HAL Id: hal-01611993
https://hal.archives-ouvertes.fr/hal-01611993
Submitted on 20 Oct 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Novel one-step synthesis and characterization of
bone-like carbonated apatite from calcium carbonate,
calcium hydroxide and orthophosphoric acid as
economical starting materials
Doan Pham Minh, Ngoc Dung Tran, Ange Nzihou, Patrick Sharrock
To cite this version:
Doan Pham Minh, Ngoc Dung Tran, Ange Nzihou, Patrick Sharrock. Novel one-step synthesis and
characterization of bone-like carbonated apatite from calcium carbonate, calcium hydroxide and
or-thophosphoric acid as economical starting materials. Materials Research Bulletin, Elsevier, 2014, 51,
p. 236-243. �10.1016/j.materresbull.2013.12.020�. �hal-01611993�
Novel
one-step
synthesis
and
characterization
of
bone-like
carbonated
apatite
from
calcium
carbonate,
calcium
hydroxide
and
orthophosphoric
acid
as
economical
starting
materials
Doan
Pham
Minh
*
,
Ngoc
Dung
Tran,
Ange
Nzihou,
Patrick
Sharrock
Universite´ deToulouse,MinesAlbi,UMRCNRS5302,CentreRAPSODEE,CampusJarlard,F-81013Albicedex09,France1. Introduction
Calcium hydroxyapatite(Ca10(PO4)6(OH)2,Ca-HA)isfoundto
bethemainmineralphaseofhumanhardtissues[1].However, pure Ca-HA neveroccursinanybiological systembutco-exists withitsderivatives[2].Theseareformedbypartialreplacementof calcium(Ca2+),orthophosphate(PO
43!)orhydroxide(OH!)groups
byotherspeciessuchasMg2+,Na+,K+,F!,Cl!,andCO
32![3,4].
Amongthem,carbonatedapatite(CAP)istheclosestbiomimetic solid resembling the minerals in calcified tissues [4–6]. The amount of carbonate present in human bone mineral reaches typically4–8wt%[3,7].Recentstudiesshowedthatsubstituted Ca-HA,suchasCAP,weremoreeffectivethanpureCa-HAforskeletal implants[7–9].Thisleadstoagrowinginterestinthedevelopment ofsuchsubstitutedmaterials.
Manyhydroxyapatitetypeshavebeensynthesized, character-ized and examined for use as implantable bone-compatible biomaterials [10–12].Insynthesis proceduresfor pure Ca-HA, atmospheric carbon dioxide can be incorporated in the precipitated solids which lead tophosphate substitution with carbonateions.ThisyieldsCa-HAwithlowcarbonatecontentand
non-stoichiometric Ca/P ratios [13,14]. Onlythe more closely stoichiometricCa-HAwithstandsthehightemperaturerelatedto ceramic sintering or plasma-spraying, without decomposition intolimeortricalciumphosphate(TCP).CAPcontaininghigher carbonateamountscanbesynthesizedbymulti-stepsynthesis proceduresincludingtheprecipitation ofsoluble calciumsalts (Ca2+)withPO
43!inthepresenceofCO32!(liquidroute)[15]or
theheatinguptomorethan10008Cofacalciumphosphateunder carbondioxide flux (thermal route)[16]. These methods have disadvantages of added manipulations for the elimination of waste counter ions or heating step under controlled carbon dioxide.CAPstartingfromcalciumcarbonateasthesourceofboth calcium cations (Ca2+) and carbonate anions (CO
32!) and
potassium dihydrogen orthophosphate (KH2PO4) under
atmo-sphericpressurewasbrieflymentionedbyLomo[17].Thisroute has recently been investigated under higher pressures of hydrothermalconditions[15,18,19].Despitetheseverereaction conditionsused(hightemperature,longreactiontimeandwater rinsing),theconversionofcalciumcarbonatewasnotcomplete
[19]. So, it is of current interest to find simple synthesis proceduresthatleadtoCAPwithcontrolledcarbonatecontents. In this study, the synthesis of CAP using calcium carbonate (calcite),calciumhydroxideandorthophosphoricacidwas investi-gatedundermoderateconditions.Orthophosphoricacidwaschosen becauseofitsstrongestacidityamongtheavailableorthophosphate sourcesandtheabsenceofalkalicationsinthefinalCAPproduct.
Keywords: A.Structuralmaterials B.Chemicalsynthesis C.Thermogravimetricanalysis D.Microstructure D.Thermalexpansion ABSTRACT
There is growing interest in the development of substitutedcalcium hydroxyapatite (Ca-HA) for biomedicalapplications.Carbonatedapatite(CAP)appearsasanimportantsubstitutedCa-HAbecauseof itsbetterbiocompatibilitycomparedtopureCa-HA.Thispaperreportsanovelpressure-mediated one-stepsynthesisofCAPstartingfromorthophosphoricacid,calciteandcalciumhydroxideasavailableand high-puritystartingmaterials.Undermoderatesynthesisconditions(808Cand<13bar),CAPwith differentcarbonatecontentscouldbeobtained.Theratioofcalcite/calciumhydroxidemixtureplayeda crucialroleforboththeadvancementofreactionandthecarbonatecontentinsertedinCAP’sstructure. At808C,thetotaldecompositionofcalciterequiredacalcite/calciumhydroxidemixturecontainingat leastahalfofcalcite.CAPscontaining2.25–4.17wt%ofcarbonateinsertedinitsstructurewereobtained asafunctionofthecompositionofcalcite/calciumhydroxidemixture.Theresultsopenasimplebut effectivewayforthesynthesisofhighqualitybiomimeticCAP.
* Correspondingauthor.Tel.:+33563493258;fax:+33563493043. E-mailaddresses:doan.phamminh@mines-albi.fr,doanhoa2000@yahoo.fr
Variousmolar ratiosof calcium carbonatetocalcium hydroxide wereusedinordertocontroltheamountofcarbonateinsertedinthe apatitic structure. The results showed that CAP with different carbonatecontentscouldbeobtainedina one-stepsynthesisat 808Candpressuresbelow13atm,withoutawashingstep. 2. Materialsandmethods
Calcitepowder(CaCO3,98wt%)fromFisherScientific,calcium
hydroxide powder (Ca(OH)2, 98wt%) from Acros Organics and
orthophosphoricacid(H3PO4,85wt%inwater)fromMerckwere
used as received. CAP synthesis was carried out in a 250mL stainlesssteelreactor(TopIndustrial)whichwasequippedwithan electricalheatingjacketandamagneticstirrer.Foreachreaction, calcite or mixture of calcite/calcium hydroxide containing 100mmol of calcium and 45mL of water were fed into the reactor.After closing, 66.7mmol of orthophosphoric acid were quicklyinjectedintothereactor.Thisledtothestartingmolarratio ofcalciumtophosphorusof1.67(Table1).Duringthereaction,the stirringratewassetat800rpmandthereactorwasthermostated at808Cfor48h.Thisreactiontemperaturewaschosenbecause supplementaryexperimentsshowedthatthetemperatureslower than808Cwerenotsufficientforagooddecompositionofcalcite particles.After48hofreaction,thereactorwasfreelycooleddown toroomtemperature.Solidproductswereseparatedfromliquid phase by filtrationusing 0.45
m
mfilterpaper. Then they were driedat508Cfor48hbeforefurthercharacterizations.Bothsolidandliquidphasesobtainedfromthefiltrationstepwere analyzedusingdifferentanalysisandcharacterizationtechniques. Elementalanalysiswascarriedoutusinginductivelycoupledplasma atomicemissionspectroscopy(ICP-AES,HORIBAJobinYvonUltima2 apparatus).Thermogravimetry(TG)wasperformedinaTA Instru-mentsSDTQ600analyzerwithaheatingrateof58Cmin!1underair
flux(100mLmin!1).Thermo-mechanicalanalysis(TMA)wascarried
outinaSETARAMSetSys16/18analyzerwith5gconstantloadonthe powdersample.X-raydiffraction(XRD)dataofthesolidscollected usingaPhillipsPanalyticalX’pertProMPDdiffractometerwithaCu K
a
(1.543A˚) radiation source. Fourier transform infrared (FTIR) spectroscopywasperformedusingaShimadzu8400Sspectrometer. Particlesizedistribution wasdeterminedbylaserscatteringin a Mastersizer2000(MalvernInstruments Ltd.,Malvern,UK) inthe particlesizerangeof0.020–2000m
m.Scanningelectronmicroscopy (SEM) was carried out on a Philips XL30 ESEM apparatus (FEI Company) which was coupled with an energy-dispersive X-ray spectroscopy(EDXanalysis).3. Results
3.1. Elementalanalysis
Theliquidseparatedfromthefiltrationofthereactionmixture wasacidifiedwithconcentrated nitricacidtoavoidall further
re-precipitation. Solidproducts weremineralized using concen-trated nitric acid. The resulting homogeneous solutions were analyzed by ICP-AES technique and results are presented in
Table 2. In all cases, the contents of soluble calcium and phosphorusintheliquidphasewerelowerthan1.4%oftheinitial quantitiesofcalciumandphosphorusintroducedinthereactor.So, theprecipitationof orthophosphatespecieswasquitecomplete and most of the calcium existed in solid phases after 48h of reaction.Becauseoftheabsenceofanycounterionsintheliquid phase,nowashingstepwasrequiredforthepurificationofthefinal solidproducts.Thisbringsasignificantadvantageofthepresent synthesisprocessincomparisonwiththesynthesisusingsoluble calcium salts and/oralkali orthophosphates, since thewashing stepisusuallyarduouswhensmallparticlesareformed.
Themolarratiosofthebulksolidswerehigherthanthatofthe stoichiometric Ca-HA (1.67). This result is discussed in more detailsinthesectionofFTIRanalysis.
3.2. Decompositionofcalciumcarbonate
Thereaction ofcalcite withorthophosphoric acidledtothe formationofcarbondioxidewhichcanexistinbothgasandsoluble states. Pressure in the reactorincreasedwith theformation of carbondioxideingasphase.Takingintoaccountthevolumeofgas phaseinthereactor,thepartialpressureofwatervaporat808C andusingtheidealgaslaw,thequantityofcarbondioxideingas phase could be calculated from the final pressure at 48h of reaction.Fig.1indicatestherelativeadvancementofthereaction viathecalculatedamountsofcarbondioxide.Thedecomposition ofcalcitereachedatleast43,89,83and88%inthesynthesesusing 25, 50, 75 and 100% of calciteas calcium source, respectively,
Table1
Compositionofthestartingreactantmixturesanddesignationofsolidproducts; othercondition:synthesistemperatureof808C;stirringrateof800rpm;initial waterof45g.
Calciumsource Phosphate
source Solidproduct designation CaCO3 powder,mmol Ca(OH)2 powder,mmol H3PO4, mmol 100 0 66.7 CAP100/0 75 25 66.7 CAP75/25 50 50 66.7 CAP50/50 25 75 66.7 CAP25/75 Table2
ResultsofelementalanalysisusingICP–AEStechnique;CaLiqandPLiq:amountof calciumandphosphorusremainedintheliquidphase;Ca/P:molarratioofbulk solidproducts.
Solid CaLiq(%) PLiq(%) Ca/P
CAP100/0 1.4 1.1 1.88 CAP75/25 0.9 1.2 1.84 CAP50/50 1.4 1.2 1.82 CAP25/75 1.3 1.2 1.74
10.9
44.4
62.3
87.9
0
10
20
30
40
50
60
70
80
90
100
CO
2 ga s, m
m
ol
Fig.1.Quantityofcarbondioxideingasphase(COgas2 )calculatedfromthefinal reactionpressureasafunctionofthemolarpercentageofcalciteintheinitial mixtureofcalciteandcalciumhydroxide.
whichdoesnotconsidertheamountofcarbondioxidedissolvedin theliquidphase.
3.3. XRDcharacterization
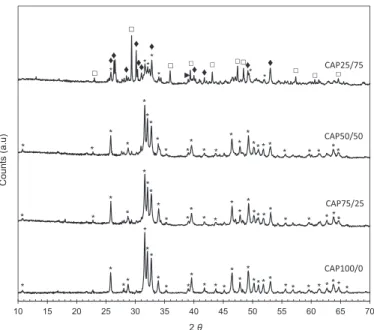
Inordertoidentifycrystallinephasesofthesolidproducts,XRD characterizationwasperformedandtheresultsarepresentedin
Fig.2.Forthesolidproductstartingfrom100%calciumcarbonate (CAP100/0), alldiffractionpeakscouldbeattributedtocalcium hydroxyapatite (Ca-HA). CAP might be also present but they seemedtoberelativelylowincomparisonwiththatofCa-HA.No
evidenceofcalciumcarbonatewasobserved,indicatingthatthe decompositionoftheinitialcalcitemustbequitecompleteforthis synthesis.
BothCAP75/25andCAP50/50products,whichwereformedfrom the mixture of calcite and calcium hydroxide, had similar XRD patternscomparedtothatofCAP100/0.Mostoftheirpeakscouldbe alsoattributedtoCa-HA.Traceamountsofoctocalciumphosphate (OCP,Ca8(HPO4)2(PO4)4"5H2O)couldbealsofound.Infact,thefinal
pHofthereactionmixturewasaround7–8.ThispHisfavorablefor theformationofOCPbecauseinthispHrange,themainsoluble phosphatespeciesareH2PO4!and HPO42![26].Thisresultwas
supportedbypreviousworkonthesynthesisofCa-HAundersimilar conditions,atatmosphericpressure[26].Asobservedpreviouslyfor CAP100/0,calcitewasalmostabsentinthesetwosolidproducts, whichwasinagreementwiththeresultsinFig.1.
Ontheotherhand,theresultschangednotablyforCAP25/75. Calcite, Ca-HA and OCP werefound tobe themain crystalline phases present in this sample. A small amount of crystalline calciumhydroxidewasfound,asillustratedbythelowintensityof itsmaindiffractionpeakat39.068.Calciumhydroxidehashigher solubility and basicity than calcite. Thus, orthophosphoric acid introduced in the mixture of calcite and calcium hydroxide preferentially consumes the calcium hydroxide. When a high amountofcalciumhydroxideisusedsuchasforthesynthesisof CAP25/75,thepHofreactionmixtureremainsathighvalue,which disfavorsthedecompositionofcalcite.
Inallcases,XRDresultsrevealedthepresenceofCAPatlevels muchlowerthanthoseofCa-HA.TheidentificationofCAPbyXRD isdelicatebecauseinmostcases,themaindiffractionpeaksofCAP areclosetothoseofCa-HAandwouldrequireRietveldrefinement forincreasedresolution.Othercharacterizationtechniqueswere usedtohighlighttheformationofCAP.
3.4. IRcharacterization
IRspectraofthesolidproductsarepresentedinFig.3.Inthe wavenumberrangefrom4000to1700cm!1(notpresented),there
10 15 20 25 30 35 40 45 50 55 60 65 70 Counts (a.u) 2 CAP100/0 CAP75/25 CAP50/50 CAP25/75 * * ** * ** * ** * * * * * ***** * * * * * ** * * ** * * * * * * * * * * * * * ***** * * * * * * * * * * * * * *** ** * * *** * * * * * * * * * * * * * *
Fig.2.XRDpatternsofthesolidproducts;(*)Ca-HA(JCPDSstandardNo 01-072-1243);(&)calcite(JCPDSstandardNo00-047-1743);(^)OCP(JCPDSstandardN0 00-026-1056);(")calciumhydroxide(JCPDSstandardNo00-050-0008).
550 750 950 1150 1350 1550 Trans m itt ance (a. u. ) Wavenumber (cm1) CAP100/0 CAP75/25 CAP50/50 CAP25/75 1415 1450 CO CO Phosphates Phosphates CO 1545 870 880
wasonly a very broadweak peak around 3500cm!1, which is
attributedtothestretchingofmolecularwater.Thevibrationsof orthophosphategroupsarecharacterizedbytheabsorptionbands inthewavelengthrangesof 650–550cm!1and 1300–910cm!1
[20].AccordingtoXRDresults,calcitewasonlynotablypresentin CAP25/75asconfirmedbyitsnetabsorptionbandat711cm!1.
Thisisoneprincipalbandofcalciteandisnotsuperimposedwith bandsoforthophosphatesgroupsorcarbonatesgroupspresentin apatiticstructure[21].
The presence of carbonate groups inserted in the apatitic structure to form CAP wasclearly revealed by IR spectra. The replacement of PO43! groups present in Ca-HA’s structure by
CO32! groups led to the formation of B-type CAP, which is
characterizedbythebi-modalpeak at1450/1415cm!1and the
peakat870cm!1[22,23].Thus,B-typeCAPwaspresentinallsolid
productsbutitscontentwashigherinCAP100/0,CAP75/25and CAP50/50 than that in CAP25/75. The higher intensity at 1415cm!1comparedtothatat1450cm!1ofCAP25/75wasdue
totheprincipalpeakat1389cm!1ofcalcitethatremainedinthis
product.TheformationofB-typeCAPexplainedthemolarratioof Ca/P in Table 1, which was slightly higher than that of the stoichiometricCa-HA. Ontheotherhand,A-typeCAPis formed when OH! groups of Ca-HA’s structure are replaced by CO
32!
groups,whichhavethecharacteristicbandsat1545and880cm!1
[22,23].So,A-typeCAPwasonlyformedinCAP100/0,CAP75/25 andCAP50/50,andwaspracticallynotpresentinCAP25/75. 3.5. Thermalanalysis(TG)
Fig.4showstheresultsofthethermalanalysisoftheproducts inthewiderangeoftemperaturesfrom25to14008C.Aweightloss smallerthan1wt%tookplacebelow1008Cwhichcorrespondsto theremovalofsurfacemoisture.Thenextweightlossof1–2wt%, which occurredcontinuouslyfrom 1008C toabout 340–3808C, could be attributed to the removal of lattice molecular water
[24,25].Theweightlossinthetemperaturerangeof380–5008C wasattributedtothecondensationofHPO42!groups,presentin
nonstoichiometricapatitesuchasOCP,toformcalcium pyrophos-phate(Ca2P2O7),whichwasobservedforallproducts.Remaining
calcite decomposedat 610–6708C [26]. DTGsignalof both the condensationofHPO42!groupsandthedecompositionofcalcite
increasedwiththeincreaseofcalciumhydroxidecontentinthe initial reactant mixture. This showed the unfavorable effect of calcium hydroxide contentfor the decomposition of calcite,as explained in XRD section. According to XRD results, unreacted calciumhydroxideremainedonlynotablyinCAP25/75.Itsthermal decompositionischaracterizedbytwoseparatedpeaks[27].The firstweightlossstartingat3258Cwaspartiallysuperimposedwith thecondensationofHPO42!groups,andthesecondweightloss
startingat6008Cwaspracticallysuperimposedwiththethermal decompositionofcalcite,highlightedbyitsDTGcurve.
ThedecarbonationofCAPtookplaceinthetemperaturerange of670–12508C.AccordingtoIRresults,A-typeCAPwaspractically not present in CAP25/75. Thus, we deduce that the thermal decarbonationofA-typeCAPandB-typeCAPtookplaceat870– 12508Cand670–8708C,respectively.Finally,thelastweightloss at1250–13408CwasattributedtothedehydrationofOH!groups
presentinCa-HA’sstructure[28].
86 88 90 92 94 96 98 100 0 200 400 600 800 1000 1200 1400 TG (w t% ) Temperature (°C) CAP100/0 CAP75/25 CAP50/50 CAP25/75 (A) 0 200 400 600 800 1000 1200 1400 DT G (a. u) Temperature (°C) CAP100/0 CAP75/25 CAP50/50 (B) CAP25/75
Fig.4.TG(A)andDTG(B)curvesofthesolidproductsanalyzedinthetemperaturerangeof25–14008Cunderairatmosphere.
Table3
CarbonatecontentsinthesolidproductsdeterminedbyTGanalysis. Product CO32!(wt%) CaCO3 (610–6708C) B-typeCAP (670–7808C) A-typeCAP (780–12508C) CAP (670–12508C) CAP100/0 0.30 0.63 3.5 4.17 CAP75/25 0.75 0.69 2.73 3.42 CAP50/50 1.20 0.63 1.62 2.25 CAP25/75 <4.7a 0.54 0 0.54
a SuperimpositionofTGsignalsofCaCO
3decompositionandCa(OH)2 dehydra-tion,givinganapproximatevalue.
From TG curves in Fig. 4, the content of different types of carbonate present in the solid products could be calculated (Table 3). Asobservedby XRDcharacterization,theincrease of calciumhydroxidecontentintheinitialmixtureofcalciumsource ledtotheincreaseofremainingcalcitecontent.Inallcases,the carbonatecontentsofB-typeCAPwereclosetoeachother.Onthe otherhand,thecarbonatecontentofA-typeCAPdecreasedwith theincreaseofcalciumhydroxidecontentintheinitialmixtureof calciumsource,whichwasingoodagreementwithIRresults.The totalamountof carbonateobtained inCAP100/0and CAP75/25 approximated theamount of carbonate present in themineral phaseofbone,dentinandenamel,whichisintherangeof4–8%
[3,7,22].Itisevidentthatthecompositionoftheinitialmixtureof calciumsourceisanefficientparametertocontrolthecarbonate contentfortheone-stepsynthesisofCAP,sincetheconversionof bothinitialcalciumandorthophosphatesourcesintodesiredsolid productswaspracticallytotalinthesynthesisofCAP100/0,CAP75/
25,CAP50/50.Takingintoaccountthequasi-complete decompo-sitionofcalcite,thehighselectivityinapatiticcompoundsandthe weightofthesolidproductsrecoveredafterfiltrationanddrying steps,theyieldofthesynthesisprocedureindesiredCAPpowder could be calculated which reached at least 97% for CAP100/0, CAP75/25andCAP50/50.
3.6. SEM,particlesizedistribution
TheresultsofSEMobservationareillustratedinFigs.5and6.In theleft-handsideofFig.5(100
m
mscale),particlesofsizesranging fromseveralnmtohundredsofm
mcouldbeobservedforCAP100/ 0,CAP75/25andCAP50/50.Theimagesontheright-handsideofFig.5(10
m
mscale)revealedthatthelargerparticlesconsistedof theagglomerationofsmallerones.Theagglomeratesofthesesolidshadaflat-needle-like morphol-ogy.Thiswaspreviouslyobservedforapatiticcalciumphosphates
synthesized underhydrothermal conditions [19,29] or at atmo-sphericpressure [30]. Theformation ofthese particles could be attributedtothegrowthofCAPparticlesalongthec-axisdirection, whereCO32!groupsreplacedOH!groups[31,32].ForCAP50/50,in
additiontothepresenceofparticlesofflat-needle-likemorphology, particles ofsheet structure couldbe alsoobserved,which were attributedtoapparentmorphologyofOCP,whichcontainsHPO42!
groups[33].
SEMimagesofCAP25/75areshowninFig.6.At100
m
mscale correspondingtoamagnificationof200times(Fig.6(A)),particle sizesvariedalsoina largerangeasobservedinFig.5 forother solids. At higher magnification, calcium phosphate particles of sheet-likestructurewereclearlyobserved,whichwasattributedto thepresenceatlargeamountofOCP(Fig.6(B)).Remainingcalcite particles were also frequently present, as confirmed by EDXanalysis(Fig.6(C)).Weobservedalsothattheremainingcalcite particleswereusuallycoveredbycalciumphosphatelayers.This explainedwhytheFTIRsignalofcalcitewasnotsignificantdespite itshighcontentinCAP25/75(Fig.3).
TobetterquantifytheresultsofSEMobservations,particlesize distribution was examined and Fig. 7 shows the volume distribution as a function of particle size. The initial calcite powderhad aGaussiandistributionintheparticle sizeranging from6to70
m
m.Ontheotherhand,theinitialcalciumhydroxide haddifferentcategoriesofparticlesizes,whichvariedfrom0.3m
m toabout240m
m.The volume distribution of all four solid products can be classified into two categories. The first one consisted of fine particlesrangingfrom0.35toabout1.5
m
m.Thevolumeoccupied bythiscategorywasnotsignificantanddidnotexceed1.4%ofthe totalvolumeofthesolids.Thesecondoneincludedparticleslarger than1.5m
m,whichstretchedupto310,400,450and710m
mfor CAP100/0, CAP75/25, CAP50/50 and CAP25/75, respectively, indicatingdifferentlevelsofagglomerationofparticlesforeach product. The higher the calcium hydroxide content was, the higherthepHofcalcite/calciumhydroxidemixturewas,andthe more the acid–base reaction was violent leading to stronger agglomeration. In all cases, these results confirmed SEM observationsofFigs.5and6.3.7. Thermo-mechanicalanalysis
Used as biometerials, apatitic calcium phosphate based productscanbestabilizedathightemperatureinordertoavoid allfurtherevolutionofmaterials.Thissectionwasdedicatedtothe study of the thermo-mechanical behavior of CAP100/0 as the selectedproduct.
Thermalshrinkageisdefinedas(L!L0)/L0or
D
L/L0,whereL0istheinitiallengthofsample,andListhelengthofsamplemeasured at temperatureT or timet.Fig.8(A)shows thenon-isothermal analysisoftheCAP100/0sampleinthetemperaturerangeof30– 10008C.Thetemperatureof8808Cwasfoundtobethecritical pointforthethermalshrinkageofCAP100/0wheretheshrinkage accelerated. Only a slight thermal effect on the shrinkage was observedbelow8808Cwhichwassmallerthan0.02%.Ontheother hand,itbecamesignificantabove8808Candreached!0.46%when thetemperatureroseto10008C(Fig.8(A)).
Fig. 8(B) illustrates the isothermal shrinkage at different temperaturesbeforeandafterthecriticalpoint.Theinitialvalue of
D
L/L0ofeachcurvecorrespondedtotheshrinkageofthesampleFig.6.SEMimagesofCAP25/75.
0 10 20 30 40 50 60 70 80 90 100 0.1 1 10 100 1000 Vo lu me a cc umu la tio n, % Particle size, µm CAP100/0 CAP75/25 CAP50/50 CAP25/75 Initial CaCO3 Initial Ca(OH)2
Fig.7.Volumedistributionasafunctionofparticlesize(inbase10logarithmic scale).
during the heating time to reach the desired temperature. No significantthermaleffectwasobservedat600and8008Cforan isothermal time of 300min, confirming that CAP100/0 was thermo-mechanicallystableatthesetemperatures.Asexpected, theisothermalshrinkagetookplaceat10008Candreached!3.85% after300min.
The thermal shrinkage can be attributed to the sintering phenomenon wherein denser mass is formed and chemical reaction, i.e. the decarbonation or dehydration. Fig. 9 shows SEMimagesofCAP100/0sampleafterTMA.Theisothermaltimeof
300minat600and8008Chadnosignificanteffectonthesurface morphologyofCAPparticles,comparedtothesolidbeforethermal treatment(Fig.5fortheCAP100/0sample).At10008C,particlesof flat-needle-like morphology became only partially rounded. Probably, this morphology distortion of CAP particles corre-spondedtothefirststep ofsintering wherein theformationof thecontactareasbetweenadjacentparticlesstarted[34].
TG analysis showed that the heating of CAP solids led to different chemical reactions. We are interested in the thermal stabilityofcarbonateanionspresentinapatiticstructureofCAP.As
-4.0
-3.0
-2.0
-1.0
0.0
0
200
400
600
800 1 000
Sh
rin
ka
ge
(
%
)
Temperature (°C)
880 oC(A)
-4
-3
-2
-1
0
0
50
100 150 200 250 300
Sh
rin
ka
ge
(
%
)
Time (min)
(B) 600oC 1000 oC 800oCFig.8.TMAofCAP100/0;(A)non-isothermalanalysiswiththeheatingrateof108Cmin!1;(B)isothermalanalysis.
showninFig.10forIRspectraofCAP100/0samplebeforeandafter TMA, the decarbonation of both A-type (bands at 1545 and 880cm!1)andB-type(bandsat1450,1415and870cm!1)CAP
wasonly partialafter theplateau time of 300min at different temperatures.Thisdemonstrates thepossibilitytostabilizeCAP usingthermaltreatmentwhileconservingitscarbonatecontent. 4. Conclusions
Forthefirsttime,carbonatedapatite(CAP)ofhighpuritywas successfullysynthesized by a one-step synthesis process using orthophosphoric acid, and a mixture of calcite and calcium hydroxideassimplestartingreactantsundermoderateconditions (808C, <13bar). Carbon dioxide was formed as the only by-productandnowashingstepwasrequiredforthepurificationof finalproducts.BothA-typeandB-typeCAPwereformedunderthe synthesisconditionsused.
Thetotal contentof carbonateinserted in apatitic structure couldbecontrolledby varyingthecalcitecontentin theinitial mixture of calcium source. We obtained CAPcontaining 2.25– 4.17wt%ofcarbonatewiththeinitialmixtureofcalciumsource containing 50–100% calcite. For a complete decomposition of
calcite, an initial mixture of calcite and calcium hydroxide containingatleast50%ofcalcitewasmandatory.
TheCAP100/0samplepreparedfrom100%calciteand contain-ing4.17%carbonate,wasfoundtobethermo-mechanicallystable upto8808C.ThisoffersagoodpossibilityforthesynthesisofCAP forbiomedicalapplications.
Acknowledgments
Theauthorsacknowledge gratefullycolleaguesatRAPSODEE center,NathalieLyczko,ChristineRollandandPhilippeAccartfor differentcharacterizationmeasurements.
References
[1]R.Z.LeGeros,Chem.Rev.108(2008)4742–4753.
[2]S.V.Dorozhkin,M.Epple,Angew.Chem.Int.Ed.41(2002)3130–3146.
[3]N.Nassif,F.Martineau,O.Syzgantseva,F.Gobeaux,M.Willinger,T.Coradin,S. Cassaignon,S.Padilla,I.Izquierdo-Barba,M.Vallet-Regı´,Chem.Mater.22(2010) 3653–3663.
[4]S.Padilla,I.Izquierdo-Barba,M.Vallet-Regı´,Chem.Mater.20(2008)5942–5944.
[5]H.Morgan,R.M.Wilson,J.C.Elliott,S.E.P.Dowker,P.Anderson,Biomaterials21 (2000)617–627.
[6]K.Onuma,Prog.Cryst.GrowthCharact.Mater.52(2006)223–245.
[7]Y.S.Kim,H.K.Kwon,B.I.Kim,J.Dentistry39(2011)636–642.
[8]A.E.Porter,N.Patel,J.N.Skepper,S.M.Best,W.Bonfield,Biomaterials24(2003) 4609–4620.
[9]E.S.Thian,Z.Ahmad,J.Huang,M.J.Edirisinghe,S.N.Jayasinghe,D.C.Ireland,R.A. Brooks,N.Rushton,W.Bonfield,S.M.Best,Biomaterials29(2008)1833–1843.
[10]S.Peroos,Z.Du,N.HenriettedeLeeuw,Biomaterials27(2006)2150–2161.
[11]Q.Bao,C.Chen,D.Wang,J.Liu,Cryst.GrowthDesign8(2008)219–223.
[12]L.Niu,H.Kua,D.H.C.Chua,Langmuir26(2010)4069–4073.
[13]S.R.Radin,P.Duchyne,J.Biomed.Mater.Res.27(1993)35–45.
[14]A.Afshar,M.Ghorbani,N.Ehsani,M.R.Saeri,C.C.Sorrell,Mater.Design24(2003) 197–202.
[15]O.Frank-Kamenetskaya,A.Kol’tsov,M.Kuz’mina,M.Zorina,L.Poritskaya,J.Mol. Struct.992(2011)9–18.
[16]T.Tonegawa,T.Ikoma,Y.Suetsugu,N.Igawa,Y.Matsushita,T.Yoshioka,N. Hanagata,J.Tanaka,Mater.Sci.Eng.B173(2010)171–175.
[17]L.A.Lomo,J.Am.Chem.Soc.76(1954)3924–3925.
[18]T.I.Ivanova,O.V.Frank-Kamenetskaya,A.B.Kol’tsov,V.L.Ugolkov,J.SolidState Chem.160(2001)340–349.
[19]A.Kasioptas, T.Geisler, C.Perdikouri, C. Trepmann, N.Gussone, A. Putnis, Geochim.Cosmochim.Acta75(2011)3486–3500.
[20]D.W.Kim,I.S.Cho,J.Y.Kim,H.L.Jang,G.S.Han,H.S.Ryu,H.Shin,H.S.Jung,H.Kim, K.S.Hong,Langmuir26(2010)384–388.
[21]D.PhamMinh,M.GaleraMartinez,A.Nzihou,P.Sharrock,J.Therm.Anal.Calorim. 112(2013)1145–1155.
[22]A.Antonakos,E.Liarokapis,T.Leventouri,Biomaterials28(2007)3043–3054.
[23]M.E.Fleet,Biomaterials30(2009)1473–1481.
[24]S.Lazic,S.Zec,N.Miljevic,S.Milonjic,Thermochim.Acta374(2001)13–22.
[25]W.L. Suchanek,P.Shuk, K.Byrappa, R.E.Riman,K.S.TenHuisen, V.F.Janas, Biomaterials23(2002)699–710.
[26]D.PhamMinh,N.D.Tran,A.Nzihou,P.Sharrock,Ind.Eng.Chem.Res.52(2013) 1439–1447.
[27]M.J.Hologado,V.Rives,S.SanRoman,J.Mater.Sci.Lett.11(1992)1708–1710.
[28]C.J.Liao,F.H.Lin,K.S.Chen,J.S.Sun,Biomaterials20(1999)1807–1813.
[29]K.Ioku,S.Yamauchi,H.Fujimori,S.Goto,M.Yoshimura,SolidStateIonics151 (2002)147–150.
[30]M. Aizawa, T. Terado, F.S. Howell, K. Itatani, Mater. Res. Bull. 34 (1999) 1215–1225.
[31]J.Xiao,Y.Zhu,Q.Ruan,Y.Liu,Y.Zeng,F.Xu,L.Zhang,Cryst.GrowthDes.10(2010) 1492–1499.
[32]M.E.Fleet,X.Liu,Biomaterials28(2007)916–926.
[33]S.Mandel,A.C.Tas,Mater.Sci.Eng.C30(2010)245–254.
[34]S.Bailliez,A.Nzihou,Chem.Eng.J.98(2004)141–152.
550 750 950 1150 1350 1550 Tr an sm itt an ce ( a. u .) Wavenumber (cm-1) 105oC 600 oC 800 oC 1000 oC 1545 1450 1415 870 880
Fig.10.IRspectraofCAP100/0before(1058C)andafter(600,800and10008C) TMA.