,and77%(w/w)ofwaterforinjection (WFI) wasprepared.An aliquotof10ml ofthe suspensionand 20ml of Zirconium oxide beads(700
m
mdiametersupplied by Netzsch(Germany))wereintroducedin30mlglassvial.Thevial wasshakeninorbitalrollermillfor5daysat0.03m/sandatroom temperature.Fortheassessmentofprocessabilityusinghighshearmilling,a suspensioncontaining20%(w/w)ofAPI,3%(w/w)ofdispersant/ wettingagentsand77%(w/w)ofWFIwasprepared.Analiquotof 50mlsuspensionand50mlofPolymill1Cross-linkedPolystyrene
beadsmillingmedia(500
m
mdiametersuppliedbyAlkermes,Inc., (Waltham,MA,USA)wereintroducedinaNano-mill101millingTable1
Physico-chemicalpropertiesoftheAPI.
Averageparticlediameter 5mm Specificsurfacearea(m2g)** 1.5
Molecularweight(g/mol) 497.4 Watersolubility(mg/ml) 0.2
pKa NopKa
LogP* 6.9
Realdensity(g/ml) 1.42 Meltingpoint("C) 156.7 * Pisthepartitioncoefficientbetweenoctanolandwater. ** MeasurementisperformedusingBlainemethod(Kaye,1967).
system(AnnularmillpurchasedfromAlkermes,Inc.,(Waltham, MA,USA),havingastatorof80mmdiameterandarotorof73mm). Themillwasoperatedduring1hat20"Cand3m/s.
Forthe optimizationofthe %ofselected dispersant/wetting agent,asuspensioncontaining20%(w/w)ofAPI,thedispersant/ wettingagentsconcentrationvaryingbetween0.3and3%(w/w) andWFIqs100%(w/w).Analiquotof50mlsuspensionand50ml of Polymill1 Cross-linked Polystyrene beads milling media
(500
m
m diameter supplied by Alkermes, Inc. (Waltham, MA, USA)wereintroducedinaNano-mill101millingsystem(AnnularmillpurchasedfromAlkermes,Inc.(Waltham,MA,USA),havinga statorof 80mmdiameterand a rotorof 73mm).The millwas operatedat20"Cand3m/s.Themillingoperationwasperformed during105–240min.
3.2.2.Characterization
3.2.2.1. Measurementof specific surface areaof APIpowder. The specificsurfacearea(SSA)ofAPIpowderismeasuredusingBlaine apparatusBSA1suppliedbyACMEL,France.Themeasurementwas carriedoutatroomtemperature.TheSSAwasdeterminedbased onKozeny–Carman theory(Rigden,1947),bymeasuringtheair permeabilityofcompressedpowderbed.Therelationshipbetween thespecificsurfacearea(SSA)andtheflowtime(t)ofa known volumeofairinisgivenbyEq.(1):
SSA¼ K$
e
3=2r
s$ ð1!e
Þ$t1=2
ð0:1
h
Þ1=2 (1)whereKistheapparatusconstant[g1/2
$ cm3/2
$ s!1],
e
isporosityofthecompressedpowderbed,
h
isairviscosity(Pas),r
sistheabsolutedensityoftheAPI(g$cm3)andtistheflowtime(s). 3.2.2.2.Suspensionparticlessizemeasurement. Theparticlessize measurementwasperformedusingtwomethods:
(i)Dynamic light scattering, referred to as photon correlation spectroscopy(PCS),usingCoulterN4+1equipmentsuppliedby
Beckmancoulter(France).Themethodisbasedonmeasuring the intensity fluctuation of scattered light as the particles undergoBrowniandiffusion.Fromtheintensityfluctuation,the diffusioncoefficientDcanbecalculated,andfromwhich,the particle radius, r, is estimated using the Stockes–Enstein equation(Pecora,1985).Theparticlesizemeasurementswere carriedoutusingascatteringangleof90".Therefractiveindex was fixed at1.332. Thetemperature was fixed at20"C.The suspensionwas dilutedfrom20%(w/w)to0.1% (w/w)with purifiedwater.10
m
lofdilutedsuspensionwereaddedto1ml ofpurifiedwater.Theresultingsuspensionwasgentlymixedin DLScuvetteandthenplacedintothemeasuringcellofDLS.The measurementwasrepeated3times.(ii)Laser diffraction using Malvern Mastersizer 20001. This
methodisbasedonmeasurementofangleoflightdiffracted bytheparticles,whichdependsontheparticleradius,using Fraunhoferdiffractiontheory. Thismethodcanmeasure the particlesizesdownto1
m
m.Forsmallparticles,forwardlight scatteringismeasuredwiththeapplicationofMieTheoryof lightscattering.Table2
Listofwitting/dispersantagentusedfortheinvestigation.
Chemicalcategory Material Supplier Molarmass Expectedaddedvaluefor stabilization
Cellulosederivatives HydroxypropylmethylcelluloseHPMC(Pharmacoat1606) SEPPIC(France) Range:10,000–
1,500,000 Stericstabilization Klucel1Hydroxypropylcellulose:HPCHF Hercules(France) Average:
1,150,000
Stericstabilization
Povidones Luvitec1polyvinylpyrrolidone:PVP(K30) BASF(France) Average:50,000 Stericstabilization
PVP-VA(PlasdoneTMS-630)linearrandomcopolymerof
N-Vinyl-2-pyrrolidoneandvinylAcetate
Ashland(France) Average:27,000 Stericstabilization Phospholipids Phosal150PGcompoundof50%phosphatidylcholinefromsoybean
withpropylenglycol
PhospholipidGmbH (Germany)
775 Electrostatic stabilization Phospholipon190GPurephosphatidylcholinestabilizedwith0.1%
ascorbylpalmitate
PhospholipidGmbH (Germany)
758 Electrostatic stabilization Lipoid1S100phosphatidylcholinefromsoybean Lipoid(Germany) 787 Electrostatic
stabilization Poloxamers Poloxamer188Pluronic1F68 NFPrillBlockcopolymersbasedon
ethyleneoxideandpropyleneoxide
BASF(France) Range:7680– 9,510
Stericstabilization Poloxamer407Pluronic1F127NFPrillBlockcopolymersbasedon
ethyleneoxideandpropyleneoxide
BASF(France) Range:9840– 14,800
Stericstabilization Poly-Ethylene-Glycol
&derivatives
PolyethyleneGlycol8000PEG8000 Sigma–Aldrich(France) Average:8000 StericStabilization Solutol1HS15Macrogol15HydroxyStearate BASF(France) 813.2 Stericstabilization
VitamineETPGS1(d-AlphaTocopherylPolyethyleneGlycol
1000Succinate)
EastmanChemical Company(Netherlands)
1,513 Stericstabilization
FattyAcidsandFatty acidEsters
Cremophor1RH40Macrogol-Glycerolhydroxystearate BASF(France) Range:300–
6,000
Stericstabilization Montanov168CetearylAlcohol&CetearylGlucoside SEPPIC(France) N.A. Stericstabilization
Montanox180Ethoxylatedsorbitanester) SEPPIC(France) 1,310 StericStabilization
Gelucire14414Lauroylmacrogol-32glycerides Gattefossé(France) N.A. Stericstabilization
Simulsol1M49Polyethoxylatedcastoroil(PEG-20stearate) Seppic(France) 1,165 Stericstabilization
Others SoduimPolyacrylate1 BFGoodrichchemical(USA) 104,400 Electrosteric
stabilization Sodiumdodecylsulfate(SDS) Univar(France) 288.4 Electrostatic stabilization
By combiningtheresults obtainedwithlight diffractionand forwardlightscattering,theparticlesizedistributionsintherange 0.02–10
m
mcanbemeasured(Swithenbanketal.,1976).TheMastersizerisequippedwithlens havingfocallengthof 550mmandcellmeasurementhaving thicknessof2.4mm.The samplewasdilutedin100mlofpurifiedwaterandintroducedin MS1 sampler. The suspension was stirred at 1500rpm and recirculated through themeasurement cell. Thedilution factor wasadjustedinordertoensureanobscurationintherangeof2.5– 4.5. Themeasurements werecarried out at roomtemperature. Each measurement was performed during 20s and repeated 3times.TherefractiveindexoftheAPIandofdispersingwerefixed at1.61andat1.33,respectively.
3.2.2.3. Scanning electron microscopy (SEM) evaluation of suspension. The nanosuspension was diluted 10,000 times usingWFI.1mloftheobtainedsuspensionwasfilteredthrough Isopore1 (Polycarbonate)
filter having diameter of 13mm and porosityof0.1
m
m(suppliedbyMillipore,France).Thefilterwas thenrinsed3timeswith1mlofWFIforeachrinse.Thefilterwas then bondedtoanaluminumpadusingconductiveadhesiveon bothsides,andsubsequentlymetalizedwithgoldusingmetallizer Xenosput XE200 Edwards. The golddeposit was approximately 1.5–2nm thickness. For an overview and detailed view, nanoparticles were observed at 15kV using JEOL JSM-6300F1field emissionSEM(suppliedbySEMtechsolutionInc.,USA),at severalmagnifications(X1000,X5000,X10,000$20,000). 3.2.2.4. Suspensionstabilityassessment. The short-termstability wasmonitoredbymeasuringtheparticlesizeimmediatelyafter milling, after 7 days and after 15 days of storage at ambient temperature.
Fortheselectedformulation,thestabilitywasmonitoredover 8weeksatambienttemperature.
3.2.2.5. Zeta potential measurement of suspension. A Zetasizer NanoZS1(Malvern,UK),whichappliestheM3-PALStechnique,a
combinationoflaserDopplervelocimetry(LDV)andphaseanalysis light scattering (PALS), was used for the zeta potential measurements.TheequipmentemploysaHe–Nelaser(redlight of 633nmwavelength)which firstsplitsintotwo, providingan incidentandareferencebeam.
From the electrophoretic mobility,
m
, zeta potential,z
, is calculatedusingtheSmoluchowskiequation(Hunter,1988),thatis validwhenk$r>>1(wherek!1istheDebyelengthandristheparticleradius). Incase of smallparticlesand a lowelectrolyte concentration,theHuckelequationisapplicableforthecalculation ofzetapotential.
The sampletobemeasuredwasdilutedinpurifiedwaterto achieve a solid concentration in the range of 0.0001–0.1%. The obtained suspension was introduced in disposable cuvette (DTS1060) andgently mixed.Thecuvette was thenplaced into themeasuringcelloftheZetasizer.Thedilutionfactorwaschecked inordertogenerateaminimumcountrateof20,000countsper second.Themeasurementswereperformedatroomtemperature andrepeated3times.
3.2.2.6. Rheological measurement of unmilled suspension. Study state,shearstressversusshearratecurves,werecarriedoutusing HAAKE Viscotester1 VT550 supplied by HAAKE (Germany). A
concentric cylinderdevicewas usedfor thismeasurement. The measurement was carried out at 20"C. The shear rate was gradually increased from 0 to 1500s!1 (up curve) for over a
period of2minanddecreasedfrom1500to0s!1(downcurve)
over another period of 2min. The test samples were 25ml of unmilledsuspensioncontaining20%API(w/w),3%(w/w)stabilizer
and77%(w/w)ofWFI.Thesesampleswerehomogenizedbyusing an ultra-Turrax1 T-8 (suppliedby IMLAB France) for 10minat
6000rpm. The measurements were performed at room temperature.When it is a Newtonian system, theshear stress increaseslinearlywiththeappliedshearrate,andtheviscosityof thesuspensioncanbeobtainedfromtheslope.Inthiscase,theup and downcurves coincidewith each other. When, it is a non-Newtoniansystem,theviscosityofthesuspensiondecreaseswith theappliedshearrate.When,itisathixotropicsystem,thedown curveisbelowtheupcurveshowinghysteresis.Thelattercouldbe assessedbymeasuringtheareaundertheloop.
3.2.2.7. Surface tension measurement of wetting/dispersant solution. The surface tension
g
of selected dispersant/wetting agentwasmeasuredbyusingKRÜSSK121tensiometersuppliedbyKRÜSSGmbH(Germany).Inthesemeasurements,theWilhelmy platemethod(BiswasandMarion,2001)wasappliedunder quasi-equilibriumconditions.Therefore,theforcerequiredtodetachthe platefromtheinterface wasaccuratelydetermined.Fromthe
g
versuslogC,whereCisthetotalsurfactantconcentrationcurves, the critical micelle concentration (CMC) was determined. The measurementswerecarriedoutatroomtemperatureandrepeated 3times.3.2.2.8. Evaluation of wetting/dispersant agent. Wetting was assessed using the sinking time test method (Walker et al., 1952),aswellasbymeasuringtherateofpenetrationofwetting/ dispersantsolutionthroughapowder plugbasedonWashburn method (Aartsen, 1974; Chander and Hogg, 2007). By manual tappingaknownweighofpowderwasplacedinglasstube,inner diameterabout9.8mm(seeFig.1).Toensureaconstantpackingof thepowder,thetubewas alwaysfilledtothesameweight.The lowerendthetubewasclosedwithaglassfilter.Thehigherendof thetubewas hanging onweighingscale platform(precision of ' 0.1mg).Themassof theliquid penetratedwithinthepowder plug was measured when thelower end of the tube is placed verticallyinthewettingliquid.Theexperimentswereperformed at22' 1"C.Eachexperimentwasrepeated3times.
Fromtheslopeofthelinearrelationshipbetweenthesquare penetratedliquidweightand timethewettabilityfactor canbe calculatedusingthefollowingequation:
H2¼ 2
g
h
$ C$ R$ cosu
$ t (2) whereHistheheightoftheliquidpenetratedwithinthepowder plug,u
isthecontactangle,g
isthesurfacetensionoftheliquid,h
is theliquidviscosity,Risthemeanradiusofthecapillarywithinthe powderplug,Cisthetortuosityfactor,andtisthetime.Sinceallpowderplugswerepreparedatthesamecompressionpressure, theparameterCisassumedaconstant.
TocalculateH2,therelationshipofthemass(m)andtheheight
oftheliquidpenetratedwithinthepowderplugwasused.Itcanbe expressedbythefollowingequation:
m¼ H$ S$
e
$r
(3)where,misthemassoftheliquidpenetratedwithinthepowder plug,Histheheightoftheliquidpenetratedwithinthepowder plug,
r
isthevolumetricmassoftheliquid,Sisthesurfaceofthe powder plug, ande
is the fraction of the dead volume of the powder.CombiningEqs.(2)and(3),thefollowingequationisobtained: m2 ¼
g
$r
2h
$ S2 2 $C$ R$e
2$ cosu
$ t (4)Fromaplotofm2versustime(linearcurve),theslope(d(m2)/
dt)canbedeterminedandthewettabilityfactorcanbecalculated fromaknowledgeofthesurfacetension(
g
)andtheviscosity(h
)of theliquid.The wetability factor can be expressed by the following equation: d!m2" dt $
h
g
# $ ¼ K¼ s 2$r
2$ C$ R$ cosu
2 (5)3.2.2.9.AdsorptionisothermmeasurementofPVP. Theadsorption isothermoftheselecteddispersant,namelyPVP,wasmeasuredat roomtemperature.KnownamountsoftheAPIwereintroducedin vials at roomtemperature with various concentrations of PVP solutions.Then,thevialscontainingthevariousdispersionswere rotatedfrom a few hours to up to 15h until equilibrium was achieved.Indeed,itisreportedthattheintrinsicadsorptionkinetic ofhomopolymerisusuallyinstantaneous(lessthan1h)(Dijitand
CohenSturat,1992;Somasundara,2006)
Thentheparticleswereremovedfromthedispersantsolution bycentrifugationat3000rpmduring20min.Thesupernatantwas thenfilteredthrough0.45
m
mPVDF1filter(suppliedbyMillipore). Thedispersantconcentrationinthesupernatantwasdetermined usingUVspectrometrybyCary150(UV–visspectrophotometer
supplied by Varian Australia) at 200nm wavelength. Each measurement was repeated 3 times. To obtain the amount of adsorptionperunitareaofthepowder(
G
),thespecificsurfacearea of thepowder (A) in m2/g was determined usingthe gasflow method(Blaine).
3.2.2.10. Methodology for selection of wetting/dispersant agent. Nowadays, the general strategy (‘fast-to-patient’) in pharmaceuticalindustryistotestanewAPIina targetpatient populationasquicklyaspossible(Pritchard,2010).Thescreening methodology,basedonlyonphysico-chemistry,wouldprovidea lotofscientificinformationbutwouldbeverytimeandresources consumingandconsequentlynotalignedwiththefasttopatient strategy.Incontrary,apurelyempiricalmethodology(e.g.design ofexperiment,trialerrorapproach)mayprovideaquicksolution with poor scientific information. The typical risk of such methodologyisthatdue tothelackofscientificunderstanding, longtermstabilityorformulationrobustnessarenotanticipated and lead tounfordable formulation development. Our proposal hereistouseacompromisebetweenpurelyscientificandpurely empiricalmethodologyinordertoachievebothtimeeffectiveness andscientificinformation.
The proposed screening methodology derived from the previous theoretical considerations was divided in two major parts:
(i)Part 1 focused on qualitative screening to select a lead generation:in thispartseveralscreeningtestswereapplied tothementionedlistofwetting/dispersantagents(Table2).
(ii)Part2focusedonquantitativescreeningaimedtooptimizethe selectedlead: acustomizedquantitative optimizationof the amount of wetting/dispersant agent, based on wetting, on
adsorptionandonprocess-ability.Asinthestudiedcasethe SDS/PVPassociationwaschosenafterPart1,thePVPandSDS ratioandtheiramountwereoptimized.
ThetestsaresummarizedinFig.2anddescribedinmoredetails hereafter.
InPartIalltestedsuspensionscontained20%ofAPI,whichis sufficient from a process productivity point of view,and 3% of wetting/dispersantagentareused(3%ofwetting/dispersantagent should besufficient tomeet a fullcoverageof particles having approximately80nmmeandiameterassumingatypical
adsorp-tion of 3mg/m2 (Tadros,2012)). In this partthe selection was
performedbythefollowingstepbystepapproach:
(i)Step#1.1:Atthissteptherollermillisusedinordertoperform severaltestsinparallelwithareducedamountofproduct.Two criteriawereusedtoselecttheoptimumstabilizer.Thefirst criterionisthattheAPIparticlemeandiameterhastobeinthe rangeof100–500nmaftermilling(typicalnanocrystalssize
(Leena Peltonen, 2010)). The second criterion is that the
formulation should be free of flocculation upon at least 2weeksstorageatroomtemperature.Theprepared suspen-sions were assessed by visual observation, particle size measurement and stability after 2 weeks storage at room temperature.
(ii)Step#1.2:Atthisstepthemeasurementofzetapotentialof
selectedsamplesfromstep#1.1werecarriedout.Toensure electrostaticrepulsion,anabsolutevaluegreaterthan15mV wasfixedascriterion.
(iii) Step#1.3:Atthisstep,theprocessabilityofselectedsamples fromstep#1.2wasassessed.Thisevaluationwasdonebased onthefollowingtests:
a)Step#1.3a:Atthisstep,theviscosityof unmilledsuspension wasmeasuredasafunctionofshearrateaswellasofthixotropy. Thesamplesthatgaveviscositygreaterthan10mPasatshear rateof1000s!1wereexcluded.Indeedourinternalobservations
evidencedthatthiscriterionisessentialtoensurefastermilling kineticsaswellasmanufacturing-abilityatindustrialscale.
b)Step#1.3b:Atthisstep,themillingabilityusingthehighshear mill,namelyNano-mill101millingsystemofselectedsamples
fromstep#1.3awasassessed.Thisstepisessentialtoensurethe preparation of nanosuspension atindustrial scale usinghigh speedmilling.Allthesamplesthathadparticlesizegreaterthan 500nmorthatshowedinstabilityduetoflocculationorOstwald ripeningwereexcluded.
FromthepartI,thecombinationofSDS/PVPappearedassuperior totheothertestedagents.Therefore,thiscombinationwasselected forfurtherevaluationinthepart2asdescribedbelow:
(i)Step#2.1:AtthissteptheSDS–PVPratiowasoptimized.The synergistic effect of the combination was confirmed by performingthefollowingtests:
c) Step#2.1a:Atthisstep,bothsurfacetensionandcriticalmicelle concentration(CMC)ofsolutionsmadeofSDS–PVPatdifferent ratioweremeasured.Indeed,Cabane(1977)demonstratedthat the poly(ethy1ene oxide) chains are able to capture SDS monomerand micelles.Theyhaveshowntheexistenceofan optimalSDS/PEOratiowhichmaximizestheinteractionofboth components.Thebestassociationcanbeeasilydeterminedby measuringthesurfacetension.
d) Step#2.1b:Atthisstepthezetapotentialofsuspensionsmade ofSDS–PVPatdifferentratioweremeasured.
(ii)Step#2.2:Atthisstep,theamountofselectedSDS–PVPratio was optimized. This optimization was done based on the followingtests:
a) Step#2.2a:AtthisstepthewettabilityofselectedSDS–PVPratio was evaluated in order to select the amount of SDS–PVP allowinga maximumreduction in thesurface energy ofthe powder.
b)Step#2.2b:Atthisstep,ahighshearmillingofsuspensionmade ofselectedSDS–PVPratioatdifferentamountwasevaluated. Fromthisstepanoptimalamountthatgavesuspensionwitha highimplicitspecificsurfaceareawasselected.
c) Step#2.2c: Atthisstep,theadsorptionisothermofPVP was measuredtoensure the strong adsorption of thedispersant (PVP)ontheparticlessurface.
(iii)Step#2.3:Atthisstep,thephysical stabilityof theselected formulation wasevaluated. This wasassessed by monitor-ing the particle size distribution during 8 weeks at room temperature.
4. Resultsanddiscussions
4.1.Part1:Qualitativescreeningevaluation(leadgeneration)
(i)Step#1.1:Assessmentofmillingabilityusinglowshearmill (rollermill).
After roller milling, all samples were inspected for API suspendability. HPC, PEG 8000, Montanov1 68 and Sodium
polyacrylate1resultedinanobvious
flocculationandin appear-anceofa‘dry’sample.Theparticlessizeofthesesuspensionswas notmeasured.
The remaining samples were assessed by measuring the particlessizeattime0,7and14days.Theresultsareshownin
Fig.3.Suspensionswithaparticlessizegreaterthan500nmand/or showing flocculation after 7 days were not evaluated further. Discarded samples were those made with HPMC, Poloxamer 188andPoloxamer407.
(ii)Step #1.2: Assessment using surface charges measurement
(Zetapotential).
Fig.3. Lowshearmillingevaluationusingrollermillduring5daysat0.03m/sand atroomtemperature.Thefigurerepresentstheaverageparticlessizeatinitialtime, at7andat14daysstorageatroomtemperature(mean'S.D,n=3).
In the present study, the results of the zeta potential measurements of selectedwetting/dispersant agents from step #1.1 were collected and compared. Fig. 4 shows that all the wetting/dispersantagents,excepttheCremophor1RH40,gavean
acceptablezetapotentialvalue.Onecanobservethatthecharged species(SDS,PVP–SDS)leadtoa highabsolutevalue.Therefore, uponthissteptheCremophor1RH40wasexcluded.
(iii)Step#1.3Processabilityassessment.
The evaluation of processability was done in two steps: Rheological evaluation to select the wetting/dispersant agents thatwillpromotethemillingprocessandhighshearmillingability ofsuspensions madefromselectedwetting/dispersantagentsto selectthosethatwillleadtostablesuspension
a)Step#1.3a:Assessment usingtherheologicalbehaviorofthe suspension(Step#3a).
Fig. 5 and Fig. 6 show typical flow curves of unmilled suspensions prepared using Solutol1 HS15 and Phosal1 50PG.
ThepreparedsuspensionusingSolutol1HS15showsaNewtonian
behavior with a low viscosity of 2.8mPas. In contrast, the suspension using Phosal1 50 PG showed a non-Newtonian
behaviorwithaclearthixotropy,indicatingaflocculationofthe suspension.
The Table 3 summarizes the rheology results of various
dispersants.Suspensionswithviscositygreaterthan10mPasat highshearrateof1000s!1wereexcludedfromfurtherevaluation.
In fact, we have demonstrated that milling a suspension at differentviscosityleadstothefollowingobservation:higherthe viscosityofthesuspension,sloweristhemillingkinetic (unpub-lisheddata).Thismayimpacttheproductivityatindustrialscale. From this step, the suspensions made of Phosal1 50 PG,
Phospholipon190andLipoid1S100wereexcluded.
b)Step#1.3b:Assessmentofmillingabilityusinghighshearmill (Nano-mill101millingsystem:Step#3b).
After highshear milling, the suspensions were assessed by measuringtheparticlessizeattime0,7and14days.Theresultsare showninFig.7.
Twosystems,SDS/PVPataratioof70/30andVitaminETPGS1
provided the highest stabilization of the nano crystalline formulations.TheseresultswereconfirmedbySEMmeasurement as illustratedin Fig.8 for suspensions preparedusing SDS/PVP (Fig.8A)andusingMontanox180(Fig.8B).TheseSEMpictures
showed significantdifferences betweenthe2formulations.The needlesshapedcrystalsobservedwithMontanox180formulation
depict an anisotropic crystal growth (Ostwld repining). This is likelylinkedtopreferentialadsorptionofMontanox180oncrystal
faces which are parallel to the crystal axis. However, the suspension made of SDS/PVP presents a small, but irregular shapedparticles.Furthermore,itcanbeobservedthattheparticles
Fig.5.Flowcurves(shearstressasfunctionofshearrate)ofunmilledsuspension preparedusingSolutol1HS15.ThesuspensionshowsNewtonianbehaviourwitha
lowviscosityof2.8mPas(n=1).
Fig.6.Flowcurves(shearstressasfunctionofshearrate)forunmilledsuspension preparedusingPhosal150PG.ThesuspensionshowsnonNewtonianbehaviour
withclearthixotropy,indicatingflocculationofthesuspension(n=1).
Table3
Summaryofrheologicalresultsfordifferentdispersant/wettingagents. Dispersant/wettingsystem Viscosityat1000s!1(mP)
Phosal50PG 11 Phospholipon90G 12 LipoidS100 15 SolutolHS15 2.8 VitamineETPGS 4.5 PVP 4.6 SDS–PVP30/70 3.6 PVP/VA 4.0 Montanox80 2.3 Gelucire44/14 3.4 SimulsolM49 2.8 Fig.4.Resultsofthezetapotentialmeasurementscarriedoutonselectedwetting/
dispersantagentsfromstep#1.1.Cremophor1RH40showsasmallvalueofzeta
sizeofthesuspensionusingPVP-VAincreasedfrominitialtimeto 7 days andthen decreased at14 days.Thismaybedue tothe relaxation of the suspension. Indeed, it was reportedfor some formulationsthatwhenmillingisstopped,nanoparticles agglom-eratedwithinafewdays.Theparticlessizecontinuedtoincrease untilreachingamaximumsize.Thereafter,thesuspensionrelaxed spontaneouslywithanapparentsizereduction(Dengetal.,2008) Insummary,PVPalone,Mantanox180,andSimulsol1M49,as
they did not meet the optimization selection criteria, were discontinuedfromthisstudy.
Attheendofthisscreeningpart,twoformulationsareclearly superior:SDS/PVPandVitaminETPGS1formulations.Inorderto
illustratetheleadoptimizationpart,theSDS/PVPmadefromionic surfactant (SDS) andpolymer (PVP) associationwas considered morerelevant.Therefore,itwasselectedforthesecondpartofthe methodology.
4.2.Part2:Quantitativescreeningevaluation(leadoptimization) Inthispart,theSDS–PVPratioisfirstlyoptimizedbasedonboth wettabilityandzetapotential.TheamountofSDS–PVPofselected ratioisthenoptimizedtoensurethemillingprocessabilityanda longtermstabilityunderstorageconditions.
(i)Step#2.1:OptimizationofPVP–SDSratio.
a) Step#2.1a:Surfacetensionmeasurements.
Fig.9showsthe
g
-logCcurveforatypicalSDS/PVPmixture (20–80%).Thisgraphshowsatypicalbehaviorwithg
decreasing withlogCincreaseuntiltheCMCisreachedafterwhichg
shows onlyasmalldecreasewithincreaseoflogC.AplotofCMCversus%ofPVP(Fig.10)inthebinarymixtureshowsaminimumat60%of PVP.Thisresultimpliesamaximumofsurfaceactivityat60%of PVPinthebinarymixture.
b)Step#2.1b:Zetapotentialmeasurements.
TheFig.11outlinedthatintheabsenceofSDS,PVPaloneresults inalownegativezetapotentialof!20mV,whichisnotsufficient toinduceelectrostaticstabilization.Inthiscase,themainstability isobtainedfromastericrepulsionasaresultoftheadsorbedloops andtailsofPVPmolecules.UponadditionofSDS,thezetapotential decreased sharply to !50mV, which likely contributes to the stabilitythroughelectrostaticrepulsion.Withfurtherincreaseof SDS concentration to40% (40–60SDS!PVP), thezeta potential decreased downto!54mV andremains almostconstant. Thus whenusingamixtureofSDSandPVPthestabilizingmechanismis acombinationofelectrostaticrepulsion,whichshowsamaximum energyatintermediateseparationdistance,andstericrepulsion thatoccursatshorterdistancesofseparationthatiscomparableto twiceoftheadsorbedlayerthickness.Thiscombination stabiliza-tion mechanism is referred to as electrosteric stabilization
(Napper,1982;Tadros,1985;Tadros,1982)
Basedonthesetwoevaluations,theSDS–PVPataratioof40– 60% appeared to provide the best association of the two components. Therefore, it was selected for the quantitative optimization.
(ii)Step#2.2:AmountoptimizationofPVP–SDSatratioof40–60% w/w.
(a)Step#2.2a:Wettabilitymeasurements.
Fig.7.Highshearmillingevaluationofusingselectedwetting/dispersantagents fromStep#1.3a.MillingwasperformedusingNano-mill101system(Annularmill)
at3m/sandatroomtemperature.Thefigurerepresentstheaverageparticlessizeat initialtime,after7and14daysstorageatroomtemperature(mean'S.D,n=3).
Fig.9.SurfacetensionasfunctionofSDS/PVP(80–20%w/w)concentration(g-logC curve)obtainedusingWilhelmyplatemethod.Theadditionwetting/dispersant agentdecreasesthesurfacetensionofthesolution,asafunctionofthetotalits concentration.Abovethecriticalmicelleconcentration,nofurtherdecreasein surfacetensionisdetected.
Fig.8. Scanningelectro-microscopypicturesofsuspensionsproducedusingPVP–SDS(70/30%w/w)(A)andMontanox180(B)at3%w/wusingNano-mill101millingsystem
Fig.12showsthevariationofthesquareofpenetratedliquid weightversustime forthe40–60%(w/w)SDS–PVPsystem ata totalconcentrationof1.2%.Fig.13showsaplotofwettabilityfactor K versus SDS–PVP concentration. For comparison, the results obtainedusingSDSaloneareshowninthesamegraph(triangle scatter).ItcanbeseenonFig.13thatKincreaseswithincreaseof surfactantconcentration,reachingaplateauatacertainsurfactant concentration.FortheSDS/PVPsystem,thisplateauisreachedat 1.2%consisting of 0.72%PVP and 0.48% (w/w). Using thesame concentrationofSDSalone(0.48%w/w),theKvaluewasfoundto
bemuchlowerthanthatobtainedwiththecombinationsystem. Thisclearlydemonstratesthesynergisticeffectobtainedwhena polymer surfactant mixture is used. The latter is much more effectivewettingsystemascomparedtoindividualcomponents. By using 1.2% of SDS–PVP at the ratio of 40–60%, maximum reductioninsurfaceenergycanbeexpectedforthepowder-liquid interface, which will lead to an enhanced cracks propagation (Rehbindereffect),andanenhancedbreakageoftheparticlesupon wetmillingprocess.
(b)Step#2.2b:Millingabilityasfunctionof%ofPVP–SDSatratio of60–40%(w/w).
Themillingabilitywasinvestigatedusingakineticexperiment wherethereductioninparticlessize,ortheequivalentincreasein implicitspecificsurfacearea,wasmeasuredasfunctionofmilling time.AtypicalresultisillustratedinFig.14using1.2%w/wPVP– SDS at the ratio of 40–60%. The results obtained show an exponential increase in the implicit specific surface area (or decreaseinparticlesizes)reachingasteadystatevalueat100min of milling. The results can be represented by the following equation: 6 d50¼ 6 d50 # $ 1$ ð1! e!t=tÞ (6)
where,6/d50istheimplicitspecificsurfacearea,d50isthemean
particlesdiameterattimet,(d50)1isthemeanparticlesdiameter
steadystatevalueovertime,(6/d50)1istheimplicitspecificsurface
Fig.10. CriticalmicelleconcentrationasfunctionofPVP%inthebinarymixture SDS/PVP.Thefigureshowsaminimumvalueat60%ofPVPsuggestingamaximum ofsurfaceactivityofPVPSDSassociation(mean'S.D,n=3).
Fig.11.ZetapotentialoftheSDS/PVPsystemasafunctionofSDSconcentration (mean'S.D, n=3). By increasing the SDS concentration, the zeta potential decreasedsharplyto!50mV,whichlikelycontributedtothestabilitythrough electrostaticrepulsion.
Fig.12.Liquid penetration rate of SDS/PVP (40–60% w/w) mixture at total concentrationof 1.2%. A linear relationship is observedbetween the square penetratedliquidweightandtime.Fromtheslopeoftheline,awettabilityfactor(K) canbeobtained.
Fig.13.Wet-abilityfactorasfunctionofstabilizerconcentrationofSDS/PVPbinary mixtureandSDSalone.FortheSDS/PVPsystem,thisplateauisreachedat1.2% consistingof0.72%PVPand0.48%(w/w)(mean'S.D,n=3).
Fig.14.Milling kinetic of suspension using SDS/PVP (40–60% w/w) at total concentrationof1.2%.Themillingkineticisillustratedbyanexponentialincreasein theimplicitspecificsurfaceareareachingasteadystatevalueat100minofmilling.
areasteadystatevalueovertimeand
t
isthedurationto reach 63%of themaximumspecificsurfacearea.Valuesfor(6/d50)1andt
weregeneratedatvariousstabilizerconcentrationsandtheresultsare showninFig.15.Theseresultsshowaninitialincreasein(6/d50)1
and
t
withincreasingstabilizerconcentrationreachingaplateauat 1–1.2%,andareconsistentwiththoseobtainedfromthewettability evaluation.Theresultsshowtheexistenceoftwodistinctregimes. Below 1–1.2%, the (6/d50)1 increases dramatically when theconcentrationofSDS–PVPisincreased.Incontrast,above1–1.2%, aplateau((6/d50)1=50correspondingtod50of120nm)isreached.
Asimilarbehaviorwasobservedwhenmanufacturingsubmicron emulsion using high pressure homogenization (Laurent Taisne, 1996).Theproposedmechanismisthatatlowstabilizer concen-tration“poorregime”,theparticlessizeislimitedbythestabilizer amountcorrespondingtofullcoverage.Athighstabilizer concen-tration“richregime”,theparticlessizeislimitedbythemechanical energyofmillingsystem.Theexcessofstabilizerwillaccumulatein thesupernatantphase.Thisdemonstratesthat1–1.2%issufficient toensurethesuspensionstabilization.Anyexcessisundesiredasit mayincreasemicellarsolubilityandthereforepromoteOstwald repining
(c)Step#2.2c:AdsorptionisothermmeasurementofPVP.
Fig.16showstheadsorptionisothermofPVPontheAPIpowder surface.Asindicatedbythecompleteadsorptionofthefirstadded PVPmolecules,theresultsshowahighaffinitytypeisotherm.The greatdealofscatterobtainedathighPVPconcentrationislikely
duetothevariabilityoftheUVmethodusedfordeterminingthe remaining PVP concentration. At high PVP concentration, the instrumentmeasuresthedifferencebetweenlargequantities,and any uncertainty in the estimated concentration using the UV methodcan therefore result in a largeerror in the amountof adsorbedPVP.Henceitis difficulttoascertainanexact plateau valueoftheisothermwhichappearedasbetween0.6and0.9mg/ m2. Assuming a plateau value of 0.7mg/m2, the required
concentrationofPVPtocompletelysaturatetheparticlescanbe roughlyestimated.FromFig.14 thesmallestparticles diameter obtainedis about120nm.This gives a specific surface areaof 35.2m2/g. For a 20% suspension the total surface area was
calculated by using the following equation and estimated as 704m2:
$20/
r
$ d50 (6)Thetotalsurfaceareacoveragerequired493mgor0.493%of PVPwhichcorrespondsto0.82%ofSDS/PVP40–60%w/w.These resultsarewithintheorderofmagnitudeofthevaluesobtainedin millingabilityandwettabilitytests.
(iii)Step#2.3:Stabilityresultsofselectedformulation.
UsingtheoptimumSDS/PVPratioof40–60ataconcentration of1.2%,thestabilitydatawereobtainedbyassessingtheparticles sizeasafunctionoftimeatroomtemperature(forthe20%w/w APInanosuspension).Fig.17showstheevolutionofd10,d50and
d90 along a storage period of 57 days. It can be seen that no
significant change in particle size occurred during the storage period, further confirming the high colloidal stability of the definednanosuspensionfollowingthedevelopedmethodology.
Fig.15.Infiniteimplicitsurfacespecificarea(6/d50)1andcharacteristictime(t)as
functionofstabilizer(SDS/PVP(40–60%w/w))concentration.Asteadystatevalue overconcentrationvalueisobservedstartingfrom1to1.2%(w/w)(mean'S.D, n=3).
Fig.16.Adsorptionisotherm(highaffinity)ofPVPatroomtemperature.Aplateau valueappearedatapproximately0.7m2/g(mean' S.D,n=3).
Fig.17. Stabilityatroomtemperatureofnano-crystallinesuspensionstabilized withSDS/PVP(40–60%w/w)attotalconcentrationof1.2%w/w.variationofd10,d50
andd90withstorageduration.Notsignificantchangeisobserved(mean'S.D, n=3).
5. Discussion
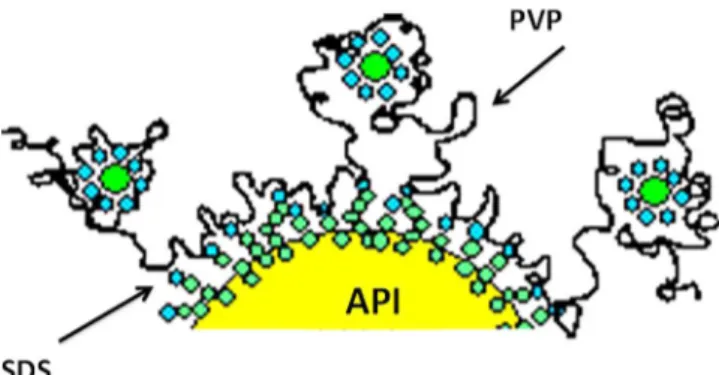
Itisinterestingtoobservethattheselectedformulationis a synergiccombinationofanionicsurfactant(SDS)andapolymer (PVP).Indeed,it achievesastrong electro-stericprotection.The trappedSDSmicellesbythePVPchainsprovideahighelectrostatic barrier(value)andfurthermorethemicellesareplacedoutsidethe surfaceofAPIasillustratedinFig.18 (BernardCabane,personal communication)
6. Conclusion
Using a combination of empirical and colloidal–interfacial fundamentalapproach,anoptimumwetting/dispersantagentwas selectedfor preparingnano-suspensions witha d50 lowerthan
150nm.Usingasystematicapproach,alargenumberofwetting/ dispersantagentswasinvestigated.Theunsuccessfulagentswere excludedfollowingeachstep.Usingasimplemillingprocedure, namelyrollermillcombinedwithparticlesizemeasurement,the agentsthatresultedinaparticlediametergreaterthan500nmor thatfailedtopreventflocculationwereexcluded.Theremaining agentswerefurthersegregatedusingzetapotentialand rheologi-calmeasurements.Samplesthatresultedinalowabsolutezeta potentialvalue(<15mV)andaviscosityhigherthan10mPasata shear rate of 1000s!1 were also excluded. Among remaining
samples,those showinga Newtonianflowwerefurther investi-gatedusinghighshearmillingtoselectthebestwetting/dispersant system. The SDS/PVP mixture was selected for composition optimization using wettability, adsorption isotherm and zeta potentialmeasurement.Anoptimumstabilizercombinationthat led to maximum wettability, the best milling results and the maximumstabilitywasidentifiedasSDS/PVP40–60%withatotal concentrationof1–1.2%.Overall,thepresentapproachofstabilizer selectiondescribedinthismanuscriptisintendedtosupportthe formulatorstoselectasuitablewetting/dispersantsystemforany API to achieve a scalable industrial process leading to stable nanosuspensions.
Astepforwardwouldbetointroduceadditionalstressteststo assesstheformulationrobustnesssuchasthermalstability, freeze-thawingstability,centrifugationstability,ionicstrengthimpactor effect,and/oradilutioninbio-relevantmedia.
Acknowledgments
TheauthorsgratefullyacknowledgeJ.L. Laly(Global Headof PharmaceuticalSciencesOperationsatSanofiR&D)forhissupport. The authors also acknowledge OtmaneBoussif (Sanofi Pasteur, France),Yanhe(LGCR,SanofiUSA),XavierPepinand Harivardhan-ReddyLakkireddy(LGCR,Sanofi,France)fortheircontributionto pre-reviewthismanuscript.
AppendixA.
SeeTable2.
References
Aartsen,B.G.B.,1974.Thedeterminationofcontactanglesofaqueoussurfactant solutionsonpowders.ColloidPolym.Sci.252,32–38.
Adler, J.,Patist,A., Rabinovixh,Y., Shah,D.,Moudgil, B.,2000.Correlation of particulatedispersionstabilitywiththestrengthofselfassembledsurfactant films.Langmuir16,7255–7262.
Bazile,D.,2011.Nanotechnologiestoolsincandidateselectionfortheintravenous route.SMi–8thConferenceonControlledRelease,London.
Biswas,S.C.,Marion,D.,2001.Interfacialbehaviorofwheatpuroindolines:studyof adsorptionattheair–waterinterfacefromsurfacetensionmeasurementusing wilhelmyplatemethod.J.ColloidInterfaceSci.244,245–253.
Borm,P.,Klaessig,F.C.,Landry,T.D.,Moudgil,B.,Pauluhn,J.,Thomas,K.,Trottier,R., Wood,S.,2006.Researchstrategiesforsafetyevaluationofnanomaterials,part V:roleofdissolutioninbiologicalfateandeffectsofnanoscaleparticles.Toxicol. Sci.90,23–32.
Cabane,B.,1977.Structureofsomepolymer-detergentaggregatesinwater.J.Phys. Chem.81,1639–1645.
Chander,S.,Hogg,R.,2007.Characterizationofthewettinganddewettingbehavior ofpowders.KONA25,56–75.
Cosgrove,T.,2010.ColloidSciencePrinciples,MethodsandApplications,seconded. Wiley.
Deng,Z.,Xu,S.,Li,S.,2008.Understandingarelaxationbehaviorinananoparticle suspensionfordrugdeliveryapplications.Int.J.Pharm.351,236–243.
Dery,R.,2012.CeramicThickFilmforMemsandMicrodevice.ElsieverInc..
Dijit,J.,CohenSturat,M.F.,1992.Kineticofpolymeradsorptionincappilaryflow. Macromolecules25,5416–5423.
Fisher,E.W.,1958.Elektronenmikroskopischeuntersuchungenzurstabilitätvon suspensioneninmakromolekularenlösungen.Kolloid-Z160,120–141.
Gordana Matijasic, G., Glasnovic, A., 2008. Suspension rheology during wet comminutioninplanetaryballmill.Chem.Eng.Res.Design86,384–389.
Hiemenz,P.C.,Rajagopalan,R.,1997.PrinciplesofColloidandSurfaceChemistry, thirded.MarcelDekkerInc.,NewYork,pp.255–265.
Holthoff, H.,Egelhaaf, S.U.,Borkovec, M.,Schurtenberger, P., Sticher,H.,1996. Langmuir12,5541–5549.
Hunter,R.J.,1988.ZetaPotentialInColloidScience:PrinciplesAndApplications. AcademicPress,UK.
IwonaOstolska,M.W.,2014.Applicationofthezetapotentialmeasurementsto explanationofcolloidalCr2O3stabilitymechanisminthepresenceoftheionic polyaminoacids.ColloidPolym.Sci.
Kawabata,Y.,Wada,K.,Nakatani,M.,Yamada,S.,Onoue,S.,2011.Formulationdesign for poorly water-soluble drugs based on biopharmaceutics classification system:basicapproachesandpracticalapplications.Int.J.Pharm.420,1–10.
Kaye,B.H.,1967.Permeabilitytechniquesforcharacterizingfinepowders.Powder Technol.1.
Kesisoglou,F.,Panmai,S.,Wu,Y.,2007.Nanosizing—oralformulationdevelopment andbiopharmaceuticalevaluation.Adv.DrugDeliv.Rev.59,631–644.
Kipp,J.,2004.Theroleofsolidnanoparticletechnologyintheparenteraldeliveryof poorwater-solubledrugs.Int.J.Pharm.284,109–122.
LaurentTaisne, L.,1996.Transferof Oilbetweenemulsiondroplets. J.Colloid InterfaceSci.184,378–390.
Lauten,R.A.,Kjoniksen,A.-L.,Nystroem,B.,2001.ColloidPolymerInteractionsand AggregationinAqueousMixturesofPolystyreneLatex,SodiumDodecylSulfate, andaHydrophobicallyModifiedPolymer:ADynamicLightScatteringStudy. Langmuir17,924–930.
Lee,J.,Lee,S.,Choi,J.,Yoo,J.Y.,Ahn,C.-H.,2005.Amphiphilicaminoacidcopolymers asstabilizersforthepreparationofnanocrystaldispersion.Eur.J.Pharm.Sci.24, 441–449.
Lee, E.M., 2002. Nanocrystals: resolving pharmaceutical formulation issues associatedwithpoorlywater-solublecompounds.In:Marty,J.J.(Ed.),Particles. MarcelDekker,Orlando.
LeenaPeltonen,J.H.,2010.Pharmaceuticalnanocrystalsbynanomilling:critical processparameters,particlefracturingandstabilizationmethods.J.Pharm. Pharmacol.62,1569–1579.
Monteiro,A.,Afolabi,A.,Bilgili,E.,2013.Continuousproductionofdrugnanoparticle suspensionsviawetstirredmediamilling:afreshlookattheRehbindereffect. DrugDev.Ind.Pharm.39,266–283.
Napper,D.H.,1982.PolymericStabilizationofColloidalDispersion.Academicpress, London.
Noyes,A.A.,Whitney,W.R.,1897.Therateofsolutionofsolidsubstancesintheirown solutions.J.Am.Chem.Soc.19,930–934.
Ostwald,W.,1901.Uberdievemeintlicheisomeriedesrotenundgelben queck-silberoxyds und die oberflachen-spannung fester korper. Zeitschrift fur PhysikalischeChemie34,495–512.
Pecora,R.,1985.DynamicLightScattering:Applications ofPhotonCorrelation Spectroscopy.Springer.
Pritchard,J.F.,2010.Fast-to-patient:creativestrategiesforquickdemonstrationof clinicalproof-of-concept.Regul.Rapporteur7,7–10.
Rigden,P.,1947.Thespecificsurfaceofpowders.Amodificationofthetheoryofthe air-permeabilitymethod.J.Soc.Chem.Ind.66,130–136.
Sato,T.,1980.StabilizationofColloidalDispersionsbyPolymerAdsorption.Marcel Dekker,Inc.,NewYork.
Savjani, K.T., Gajjar, A.K., Savjani, J.K., 2012.Drug solubility: importanceand enhancementtechniques.ISRNPharm.2012.
Sharma,D.,Soni,M.,Kumar,S.,Gupta,G.,2009.Solubilityenhancement—eminent roleinpoorlysolubledrugs.Res.J.Pharm.Technol.2,220–224.
Shegokar,R.,Müller,R.H.,2010.Nanocrystals:industriallyfeasiblemultifunctional formulationtechnologyforpoorlysolubleactives.Int.J.Pharm.399,129–139.
Simonelli,A.,Mehta,S.,Higuchi,W.,1970.Inhibitionofsulfathiazolecrystalgrowth bypolyvinylpyrrolidone.J.Pharm.Sci.59,633–638.
Singh,A.,Worku,Z.A.,VandenMooter,G.,2011.Oralformulationstrategiesto improvesolubilityofpoorlywater-solubledrugs.ExpertOpin.DrugDeliv.8, 1361–1378.
Somasundara,P.,2006.EncyclopediaofSurfaceandColloidScience.Taylorand FrancisGroup3,pp.1758–1770.
Swithenbank,J.,Beer,J.,Taylor,D.,Abbot,D.,McCreath,G.,1976.ALaserDiagnostic TechniquefortheMeasurementofDropletandParticleSizeDistribution.AIAA, AerospaceSciencesMeeting.
Tadros,Th.F.,1985.In:Buscall,R.,Corner,T.,Stageman,J.(Eds.),PolymerColloids. ElsevierAppliedSciences,London.
Tadros,T.F.,1982.EffectofPolymersonDispersionProperties.Academicpress, London.
Tadros,T.F.,2005.AppliedSurfactants:PrinciplesandApplications.WILEY-VCH VerlagGmbH&Co.KGaA,Weinheim,Germany.
Tadros,T.F.,2012.DispersionofPowdersinLiquidsandStabilizationofSuspensions. WILEY-VCHVerlagGmbH&Co.KGaA,Weinheim,Germany.
Walker,P.,Petersen,E.,Wright,C.,1952.Surfaceactiveagentphenomenaindust abatement.Ind.Eng.Chem.44,2389–2393.
Wang,G.D.,Mallet,F.P.,Ricard,F.,Heng,J.Y.,2012.Pharmaceuticalnanocrystals. Curr.Opin.Chem.Eng.1,102–107.
Wong,J.,Brugger,A.,Khare,A.,Chaubal,M.,Papadopoulos,P.,Rabinow,B.,Kipp,J., Ning, J., 2008. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv. Drug Deliv. Rev. 60, 939–954.