pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
Faculté des Sciences Fondamentales et Appliquées (Diplôme National – Arrêté du 7 Août 2006)
École Doctorale: Ingénierie Chimique Biologique et Géologique Secteur de Recherche: Chimie Organique, Minérale, Industrielle
Année Nº attribué par la bibliothèque
Présentée par:
Patrícia Veríssimo Silva MATIAS
TRANSFORMATION DU METHYLCYCLOHEXANE ET DU
n-HEPTANE SUR LA ZEOLITHE MCM-22:
propriétés catalytiques globales et participation de chaque système poreux
Directeurs de Thèse: Patrick MAGNOUX et Professeur José Manuel LOPES
Soutenue le 25 novembre 2008 devant la commission d’examen
JURY
F. RAMÔA RIBEIRO, Professeur, Universidade Técnica de Lisboa Président
J.-P. GILSON, Professeur, Université de Caen Rapporteurs
J. M. ORFÃO, Professeur, Universidade do Porto
P. MAGNOUX, Directeur de Recherche CNRS, Université de Poitiers Examinateurs
J. M. LOPES, Professeur, Universidade Técnica de Lisboa M. GUISNET, Professeur, Universidade Técnica de Lisboa S. LAFORGE, Professeur, Université de Poitiers
Acknowledgements
Firstly, I would like to deeply thank Professor Fernando Ramôa Ribeiro for providing me the opportunity to carry out a Ph.D. in his research group. I also acknowledge his kindness, encouragement and his respect for my work, which he always expressed during my thesis.
To Professor José Manuel Lopes, Professor Michel Guisnet and Dr. Patrick Magnoux, I present my sincere thanks for their scientific guidance and availability throughout all the four years of this research work. Without their interest and patience, this Ph.D. would not have been possible.
In particular, I would like to demonstrate my special recognition to Professor Michel Guisnet, since his vast experience and brilliance brought much value to this thesis. I also express my thankfulness to Professor José Manuel Lopes for his support to my work and our vital scientific discussions day after day. I would also like to thank Dr. Patrick Magnoux for accepting me in his research group and providing me with the resources to conduct part of my thesis. A word of recognition goes also to Sébastien Laforge, especially for helping me with the preparation and characterization of the zeolite.
I wish to express my gratitude to the members of my Ph.D. Supervising Committee Professor Maria Filipa Ribeiro and Professor Carlos Henriques for the availability they always showed during this work.
I present my thanks to Philipe Ayrault, for his friendship and help during my stay in the Laboratoire de Catalyse en Chimie Organique and also for the Infrared Spectroscopy results; to Jean-Dominique Comparot for some of the Infrared experiments and Christine Canaff for the mass spectrometry results. I would like to demonstrate my special gratitude to Professor Manuela Carrott and Dr. Patrícia Russo for the scientific collaboration with the adsorption studies over the MCM-22 samples, to Dr. Auguste Fernandes for the X-Ray measurements, support during my laboratory work and his friendship and also to José Luís, from the Complexo Interdisciplinar, for soldering my reactors and serpentines.
I wish to thank my lab co-workers and friends in our research group at IST, which I had the luck to know during my thesis: Sandra, Isabel, Rita, Augustinho, Luís, Elisabete, Relvas, Luz, Joana F., Jorge, Joana T., Inês Matos, Renato, João, Hugo, Pedro B., J. P. Marques, Cristina, Nuno, Rodrigo, Anabela, Ana Paula, Susete, Inês Raposeiro. Thank you
for your help, good spirit and for the amusing times. In particular, I would like to thank Isabel, Sandra and Rita for their friendship and encouragement, especially in the hard moments, but mostly for the very good times we have spent together. I also want to thank my long time friends, especially Mafalda, Raquel, Lena, Zé, Daniel, Ricardo, Rita e Leila.
My deepest thanks to everyone in the Laboratoire de Catalyse en Chimie Organique, especially to Béatrice, Muriel, Mihaela, Bachar and also to Cédric, Sébastien, Pierre, John, Orlando, Filipa Madeira, Sonia, Roman, who made my stay in Poitiers so pleasant. I wish to express my gratitude to Professor Isabelle Gener for the attention and assistance she gave to me in my arrival at Poitiers and also to Maryvonne Choumil.
I would like to acknowledge the Fundação para a Ciência e a Tecnologia de Portugal for the financial support through the doctoral grant SFRH/BD/19843/2004.
My heartfelt thanks go to you, Luís, for your support and for making me happy. Most important, I wish to thank the unconditional support and patience of my parents, Leonor and Victor, and my sister, Maria João, to whom I dedicate my thesis.
Lisbon, 14 September 2008
General Index
I. General Introduction ………. 1
II. Literature Review ……….. 7
III. Experimental Part ……… 67
IV. Physicochemical Characterization of HMCM-22 zeolite ………. 87
V. MCH transformation over HMCM-22. Mechanisms and location of the reactions… 115 VI. n-heptane transformation over HMCM-22: catalytic Role of each pore system... 173
VII. MCH and n-heptane transformations over HMCM-22: comparison with large and medium pore zeolites ……… 197
VIII. Dealumination of HMCM-22 by acid leaching ……… 209
C
C
h
h
a
a
p
p
t
t
e
e
r
r
I
I
G
Zeolites are microporous aluminosilicates with well-defined crystal structures that have found widespread applications, due to their particular ion exchange properties, acidity, catalytic activity, thermal stability and shape selectivity. Owing to their remarkable characteristics, together with their commercial interest, there has been much progress in the synthesis of zeolites and several new framework topologies have been prepared.
MCM-22 zeolite (MWW framework type) was firstly synthesized by Mobil in 1990 and has a very original structure. Its framework topology is comprised of two independent bidimensional pore systems, each accessible through 10 MR openings located on the ab-plane. One of the pore systems is formed by sinusoidal channels delimited by 10 MR (4.1 Å × 5.1 Å) with intersections of (6.4 Å × 6.9 Å). The other one is constituted by large supercages with 12 MR (inner diameter 7.1 Å and 18.2 Å height) each connected to six others through 10MR windows (4.0 Å × 5.5 Å). In addition, the outer surface of crystals presents large 12 MR pockets (also denominated as hemicages or cups) which correspond to half supercages (diameter of 7.1 Å, depth of 7.0 Å).
A material with such a peculiar structure has attracted attention for catalytic purposes and has been studied in several hydrocarbon conversion reactions. Moreover, HMCM-22 is nowadays used by Mobil for cumene production, through liquid phase alkylation of benzene with propylene (Mobil-Badger cumene processes), and ethylbenzene production by benzene alkylation with ethylene (Mobil/Raytheon EBMax). The selective (high monoalkylation) and stable production of ethylbenzene and of cumene over the HMCM-22 zeolite is explained by catalysis over the protonic sites of the outer hemicages.
Since the three pore systems of HMCM-22 zeolite (supercages, sinusoidal channels and outer hemicages) contain protonic acid sites, acid catalyzed transformations of various reactants can occur in each of them with large differences in rate, selectivity and stability. Then, it is essential to know the catalytic participation of each pore system in a given reaction. In the literature a method to determine the catalytic participation of each pore system of HMCM-22 for m-xylene transformation was developed. According to this study, the catalytic role played by each of the three pore systems of MCM-22 zeolites can be quantitatively determined by the selective supercage deactivation by coking and poisoning of the external cups acid sites with 2,4-dimethylquinoline, a bulky base molecule which hardly enters the inner pore systems.
The objective of this thesis was to the determine the catalytic role of each pore system of HMCM-22, by adapting the method reported in the literature, and study the deactivation of
this zeolite during the methylcyclohexane and n-heptane transformations, which are representative of naphthenes and linear alkanes transformations, respectively. The catalytic role of the three types of micropores is specified and mechanisms are proposed for the formation of the main reaction products. This was previously done for the transformations of the three xylene isomers and for toluene alkylation with propene, which are reactions involving relatively basic molecules capable to chemisorb and react on relatively weak acidic sites. The originality, with respect to the transformation of xylenes (reactants with similar molecular sizes), is that methylcyclohexane and n-heptane are non-basic reactants and, consequently, the activity of the acidic sites of the three pore systems could depend more largely on their strength. Moreover, the acid catalyzed methylcyclohexane and n-heptane transformations systems are more complex than xylene transformations: a large number of primary and secondary reactions can occur with therefore possibility of co-participation of the acid sites of the pore systems.
Furthermore, as n-heptane is a linear alkane, its diffusion into the inner micropores is easier than that of methylcyclohexane and diffusion limitations should play a more limited role in n-heptane than in methylcyclohexane transformation. There is another difference between n-heptane and methylcyclohexane transformations through acid catalysis, related to the carbocations they involve as transition states: the former reaction is more difficult to catalyze than the second one, hence could require stronger acid sites for its catalysis. Therefore, the comparison between the rates and selectivities of these saturated hydrocarbon transformations could provide useful information on both the shape selectivity of the pore systems and on the strength of their acid sites.
Adsorption studies were carried out with different probe molecules, specifically nitrogen, mesitylene, p-xylene, o-xylene and methylcyclohexane over fresh and deactivated HMCM-22 samples, to study the micropore volume accessible to these molecules, but also the effect of coke on each pore system of HMCM-22 zeolite.
The difference of shape and size of the pore systems of HMCM-22 can explain why the catalytic properties of this zeolite have been found intermediate between those of large and intermediate pore size zeolites in different reactions. In this work, in order to inspect the intermediate behavior of HMCM-22 between large and intermediate pore zeolites, the catalytic results of HMCM-22 during methylcyclohexane and n-heptane transformations were compared to those obtained over HUSY (FAU) and HZSM-5 (MFI), used as references of large and intermediate pore size zeolites, respectively. Moreover, as it was possible to
determine the catalytic role of each pore system in both transformations, their product distributions were also compared with HUSY and HZSM-5 zeolites.
The effect of the partial pressure of methylcyclohexane over HMCM-22 was studied with the objective to investigate the influence of this parameter in the overall activity of the zeolite. The influence of temperature on the product distribution obtained during methylcyclohexane transformation over HMCM-22 was also analyzed.
Dealumination by acid leaching was performed over the HMCM-22 zeolite and its influence on the physicochemical characteristics of the zeolite was studied by different techniques, in particular, X-Ray diffraction, nitrogen adsorption and infrared spectroscopy measurements with and without pyridine adsorption. Methylcyclohexane transformation was carried out over the dealuminated samples in order to investigate the effect of dealumination in the global and in each pore system catalytic properties of the zeolite.
Outline of the thesis
After this introductory chapter, the literature review is presented in chapter II and the experimental procedures employed in this work are described in chapter III.
The results obtained are presented in chapters IV-VIII. Chapter IV presents the physicochemical characterization by different techniques of the studied HMCM-22 sample. Chapter V is dedicated to the methylcyclohexane transformation over the HMCM-22, in which it will be established the catalytic role of each pore system of HMCM-22 in this reaction. Furthermore, a thorough study of deactivation and coke location is presented. The effect of the operating conditions, specifically temperature and partial pressure of methylcyclohexane, are also presented. Chapter VI concerns the n-heptane transformation over HMCM-22, for which the catalytic role is also determined. Chapter VII covers the comparison of the catalytic data obtained for both reactions over HMCM-22 with large (HUSY) and intermediate (HZSM-5) pore size zeolites. In Chapter VIII there are presented the results obtained for the dealumination through acid treatment on the zeolite HMCM-22, being discussed its influence in each pore system. Chapter IX summarizes the conclusions of this work and some future work is proposed.
C
C
h
h
a
a
p
p
t
t
e
e
r
r
I
I
I
I
L
Chapter II Index
1. ZEOLITES ... 9 1.1. STRUCTURE...10 1.2. ACIDITY...11 1.3. SHAPE SELECTIVITY...14 1.4. CHARACTERIZATION OF ZEOLITES...18 1.4.1. Structure Identification ...181.4.2. Morphology and Crystallite size ...18
1.4.3. Chemical Composition ...19
1.4.3.1. Framework Chemical Composition ...19
1.4.4. Determination of Zeolite Acidity ...19
1.4.4.1. Hydroxyl groups vibration bands ...20
1.4.4.2. Probe Molecules ...21
1.4.5. Adsorption properties ...22
2. MCM-22 ZEOLITE ... 24
2.1. STRUCTURE...24
2.2. CATALYTIC PROPERTIES...27
2.2.1. Catalytic role of each pore system ...29
2.2.2. Industrial Application...30
2.3. ADSORPTION PROPERTIES...31
3. REACTIONS AND MECHANISMS ON ACID ZEOLITES ... 33
3.1. CRACKING MECHANISMS...33
3.1.1. β-scission cracking ...34
3.1.2. Protolytic cracking mechanism ...37
3.1.3. Oligomerization reactions ...40
3.1.4. Hydrogen transfer ...41
3.1.5. Model compounds reactions...42
3.1.5.1. Methylcyclohexane cracking mechanism ...44
3.1.5.2. n-Heptane cracking mechanism...46
4. DEACTIVATION OF ACID ZEOLITES... 48
4.1. COKE FORMATION...48
4.2. COKE FORMATION MECHANISMS...52
4.3. COKE CHARACTERIZATION...53
4.4. COKE ELIMINATION: REGENERATION...54
1. Zeolites
Zeolites are microporous crystalline aluminosilicates discovered in 1756 by Baron Axel Fredrik Crönsted. This Swedish mineralogist observed that stilbite, a natural mineral, released water when heated. This new class of materials was designated as zeolites, from the Greek words ‘‘zeo’’ (to boil) and ‘‘lithos’’ (stone) meaning ‘‘stone that boils’’ [1-6]. Each zeolite framework topology is identified by a three capital letter code, established by the Structure Commission of the International Zeolite Association (IZA) [7].
Many zeolites occur naturally (about 40) but, until now, more than 170 (zeolite or zeolite-like materials) have been synthesized [6,8-10]. Although many structures exist, only about 17 are of commercial interest: AEL, AFY, BEA, CHA, EDI, FAU, FER, GIS, LTA, LTL, MER, MFI, MOR, MTT, RHO, TON and MWW [6]. The different applications found for these materials are related to their properties of ion exchange, acidity, high activity, thermal stability and shape selectivity [1-6, 8-10]. Zeolites are used as:
¾ Catalysts: the main industrial catalytic applications are in petroleum refining (e.g.
catalytic cracking of vacuum distillates for the production of gasoline and light olefins), petrochemicals production (e.g. benzene alkylation) [1-6,8-10] and fine chemistry processes (e.g. acetylation of anisole and veratrole with acetic anhydride) [2,9,11,12].
¾ Adsorbents: drying, purification and separation. They can be used as very effective
desiccants, for gas purification (e.g. removal of volatile organic compounds), and in separation processes (e.g. p-xylene from its isomers and n-paraffins from branched paraffins) [2,3,6].
¾ Ion exchangers: detergents. Zeolites have replaced phosphates, harmful for the
environment, as water-softening agents, by exchanging the sodium in the zeolite for the calcium and magnesium present in the water [2-3,6,9].
From the applications referred above, it can be said that zeolites contribute to a cleaner environment. They make chemical processes more efficient, thus saving energy, producing fewer by-products, and waste, and consequently reducing pollution. Other examples are their use in removing atmospheric pollutants and harmful organics from water [2,8,9].
1.1.
Structure
The zeolite framework is a three-dimensional arrangement of tetrahedral coordinated silicon or aluminium atoms (TO4, T = Si, Al, primary structural units), linked to each other by
the sharing of the oxygen atoms, forming secondary building units (SBU, well defined-shapes such as cubes or hexagonal prisms). These units will be assembled to originate large lattices by repeating identical building blocks (unit cells), as shown in Fig. 2.1 for the FAU zeolite.
Fig. 2.1 Tetrahedra arrangement forming the sodalite cage and finally the structure of the zeolite FAU.
Each aluminium (T-site) introduces a negative charge in the framework (AlO4-), which
is balanced by a metallic cation (M+, such as Na+, Mg2+, Ca2+, K+) or by a proton (H+). These cations are mobile and can undergo ion exchange by other cations. The structural formula of a zeolite is based on the crystallographic unit cell, the smallest unit of the structure, represented by:
Mx/n [(AlO2)x(SiO2)y].zH2O
where n is the valence of the counter cation M, z is the number of water molecules per unit cell, (x + y) is the total number of tetrahedra per unit cell and y/x the atomic Si/Al ratio. According to the Lowenstein rule, an oxygen atom can not be covalently bound to more than one aluminium atom in the framework, i.e., Al-O-Al groups do not exist in zeolite networks. Consequently, the atomic Si/Al ratio varies from a minimal value of 1 to infinite [1-3]. The
odalite os odalite os FAU zeolite Sodalite cage 24 tetrahedra
secondary building units can be assembled in different ways which is responsible for the large diversity of zeolite structures. The zeolite framework may contain one-, two- or three-dimensional uniform channels (interconnected or not) or cages [1]. Most of the zeolites can be classified into three categories, according to the number of T-atoms in their largest ring [1-4,13]:
¾ Small pore zeolites: eight-member ring pore openings (8 MR) with free diameters of
3.0 to 4.5 Å (e.g. zeolite A);
¾ Medium pore zeolites: formed by a ten-member ring pore apertures (10 MR) with
free diameters varying in the range 4.5-6.0 Å (e.g. zeolite ZSM-5, Theta-1);
¾ Large pore zeolites: twelve-member ring pore openings (12 MR) of 6.0-8.0 Å (e.g.
zeolites Y, Beta).
Besides the pore sizes of zeolites presented above, there are 6-member ring (6 MR) zeolites (e.g. sodalite, ZSM-39), which have no catalytic significance, since their pore apertures are too small, even for molecules like methane [13]. There are also other materials, similar to zeolites (designated as zeotypes), with more than 12 MR pore openings, such as the AlPO4-8 (MCM-37) [14] (14 MR, 7.9 × 8.7 Å), the VPI-5, an aluminophosphate (18 MR,
12.7 Å) [15,16], the Cloverite, a gallophosphate, which has a dual pore structure with 20 and 8 MR (6.0 × 13.2 Å) [17], synthesized by introducing elements other than Si and Al as T-atoms.
1.2.
Acidity
The acidity of zeolites is mainly due to the presence of Brønsted acid sites but Lewis acid sites may also be present, especially after high temperature treatments.
The protonic sites arise from the presence of a negative charge on each AlO4- unit of
the zeolite framework, which must be compensated by cations. When protons (H+) neutralise the framework charges, bridging hydroxyl groups are formed (Si(OH)Al), in which the proton is attached to the oxygen atom linked to Si and Al atoms. These are the Brønsted acid sites of zeolites (capable of transferring H+), responsible for important reactions in petroleum
refining, as the catalytic cracking reactions [2-4,18]. The as-synthesized zeolites are generally obtained in the Na+ form (or with other alkaline ions as counter cations). The introduction of H+ is achieved by ionic exchange with NH4+, followed by heating (calcination) at elevated
temperatures (300–500ºC) to remove the ammoniac and leave the H+ ions, forming the Brønsted acid sites, as illustrated in Fig. 2.2.
Si O O O Al -O Si O O O O O O NH4+ Si O O O+ O Si O O O O O O H Al δ+ δ− 300ºC - 500ºC -NH3
Fig. 2.2 Formation of the bridging hydroxyl groups (Brønsted acid sites) by calcination at elevated temperatures
of the NH4+-form of the zeolite [19].
Fig. 2.3 represents the bond formed between the proton and the zeolite framework. It is visible that one of the oxygen atoms forms three different bonds and has a negative charge. Since oxygen is more electronegative than hydrogen, there is a transfer of electronic density from the hydrogen atom to the oxygen. Thus, the O-H bond has a mainly ionic character and it is the hydrogen that accommodates the positive charge. Consequently, the O-H bond is very weak and can easily be broken, causing the proton to have a large mobility over the zeolite structure, as illustrated in the figure, and to be a very strong Brønsted acid site [3,18-20].
Si O O O+ O Si O O O O O O H Si O O O O+ Si O O O O O O H Al Al δ+ δ+ δ− δ−
Fig. 2.3 Mobility of the proton in bridging hydroxyl groups [19].
Brønsted acidity is also related with the framework Si/Al ratio, since the maximum theoretical number of protonic sites is equal to the number of tetrahedral aluminium atoms present in the framework. However, the number of protonic sites is smaller than the theoretical number due to incomplete ion exchange and to the occurrence of dehydroxylation, and even dealumination, during the zeolite activation at high temperatures, which creates extraframework Al species (EFAl) [2,3,18]. These EFAl species as well as the tri-coordinated
Al in structural defects can give rise to the presence of Lewis acid sites (capable of accepting a pair of electrons) in the zeolites. Several types of Lewis acid sites may be present in zeolites: Al3+, AlO+, Al(OH)2+, Al(OH)2+ (cationic Al), AlO(OH), Al(OH)3, Al2O3 (neutral
Al), ≡Si+ (silicate species) [1,21,22].
The formation of Lewis acid sites can be described by the two-step model proposed by Kühl [23]. An example of a possible dealumination scheme is illustrated in Fig. 2.4. In the first step (Fig. 2.4, I) dehydroxylation occurs at adjacent bridging hydroxyl groups. After this, the system is in a metastable state and tends to evolve into a more stable state, with the help of nearby framework aluminium. Then, the Al can be easily released from the zeolite framework, if there are next nearest neighbor (NNN) Al atoms, that makes Al atoms less stable (Fig. 2.4, II). The second step is easy to carry out on aluminium-enriched zeolites (low Si/Al ratio), such as Y [24]. However, in zeolites with high Si/Al ratio, where nearly all the Brønsted acid sites are isolated, the second step is less favored [22,25].
O O O Si O O O O Si O O O O O Al Si O O O+ O Si O O O O O H O O+ Si O O O O O H Al Al - Al Si O O O + -H2O O C Si O O O O Si O O O O O Al -Si O O O AlO+ I II O
Fig. 2.4 Two-step mechanism for the formation of EFAl species: I) dehydroxylation and II) dealumination.
Adapted from [22].
Other types of hydroxyl groups can be present such as silanol groups (Si-OH), which can be acidic, but with lower strength than the other hydroxyl groups [1-3].
The acidity of a zeolite depends on the concentration of Brønsted sites but also on their strength. Several factors can influence the acid strength of the Brønsted sites:
¾ T-O-T bond angle: the acid strength of the associated proton increases with the angle
of the T-O-T bond. Accordingly, the protonic sites of HMOR (bond angle between 143-180º) are stronger than those of HFAU (138-147º) [2-4,18].
¾ Ionic exchange degree: the acid strength of the protonic sites increases with the ionic
exchange degree [2-4].
¾ Proximity of the acid sites: the acid strength of an acid site depends on the number of
the next nearest neighbor (NNN) Al atoms [2-4]. The acid strength of a given site increases when the number of tetrahedral Al in the NNN position of the Al that supports the acid site decreases and will be maximum at zero Al as NNN. This fact can be explained by the higher electronegativity of Si compared to Al. Indeed, there is a higher electronic density transfer from the O to the Si and, consequently, from the H to the O, than with the Al atom, which increases the ionic character of the OH bond, hence the acid strength. This also means that the acid strength increases with increasing framework Si/Al ratio, because there is a lower density of acid sites and they will be more isolated [26].
¾ Interaction with Lewis acid sites: The acid strength of Brønsted sites can be
enhanced through the interaction with neighboring extraframework aluminium species (Lewis acid sites) due to an inductive effect [2,18,27-33]. This effect can be responsible for the increase in activity of some zeolites [18,29,30,33]. However, EFAl species can also have disadvantageous effects: i) they may block the access to the active acid sites and ii) the cations could neutralize the charge in the framework, which causes a decrease in the number of Brønsted acid sites [3,34].
1.3.
Shape Selectivity
Most of the active sites are located in the intracrystalline pore structure, where there may exist spatial constraints in the formation of transition states, or reaction intermediates, and in the diffusion of molecules. The diffusion in the pores depends on the sizes and shapes both of the molecules and of the zeolite pore structure. Consequently, zeolites can permit or reject the entry of molecules into their pores, hinder the formation of some molecules within them or prevent their exit from them. This characteristic is denominated shape selectivity and this is why zeolites are also usually designated as molecular sieves [13,35-40].
Indeed, the zeolite structure can be selected for the separation of molecules of different sizes and shapes or to favour or prevent a given reaction. Hence, a comparison between the pore openings of the zeolites and the kinetic diameter of the molecules can give information about the molecules that are allowed or not to enter the zeolite. However, the zeolite
framework should be viewed as flexible (as well as the molecules) and it should be pointed out that the pore apertures depend on the temperature [2]. Temperature increases the flexibility of the molecules but also the vibrations of the pore mouth and framework bonds of the zeolite.
Weigel and Steinhoff reported, in 1925, the first molecular sieve effect with dehydrated chabazite [1,41]. They observed that this zeolite rapidly adsorbed water and formic acid but excluded acetone, ether and benzene. In 1960, Weisz and Frilette observed the first shape selective effect on catalysis with the zeolite LTA (CaA), which could not convert isobutanol but was able to dehydrate 60% of 1-butanol [42]. Since then, the research in this area grew rapidly and shape selectivity became the basis of many industrial processes, namely in oil refining and petrochemical processes [5,13,35,38,39]. The three well-accepted types of molecular shape selectivity are presented below and are related to size exclusion [1-3,13,35,38-40]:
¾ Reactant Shape Selectivity: the zeolite pores only allow some of the reactant
molecules to enter and diffuse through the channel system. An example of this selectivity is the dehydration of n-butanol but not of iso-butanol in Linde 5A zeolite, because the branched reactant can not enter the pores of this zeolite. Another example is the Selectoforming [43], the first commercial process based on reactant shape selectivity (1968), in which there is the selective cracking of n-paraffins (4.3 Å) in the presence of isoparaffins (5.3 Å), since the isoparaffins, although easier to crack than n-paraffins, cannot penetrate the pores of the erionite zeolite. (see Fig. 2.5)
ERIONITE
+
∗ x
Fig. 2.5 Example of reactant shape selectivity: selective cracking of n-paraffins over NiH-Erionite zeolite.
Adapted from [39].
¾ Product shape selectivity: based also on the principle of size exclusion, certain
product molecules, formed inside the pore structure, are too bulky and can not diffuse out of the zeolite. These larger product molecules may be formed in the cages or in the larger intersections of the channels but can not exit from the zeolite network due to their sizes and
shapes. The selective toluene disproportionation to produce p-xylene over H-ZSM-5 zeolite, illustrated in Fig. 2.6, is an example of this type of selectivity.
2
+
Fig. 2.6 Example of product shape selectivity: selective toluene disproportionation into p-xylene over ZSM-5
zeolite. Adapted from [39].
Besides size exclusion, reactant and product shape selectivity can be observed if there are differences in the diffusion and/or in the reactivity of the molecules. Indeed, even when both molecules can enter the pores, if the diffusivity of one is significantly lower than the other, then its access to the active sites and thus, its reactivity, is disfavored against the other.
Transition state shape selectivity or spatioselectivity: the space available in the cages and channels around the active sites sterically constraint the formation of some reaction intermediates or transition states and only certain conformations are allowed. An example is the disproportionation of m-xylene over HMOR (Fig. 2.7). In the case of reactions that can occur trough mono or bimolecular mechanisms, the decrease in the space available near the active sites decreases the participation of the bimolecular mechanism.
2
Fig. 2.7 Example of transition state shape selectivity: 1,3,5-trimethylbenzene can not be formed because the
Another type of shape selectivity is the concentration effect related to the increase in concentration of the reactants when they are inside the pores of the zeolite, which has a positive effect on the reaction rates, favoring bimolecular reactions over monomolecular ones [1-3]. Other types of shape selectivity were proposed but are still subject to debate [2,13,39]: inverse shape selectivity [44,45], nest effect [13], pore mouth and key-lock catalysis [46,47], molecular traffic control [48] and the window (or cage) effect [49].
A particular case of the product selectivity is the inverse shape selectivity (ISS), proposed by Venuto et al. [44,45], for the coke formation in the FAU zeolite, a large pore zeolite. The bulky molecules formed in the supercages (13 Å) cannot escape since they are too bulky to diffuse out through the narrow connecting windows (7.0 Å) and, consequently, are trapped inside them, leading to the deactivation of the catalyst. Further studies led to the conclusion that coke formation is a shape selective reaction directly controlled by the zeolite pore structure. The pore structures of medium pore zeolites, such as ZSM-5, which does not contain supercages, present low coke formation which is ascribed to its porous network.
The nest effect [2,13,39] is related with the selectivity of the non-shape-selective catalytically active external surface sites. If these sites are more than or as active as the intracrystalline sites, then the shape selectivity of a zeolite can be influenced by these external sites. The nest effect has been proposed to explain the high selectivity in ethylbenzene and cumene obtained with HMCM-22 zeolite, a zeolite with protonic sites in the pockets of the external surface, in the benzene alkylation by ethylene or propylene [39,50]. The monoalkylated products (cumene and ethylbenzene) are formed on the acid sites of the external cups under this effect.
The pore mouth and key-lock selectivity (PMKLS) are also related with the shape selectivity on the external surface of zeolites. They were recently proposed by Martens et al. [46,47] to explain the particular selectivity of one-dimensional, non-intersecting medium-pore zeolites, such as ZSM-22 (TON), in the hydroisomerization of long chain n-alkanes, an important reaction in changing the cold flow properties of transportation fuels and lubricants. A high selectivity into monobranched isomers is obtained with ZSM-22 zeolite and is ascribed to pore mouth catalysis, since these isomers can not desorb from the narrow channels of this zeolite. In key-lock catalysis, the opposite ends of long alkanes adsorb into two different pores leading to branching near the central atom. The distance between the pore openings on the external surface of the zeolite was proposed to be responsible for the branching of n-alkanes that occurs only in certain positions [40].
1.4.
Characterization of zeolites
As said before, zeolites are crystalline aluminosilicates with micropores of uniform size widely used in adsorption and catalytic applications. The adsorption and catalytic properties of zeolites depend not only on the operation conditions of the transformations but also on their physicochemical characteristics. There is no doubt that it is important to have a good knowledge of the pore structure of these materials in order not only to explain but also to predict their adsorption and catalytic behavior. Zeolites present different physicochemical properties which are very important to characterize, such as: i) the structure and the cristallinity of the sample, ii) the shape and size of the crystallites, iii) the chemical composition and, when possible, the framework Si/Al ratio, iv) the nature, concentration and acid strength of the acid sites, v) the adsorption properties. Several techniques are applied in the characterization of zeolites and can be found extensively described in the literature [3,51-56]. The more common are briefly presented below.
1.4.1. Structure Identification
X-Ray Diffraction (XRD) is used to check the structure of the zeolite. This technique is based on the fact that every crystalline material has its own characteristic X-Ray pattern. When the X-Ray diffraction pattern is compared to the standard, if extra peaks are observed, this indicates that other crystalline phases are present and the obtained sample is not pure; or if peaks are missing, the structure is different. The cristallinity of a sample can be determined by comparison of the intensities of the same peak(s) of the standard [51-53].
1.4.2. Morphology and Crystallite size
Scanning Electron Microscopy (SEM) is the most adequate technique to determine the morphology and the crystallite size. However, a less accurate method that can also give information about the crystallite size is the XRD using the Scherrer equation [3,51].
1.4.3. Chemical Composition
The chemical composition of a zeolite is usually defined by its molar Si/Al ratio. Different techniques can be used to determine the chemical composition of a zeolite. The most commonly used are Atomic Absorption Spectrometry (AAS), Atomic Emission Spectrometry (AES) and X-Ray Fluorescence Spectrometry (XRF) [3,52,55]. Prior to analysis, the samples are prepared by different methods: dissolution by acid treatment or acidic and basic fusion by heating in melts. Generally, for atomic spectrometry, samples are mainly dissolved in concentrated acids (e.g. HF, H2SO4, HNO3) and diluted. Detailed
description of these methods and techniques can be found in the literature [55].
1.4.3.1. Framework Chemical Composition
The techniques referred above only give information about the total amount of aluminium in the zeolite; they do not distinguish between the framework and non-framework aluminium. As the Brønsted acidity is due to the bridging hydroxyl groups, it is important to know the framework aluminium existent in the zeolite. 29Si and 27Al MAS-NMR (Magic Angle Spinning Nuclear Magnetic Resonance) allow the determination of the framework Si/Al ratio [51,54,55,57].
For the Y zeolite, a relation between the framework aluminium and the unit cell dimensions [51] and between the number of lattice aluminium atoms and the symmetric stretch vibration bands of the infrared spectra was also developed [51,58].
1.4.4. Determination of Zeolite Acidity
The most widely used technique to determine the acidity of a zeolite is the Transmission Infrared Spectroscopy using Fourier transform equipments (FTIR). The acidity can be determined by the study of the hydroxyls (OH) vibration bands and by analysis of their interaction with suitable probe molecules. By both methods, infrared bands (IR) ascribed only to Brønsted acid sites are produced. With the adsorption of specific probe molecules, it is also possible to identify the bands of Lewis acid sites. The concentration of the Brønsted and Lewis acid sites can then be estimated through a modified Lambert-Beer law, using the integrated absorbance of the corresponding bands (eq.1):
where νi is the wavenumber (cm
-1) corresponding to the beginning of the band,
f
ν is the wavenumber corresponding to the end of the band, Ai is the absorbance, εi is the extinction
coefficient (cm µmol-1) corresponding to the band of the species i,
l is the optical path (thickness of the wafer), S is the surface of the wafer (cm2), ni is the mol number of species i,
Ci is the concentration of i (Brønsted or Lewis sites) (µmol g-1) and m is the wafer mass (g).
The concentration of these sites is important to determine, not only on the fresh catalyst, but also on aged samples (coked) in order to study the deactivation of catalysts.
1.4.4.1. Hydroxyl groups vibration bands
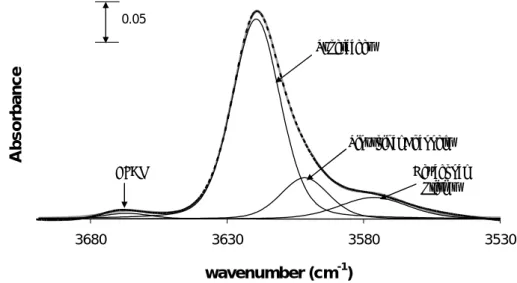
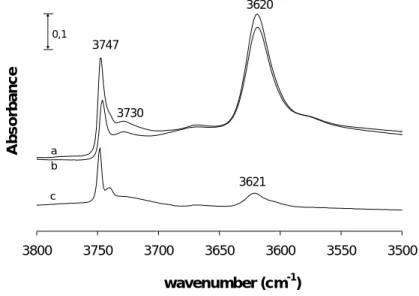
The OH bond stretching vibration originates bands in the range of 3500 cm-1 to 3800 cm-1. Generally, the weaker the OH bond, the lower the stretching frequency and the higher the acid strength. These bands can be ascribed to:
¾ OH of terminal framework silanol groups (Si-OH) at about 3745 cm-1 (non-acidic or with low acidity).
¾ OH of internal silanol groups corresponding to hydroxyl nests occurring at defect sites (~3725 cm-1), also non-acidic or with low acidity.
¾ OH groups attached to extra-framework aluminium species (Al-OH, ~3670 cm-1).
¾ Al-OH-Si, bridging hydroxyl groups, responsible for the Brønsted acidity of zeolites
(~3600 - 3650 cm-1).
The position of these bands is characteristic of each zeolite, so the values given above can vary depending on the structure of the zeolite [51,54,56]. For some zeolites, only one band corresponding to the bridging hydroxyl groups appears but for others, with more complex structures, such as the HY zeolite, different bridging OH bands appear, due to the
( )
( )
m S d A C S n d A i i i i i i f i f i ε ν ν ε ν ν ν ν ν ν × = ⇔ ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ =∫
∫
l l eq.1different location of these groups inside the zeolite structure [54,56]. The estimate of the concentration of the bridging hydroxyl groups is only possible if the extinction coefficient of this band is known. This parameter was determined for different zeolites (HY, HX, HMOR, HFER) and can be found in the literature [54].
1.4.4.2. Probe Molecules
As it was referred before, in the characterization of the acid sites it is important to know its nature, concentration and acid strength. The adsorption of probe molecules followed by infrared spectroscopy is an efficient tool for this purpose. Besides, the adsorption of probe molecules can give information about the accessibility to the acid sites. Testing bases of different shapes and sizes, followed by infrared spectroscopy, also allows the study of the acid sites accessibility. Several probe molecules can be used to study the acidity of zeolites: pyridine, ammonia, carbon monoxide, acetonitrile (and deuterated acetonitrile), nitrogen and others. However, not all of the probe molecules allow the distinction between Brønsted and Lewis acid sites. Pyridine is one of the most suitable and commonly used probe molecules to identify the nature of the acid sites and their concentration [54,56,59].
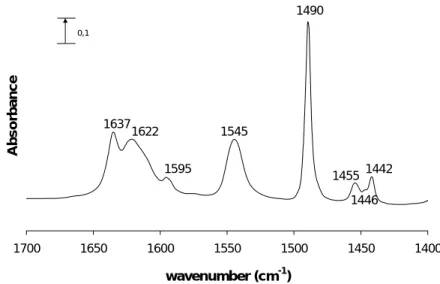
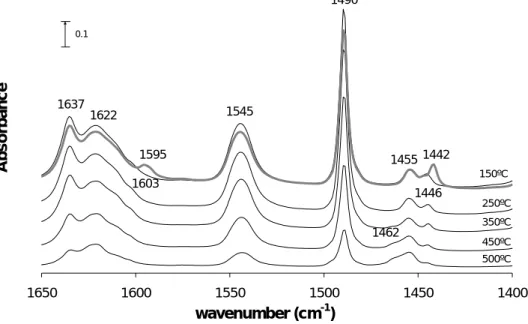
When pyridine is chemisorbed on the catalyst, it can interact with bridging hydroxyl groups (Brønsted acid sites) by protonation, forming pyridinium ions (PyH+, ~1545 cm-1 and 1640 cm-1), with Lewis acid sites by coordination (PyL), giving rise to bands around 1445-1460 cm-1 and 1600-1630 cm-1 (depending on the acid strength) [59,60] and by hydrogen bond (Py….OH) with OH groups of low acid strength (~1445 cm-1 and 1595 cm-1), as silanols [3,54,56]. The pyridine interaction with Lewis acid sites can produce bands in the range 1430-1450 cm-1 [61,62] while those of Lewis sites originated from the extra-framework aluminium species are around 1445-1460 cm-1. Another band at ca. 1490 cm-1 is also present and corresponds to the interaction of pyridine both with Brønsted and Lewis acid sites and also through hydrogen bonding. The physisorbed pyridine produces bands at ca. 1580 and 1440 cm-1 and is eliminated before the measure, under vacuum, in order to detect only the chemisorbed pyridine. The IR charactheristic vibrations of the interaction bands with pyridine are presented in Table 2.1 [56].
The integrated surface of the bands corresponding to Brønsted and Lewis sites allow the determination of their concentration, using eq.1 and the extinction coefficients of each band. The bands used for this purpose are those corresponding to the vibration modes (υ19b),
more specifically those at 1545 cm-1 (Brønsted acid sites) and 1455 cm-1 (Lewis acid sites). Several extinction coefficients for the ~1545 cm-1 band have been published for different zeolites [54,56,60-67] and for the Lewis sites [54,67-69].
Table 2.1 IR characteristic position bands of pyridine in interaction with acid sites.
Interaction Type Vibration mode
PyH+ Py-L Py….OH
υ8a υ(CC) 1640 1600-1630 1595
υ19b υ(CN) 1545 1445-1460 1445
Since pyridine is a voluminous molecule, sometimes it is not able to enter and reach all the acid sites of the zeolite structure, which depends on the shape and size of the channels and cavities (e.g. the 8-MR of FER zeolite [70]). Then, other types of probe molecules, smaller than pyridine, should be used. Ammonia and acetonitrile are probe molecules frequently used in the determination of the acidity [54,56]. Detailed description of the application of these molecules and of others can be found in the literature [54,56,59].
Another important characteristic to study is the strength of the acid sites. This can be achieved by desorption of pyridine at different temperatures, followed by infrared spectroscopy. Then, the higher the temperature for the base to be released from the acid site, the higher the strength of the acid site. Nonetheless, it should be underlined that desorption of the probe molecules can be influenced not only by the acid strength of the sites but also by the facility of desorption of the molecules from the pores. Thus, this method can only be applied to compare the acid strength of zeolites with similar porosities [3].
Another method to determine the acidity of zeolites is the Temperature-Programmed Desorption (TPD) of ammonia, which is not able to distinguish between Brønsted and Lewis sites, but allows the determination of the distribution of the acid sites strength (low temperature (LT) - lower strength and high temperature (HT) - higher strength) [3,55,56].
1.4.5. Adsorption properties
Small variations in the size of the pores can have a drastic impact on the performance of zeolitic materials as adsorbents and catalysts. Furthermore, in catalytic applications, the
type of pore structure has strong implications on the reaction mechanisms involved in the transformations and on the type of products obtained [2,13,38,39].
Nitrogen adsorption at –196 ºC is used as a routine technique for the determination of micropore volume. However, a detailed evaluation of the micropore structure of the zeolites requires adsorption measurements using other adsorbates, in particular organic compounds of different sizes: only molecules with kinetic diameter smaller than the pore openings can be adsorbed while those that are larger can not [3,51,55]. The kinetic diameters of Lennard-Jones are used to predict the accessibility of a zeolite for a given adsorbate [71]. Nevertheless, it should be pointed out that the adsorption temperature can influence the results: the dimensions of molecules and pore apertures vary with temperature. The comparison of the adsorption of the same probe molecule(s) on different zeolites can also give information on the size and accessibility of the pore structure.
Additionally, the adsorption of probe molecules can be very useful in the analysis of catalytic results if the molecules used as adsorbates are reactants or products in catalytic transformations. This type of study is also very important in aged samples of catalysts in order to evaluate the decrease of the internal micropore volume accessible to the adsorbate.
The adsorption molecules should be inert in the conditions in which adsorption is carried out, but some of them become reactive at high temperatures, in the presence of the zeolite. Others should be avoided, because even at low temperature they are reactive, such as olefins. Alkanes (e.g. 3-methylpentane, n-hexane, cyclohexane), non-reactive at low temperatures, are frequently used organic molecules in adsorption measurements [72], together with aromatics, namely xylenes (p-, m-, o-xylene) and trimethylbenzenes. Another way of probing the internal space available for catalytic reactions is with model compound reactions, which will be further discussed. Adsorption of different probe molecules, followed by infrared spectroscopy, can also give information about their accessibility to the pore structure [54].
Different methods can be used to determine the micropore and mesopore volume of the zeolite accessible to a given probe molecule and the external surface of the crystallites [73,74]. The most widely used are the t-plot [75] and the αs methods [73,74,76]. These
methods will be detailed in Chapter III. There are six types of isotherms, according to the IUPAC system [73,76]. The shape of the adsorption isotherms can identify the type of pore structure, which can be microporous, macroporous, mesoporous or microporous and mesoporous [3,73].
2. MCM-22 Zeolite
2.1.
Structure
MCM-22 zeolite (MWW structure type) was firstly synthesized by Mobil in 1990 [77]. This zeolite has a very original structure (Fig. 2.8) and is considered as an intermediate pore size zeolite. It contains two independent bidimensional pore systems each accessible through 10 MR openings located on the ab-plane: sinusoidal channels delimitated by 10 MR (4.1 Å × 5.1 Å), with intersections of (6.4 Å × 6.9 Å) (Fig. 2.9 a, Fig. 2.10 a), and large supercages with 12 MR (inner diameter 7.1 Å and 18.2 Å height) each connected to six others through 10MR windows (4.0 Å × 5.5 Å) (Fig. 2.9 b,c; Fig. 2.10 b) [78]. The supercages stack one above the other by double 6-membered ring openings (hexagonal prisms), through which molecules cannot practically diffuse. Then, there is no access to the internal pores along the c-direction through the top or bottom surface of the crystal plates [79]. Moreover, the outer surface of crystals presents large 12 MR pockets (hemicages, cups) which correspond to half supercages (diameter of 7.1 Å, depth of 7.0 Å) (Fig. 2.11) [79].
La ye r 1 Layer 2 La ye r 1 Layer 2
Fig. 2.8. Schematic representation of MWW framework structure: vertices represent silicon or aluminum atoms
and oxygen atoms are omitted for clarity. The supercage is highlighted. The shaded region shows two 12 MR cups back to back connected by a double six ring (hexagonal prism) [80].
External cups Ø 7.1 Å; Depth 7.0 Å Hexagonal prism 10 MR opening of supercages (4.0 x 5.5 Å) Sinusoidal channels (4.1 x 5.1 Å) MWW supercage
Fig. 2.9 (a) Projection along the [001] direction of the sinusoidal channels and (b) of the supercages channel
system of MCM-22. Various diffusion pathways are illustrated by arrows. (c) Supercage with its six 10-ring apertures, having an inner diameter of 7.1 Å (defined by 12-MR) and a height of 18.2 Å [79,81].
a b
Fig. 2.10 Pore size apertures of a) sinusoidal channels and b) supercages [7] viewed normal to [001].
Fig. 2.11 MCM-22 structure with the large cavities (‘‘cups’’) opened to the exterior at the termination of the
zeolite crystallite evidenced [82].
MCM-22 crystallizes as a layered precursor, generally denominated as MCM-22 (P) (Fig. 2.12). Each sheet contains the sinusoidal channels and 12 MR ‘‘external cups’’ (diameter of 7.1 Å, depth of 7.0 Å).
a) Sinusoidal Channels system b) Supercages Channel system
Supercage Channel (4.0 x 5.5 Å)
c) Supercage
Fig. 2.12 Schematic ilustration of a single sheet of MCM-22 precursor [79].
X-Ray powder diffraction analysis confirmed that the MCM-22 structure was obtained by a layered precursor. MCM-22 (P) showed a contraction of the c-unit cell of about 2 Å upon calcination, which suggests that the layers in the calcined MCM-22 are closer together than in its precursor [79]. These results were also confirmed by high resolution microscopy [79]. During the first calcination of the MCM-22 precursor (MCM-22 (P)), in order to remove the template, it occurs dehydration and condensation between facing silanols of the sheets, forming Si-O-Si interlayer bridges, which origins the supercages and the final structure of MCM-22 zeolite (Fig. 2.13). The sinusoidal channels, which already exist in the sheets of the layered precursor, are also designated ‘‘interlayer micropores’’ whereas supercages are denominated as ‘‘intralayer micropores’’.
MCM-22 (P) MCM-22
Fig. 2.13 MCM-22 structure formation from calcination of the MCM-22 (P) precursor. Projection on the bc
plane. Vertices represent the framework T atoms (Si or Al). Groups SiOH condensation, organic template, 10 MR sinusoidal channels pore apertures, 12 MR semicavities (MCM-22P) and 12 MR supercages and cups in MCM-22, 10 MR pore apertures to supercages in MCM-22. Adapted from [83].
External ‘‘cups’’ (7.1 ∅ x 7.0 Å)
10-ring windows to sinusoidal channels (4.1 x 5.1 Å)
The synthesis of a boron-containing material related to MCM-22 (similar XRD pattern) named ERB-1 was another evidence of the formation of zeolite MCM-22 from its layered precursor. According to these authors, the condensation between layers occurs from 270ºC [84].
MCM-22 as a hexagonal structure (P6/mmm) with unit cell parameters a=14.390 Å, (b=a) and c=25.198 Å [7]. Scanning electron microscopy revealed that MCM-22 is formed by platelets with 1-2 µm diameter and 0.1 µm thickness, associated in particles [84-87].
2.2.
Catalytic Properties
Due to its particular pore structure, MCM-22 has been studied in several catalytic transformations, as catalytic cracking of alkanes [88-95] and alkenes [96,97], isomerization of alkenes [98-101], alkanes aromatization [86,102,103], xylenes isomerization [86,88,104-107], aromatics alkylation [82,108-112], aromatics disproportionation [113-119], etc.
As the three pore systems contain protonic sites, acid catalyzed reactions can occur in the three locations, with however large differences in rate, selectivity and stability. This could explain why the catalytic properties of MCM-22 have been found intermediate between those of large and intermediate pore size zeolites [86,88,89,91,92,94,104,120,121] in reactions as the catalytic cracking of n-heptane [88,91,94] and m-xylene isomerization [86,88,104].
With certain reactions over HMCM-22 samples, one of the pore systems can play a major catalytic role. Thus, toluene disproportionation was shown to yield selectively the slimmer para xylene isomer, which demonstrates that this reaction preferentially occurs within the shape selective 10 MR inner micropores [113,116-118]. Moreover, as this reaction requires bulky bimolecular intermediates which cannot be accommodated within the narrow sinusoidal channels (even at their intersection), toluene disproportionation was concluded to occur within the large supercages. In contrast, a predominant role of the supercages cannot be deduced from the high para selectivity of ethylbenzene disproportionation [115,122]; indeed, in average pore size zeolites without cages (e.g. MFI [123]), hence in the MCM-22 sinusoidal channels, this reaction can proceed through a deethylation-ethylation mechanism, i.e., without participation of bulky intermediates. Likewise, selective toluene alkylation with methanol into
para-xylene [111] and selective cyclopentene hydration (limited formation of the undesired
[124] but, as again, these reactions do not require very bulky intermediates and the respective role of supercages and sinusoidal channels cannot be specified.
On the other hand, the absence of shape selectivity in the liquid phase alkylation of phenol with tert-butanol over HMCM-22 (no enhanced selective production of para-tert-butyl phenol) demonstrates that this reaction does not occur in the inner micropores, hence is catalysed by the external cups sites [125]. The selective and stable production of ethylbenzene and of cumene over the HMCM-22 zeolite is also due to catalysis on the protonic sites of the external cups (section 2.2.1.) [82,126,127]. The selectivity to cymenes obtained during the alkylation of toluene by propylene over HMCM-22 also indicates that this reaction occurs mainly in the protonic sites of the external cups [108]. Indeed, no enhanced selectivity into p-cymene is observed as it was expected from the presence of 10MR channels. The selectivity into p-cymene is close to the one obtained with large pore zeolites (28.1% with Y zeolite) and lower to the obtained over ZSM-5 (86.8%) [108]. The main participation of the external cups on the alkylation of toluene by propylene was also confirmed by J. Rigoreau et al. [109]. Moreover, it was shown that oligomerization cracking of propylene, which was the main secondary reaction, occurred in sinusoidal channels forming C4-C6 alkenes while coke
formation (nondesorbed products) ocurred in supercages.
The catalytic activity of the acid sites located on the external cups was also observed in the transformation of 1,2,4-trimethylbenzene [114], which are large aromatics with difficulty in enter the inner 10 MR pores of MCM-22, being transformed in the outer pockets. Moreover, in the toluene disproportionation, it was observed that secondary isomerization of
p-xylene into m- and o-xylene occurred in the acid sites of the external cups, whereas the p-xylene formation occurred in the supercages, as referred above [113,116-118]. It was also
observed that dealuminated catalysts presented a higher selectivity into the p-xylene isomer: an increase from 29.9% (parent) to 85.3% in a dealuminated sample was observed [113]. In fact, dealumination removed selectively the Brønsted acid sites located on the external cups, which were responsible for the secondary p-xylene isomerization [113,116,118]. Similar results were obtained in the ethylbenzene disproportionation. The external acid sites were responsible for the secondary isomerization of p-diethylbenzene (DEBz) in m- and o-DEBz [115]. The preferential remove of the Brønsted acid sites located on the external cups by dealumination [115] resulted in the increase in p-diethylbenzene with the degree of dealumination. The absence of shape selectivity of these open cups was also observed during the m-xylene isomerization: the para/ortho ratio obtained was ~1.1 [129].
A good way to confirm the preferential catalytic role of the inner micropores or of the outer hemicages is to selectively poison the external protonic sites by bulky basic molecules or to passivate or dealuminate the outer surface [82,109,113-116,118,124,128-130]. At least for reactions which are carried out at relatively low temperatures, selective poisoning is the best method. Indeed, contrary to the other methods, it allows the complete deactivation of the external acidic sites [81,129]. Therefore, the relative role played in the reaction by the inner micropores and the outer hemicages can be quantitatively determined [129].
An important remark is that deactivation, which occurs essentially by coking, affects differently the protonic sites of the three types of micropores. Thus, the alkylation of benzenic hydrocarbons with propene becomes selective in the desired products only after deactivation of the inner micropores; on the fresh sample, C4-C6 olefinic side products resulting from
oligomerization-cracking of propene can be observed [109]. Therefore, the role played by the various micropores has to be established for different degrees of deactivation, that is to say, for different values of time-on-stream.
2.2.1. Catalytic role of each pore system
The very significant difference in the sensitivity to deactivation by coking of the MCM-22 pore systems was used to develop a simple method for discriminating between the catalytic role played by the supercages and the sinusoidal channels [129]. This method, developed by S. Laforge et al. [129], consists in deactivating completely and selectively, by carbonaceous compounds (coke), the protonic sites of the supercages, which is possible by aging the zeolite during m-xylene transformation at 350ºC for 24 h. With this reaction and under these conditions, carbonaceous deposits accumulate rapidly within the supercages owing to a trapping effect [106]. Indeed, in these large supercages, m-xylene can undergo successive reactions leading to products too bulky to diffuse through the narrow apertures which therefore deactivate the protonic sites by poisoning or/and blockage. In contrast, there is practically no coke formation, hence no deactivation of the sinusoidal channels and of the outer hemicages.
In the outer hemicages, this can be related to the easy desorption of the product molecules of these open hemicages (no diffusion barriers). Successive secondary reactions leading to carbonaceous products are very difficult to occur due to the difficulty in retaining and trapping molecules in these open cups [106].
In the sinusoidal channel system, due to their size, significant steric constraints inhibit the formation of large bimolecular intermediates and products enough bulky to be trapped, as coke precursors and coke (see section 4.1). Moreover, there are limitations in the trapping of product molecules owing to the small difference between the sizes of the channel intersections and of the pore apertures. These channels have pronounced shape selective properties allowing namely the selective isomerization of m-xylene into p-xylene: para/ortho ratio of ~40 (product shape selectivity) and no formation of disproportionation products (transition state shape selectivity) [129]. The second part of this method concerns the deactivation of the acid sites located on the outer hemicages. This was achieved by poisoning with 2,4-dimethylquinolyne added to the reactant, which is a bulky base that hardly enters the inner micropores, deactivating selectively the acid sites of the external cups [81,129]. In addition, adsorption of 2,4-dimethylquinoline followed by infrared spectroscopy allows the determination of the concentration of the acid sites located on the external cups of MCM-22 zeolite [131].
By coupling supercage deactivation by coking and poisoning with 2,4-dimethylquinoline, the catalytic role played by the three pore systems of MCM-22
zeolites can be quantitatively determined [129]. This was previously done with success for the transformations of the three xylene isomers [107] and for toluene alkylation with propene [109].
2.2.2. Industrial Application
MCM-22 (MWW) zeolite is nowadays used by Mobil in the liquid phase alkylation of benzene with ethene, to produce ethylbenzene, (Mobil/Raytheon EBMax) and in the alkylation of benzene with propylene to form cumene (Mobil Badger cumene processes) [126,127,132,133]. The use of MCM-22 enables to achieve very high yields and purity in these products. The Mobyl-Raytheon cumene process produces very pure cumene (99.97 wt%) at 99.7% yield. In this process, MCM-22 catalyst has demonstrated cycle lengths higher than 2 years [126,133].
The selective and stable production of ethylbenzene and of cumene over the HMCM-22 zeolite, Mobil/Raytheon EBMaxTM and Mobil-Badger commercial processes, respectively, can only be explained by catalysis over the protonic sites of the outer hemicages (‘cups’). This particularity explains the high monoalkylation selectivity of HMCM-22
catalysts [82,126,127,134]. Moreover, it was observed that the diffusion of cumene in the two inner pore systems of MCM-22 is very constrained, which is in contrast with the high activity presented by this zeolite [135]. This is evidence that the catalytic activity is due to the acid sites located on the external cups where cumene formation occurs without diffusion
limitations. The use of collidine (2,4,6-trimethylpyridine) [128,136] or 2,6-di-tert-butylpyridine [82] to poison the acid sites located on the external hemicages
enabled to verify that the main part of the alkylation reaction takes place in the external cups. During the benzene alkylation with ethylene, the ethylene conversion decreases from 95.6% on the fresh MCM-22 to 1.4% on the poisoned zeolite [136].
2.3.
Adsorption properties
Several adsorption measurements and molecular simulation studies using adsorptives with different size, as o-xylene, toluene, 1,2,4-trimethylbenzene, were performed in MCM-22 zeolite [86,137-145].
1,2,4-trimethylbenzene presented a small adsorption over MCM-22 zeolite, which was related to large diffusion problems in entering the inner micropores and ascribed to adsorption on the entrances of micropores [137,138]. However, at this time, the presence of the external cups on the MCM-22 zeolite was not yet discovered.
In which concerns the o-xylene adsorption, it was observed that its adsorption was slightly higher than 1,2,4- trimethylbenzene and it was suggested that o-xylene could enter in the 10 MR inner micropores [137,138], but with high restrictions in diffusing inside the 10 MR channels [137,138,141,143], being its adsorption limited to the pore openings. For instance, the diffusion coefficients of o-xylene in the ZSM-5 at 30ºC were so small that the zeolite apparently could not adsorb this aromatic [146,147]. Moreover, it was shown by molecular simulation of o-xylene adsorption at 42ºC on ITQ-1 that it can not enter the 10 MR sinusoidal channels whereas some o-xylene molecules can be accommodated in certain positions of the supercages. Nevertheless, the predicted o-xylene adsorption isotherm was well above the experimental one, which was ascribed to the high limitations existent to diffusion of o-xylene through the 10 MR channels and, subsequently, in diffusing into the inner supercages [143]. From kinetic studies of the adsorption/desorption of o-xylene on MCM-22 zeolite it was suggested that the adsorption is performed by a two step process: a very fast adsorption on the external cups, with a diffusion coefficient typical of small
molecules in wide pores (> 0.4 × 10-9 cm2/s), and a very slow adsorption on the 10 MR pore system with a diffusion coefficient (0.00004 × 10-9 cm2/s) which should be in the same range as for o-xylene in MFI-zeolites [141]. This hypothesis was supported by experimental data. The o-xylene uptake in the first 12 min was 0.23 mmol g-1 while after 3 days the uptake was 0.51 mmol g-1 [141]. S. Laforge et al. [107] carried out the o-xylene isomerization over MCM-22 and observed that the acid sites of the supercages participated in the reaction whereas those of the sinusoidal channels did not, which is another evidence that o-xylene can not enter the sinusoidal channels but can reach the supercages [107]. Moreover, adsorption of
o-xylene followed by infrared spectroscopy showed that 50% of the bridging hydroxyl groups
were not affected by o-xylene adsorption, even using a pressure of 8 mbar, which are most likely those located in the sinusoidal channels [107].
Concerning the diffusion of toluene, it was observed that this molecule presents the higher uptakes when compared with o-xylene, mesitylene and m-xylene [137,138]. It was concluded that it enters well in all the pore systems of the MCM-22 zeolite, which is explained by its smaller size and supported by Monte Carlo simulations [143].
p-Xylene adsorption was also studied by simulation over ITQ-1, a purely siliceous
MWW type zeolite. It was observed that it can enter and diffuse into the sinusoidal channels and its diffusion is fast, showing the absence of diffusion limitations inside these 10 MR inner pores [139]. This easy diffusion of p-xylene can explain the high para-selectivity observed for MCM-22 zeolite in the toluene disproportionation reaction presented above (see section 2.2).
3. Reactions and Mechanisms on acid zeolites
Catalytic cracking is probably the most important conversion unit in modern refineries. It involves the C-C bond rupture of the hydrocarbons present in the feedstock to produce more valuable low molecular weight hydrocarbons including light olefins for petrochemistry, gasoline and diesel. The catalytic cracking reactions over acid zeolites are accompanied by several others. Table 2.2 lists the possible reactions for each reactant family: alkanes, alkenes, naphthenes and aromatics [148]. The reaction mechanisms involved in the cracking of hydrocarbons over zeolites [3,149-155] will be described briefly in this chapter.
Table 2.2 Catalytic cracking reactions [148].
Reagents Reactions Products Reactions Products
Alkanes alkanes+alkenes
alkenes naphtenes
branched alkenes branched alkanes
Alkenes alkanes alkenes cycloalkenes Aromatics Naphtenes Naphtenes aromatics+alkenes other aromatics Aromatics polyaromatics+H2 Coke HT-Hydrogen Transfer
3.1.
Cracking mechanisms
Cracking reactions are endothermic and involve the formation of carbocations as intermediates: carbenium ions or carbonium ions. A carbenium ion contains a tri-coordinated, positively-charged carbon atom in which the three substituents are hydrogen atoms or alkyl groups (Fig. 2.14). A carbonium ion has a penta-coordinated positively-charged carbon atom, the five substituents being hydrogen atoms or alkyl groups (see section 3.1.2.).
Isomerization Cracking Cyclization HT Cracking Cracking HT Isomerization Cracking HT HT Transalkylation Condensation Condensation
Nowadays, there are two widely accepted mechanisms for the cracking: β-scission and protolytic. The participation of each one depends on some conditions, such as temperature and pore structure of the zeolite, but this will be further discussed in section 3.1.2. In addition, thermal cracking (non-catalytic) can also occur, but its participation becomes significant only at very high temperatures (> 530 ºC) [3,149-154].
3.1.1. β-scission cracking
The β-scission cracking mechanism is also referred to as the carbenium ion, bimolecular or classical catalytic cracking mechanism and was proposed based on the works of Greensfelder [156] and on Whitmore and Church’s [157] carbenium ion chemistry. This chain mechanism involves three steps: initiation, propagation and termination. In this mechanism, the scission of the bond is made in the β position relatively to the positive charge. The mechanisms for the formation of the first carbocations depend on the reactant molecule [3,149-153]. The formation of a carbenium ion from alkenes results from the protonation of the alkene by the catalyst (Fig. 2.14):
Fig. 2.14 Protonation reaction of an olefin forming a carbenium ion (R = H, CH3).
In which concerns the cracking of paraffins the following ways to the formation of the first carbenium ions have been proposed [3,149-154]:
¾ Protonation of olefins (Fig. 2.14) that are already present in the feed (as impurities of the reactant) or are formed by thermal cracking (provided that the temperature is high enough) [156].
¾ Abstraction of hydride ions by a Lewis acid site from the catalyst [158-160].
¾ Alkanes can be protonated by Brønsted acid sites, on either a C-C or C-H bond, forming a carbonium ion, which suffers further protolytic dehydrogenation or cracking, giving a carbenium ion and H2 or a short alkane [155,161] (see Fig. 2.19 in section 3.1.2).
![Fig. 2.18 Classical cracking mechanism for an alkane molecule consisting of 1) hydride transfer, 2) isomerization, 3) deprotonation and 4) β-scission [19]](https://thumb-eu.123doks.com/thumbv2/123doknet/7983236.267506/43.892.225.667.127.574/classical-cracking-mechanism-molecule-consisting-transfer-isomerization-deprotonation.webp)
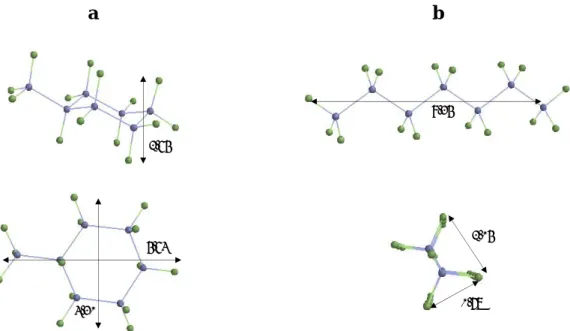
![Fig. 2.28 Location of the bulkier soluble coke molecules formed from hydrocarbons at 450ºC in the pores of a) HFAU (methylcoronene) and b) HMFI (methylpyrene) [196]](https://thumb-eu.123doks.com/thumbv2/123doknet/7983236.267506/56.892.255.643.112.426/location-bulkier-soluble-molecules-formed-hydrocarbons-methylcoronene-methylpyrene.webp)
![Fig. 3.4 Simplified scheme of the catalytic tests unit used in this work [4].](https://thumb-eu.123doks.com/thumbv2/123doknet/7983236.267506/83.892.220.658.714.1065/fig-simplified-scheme-catalytic-tests-unit-used-work.webp)