Electrostatic screening length in “soft” electrolyte
solutions
Vahid Adibnia a,b, Budha Ratna Shrestha a,‡, Marziye Mirbagheri a,b, Frederic Murschel a, Gregory De Crescenzo b, Xavier Banquy a,*
a Faculty of Pharmacy, Université de Montréal, Université de Montréal, 2900 Édouard-Montpetit,
Montreal, Quebec H3C 3J7, Canada
b Department of Chemical Engineering, Ecole Polytechnique de Montreal, P.O. Box 6079, succ.
Centre-Ville, Montreal, Quebec H3C 3A7, Canada
AUTHOR INFORMATION
‡Current address: 4700 King Abdullah University of Science and Technology, Thuwal
23955-6900, Kingdom of Saudi Arabia
Corresponding Author
*[email protected], Tel.: (514) 343-2470
ABSTRACT. Using the Surface Forces Apparatus on solutions of polymeric ions, the effect of specific ion-ion correlations is evaluated on the characteristic decay length, λ , of screened electrostatic interactions between charged surfaces. Electrolyte solutions composed of point charges surrounded by repulsive polymeric shells were used to elucidate the role of ions size and size asymmetry between co- and counter-ions on the screening of electrostatic forces. In
electrolytes composed of large polymeric cations and small point-charge anions, because of the steric and excluded volume effects, the screening length follows the simple scaling relation λ d , where d is the characteristic size of the large cation. It is also reported that both co- and counter-ion size affect the thickness of the electrical double layer, and influence the screened electrostatic interactions. In solutions of polymeric cation/anion pairs, the screening length is shown to depend on an asymmetry factor. These results provide insights into correlation effects in electrolytes, which were so far unreachable experimentally, and help elucidate such effects in electronics, energy storage devices and biomedical systems.
TOC GRAPHICS
KEYWORDS. Colloids, Debye length, Electrical double layer, DLVO.
Studying screened electrostatic potential in electrolytes near charged surfaces is crucial to understand nanoscale interactions in soft matter. Such interactions are of interest in renewable
energy1,2, electronics3 and biomedical applications4,5. In the dilute electrolyte regime and when
the ion-ion correlations are negligible (including the effect of ion size), the well-known Debye-Hückel theory predicts that these interactions decay exponentially away from the charged surface with a decay length
λD=
(
ε kBT∑
i nie 2 zi)
0.5 , (1)known as the Debye length. Here ε is the dielectric constant of the solvent, kB is the Boltzmann constant, T is the absolute temperature, ni and zi are the number density and the ion valency of ion type i , respectively, and e is the elementary charge. For 1:1 aqueous electrolytes at room temperature Equation (1) becomes λD=0.304/
√
c , where c is the ion concentration in mol L-1 and λD in nm. The width of the region with variable electrostatic potential, from the charged surface to the bulk electrolyte, is of the order of 3 λD to 4 λD 6. The Debye-Hückel framework has been successfully tested on a myriad of systems
over the past few decades. Nevertheless, there are many natural or man-made systems that deviate from this limiting regime because of ion-ion correlations7,8, such as concentrated
electrolytes including ocean’s water, blood plasma, and many electrolytes used in energy storage applications and electronics. In such systems, although the electrical potential is still theoretically expected to decay exponentially, the characteristic decay length is often different from the Debye length9–13, and the electrostatic interaction between charged surfaces is governed according to a
more general decay length. In fact, it has been shown experimentally that in several pure ionic liquids and concentrated electrolyte solutions, the decay length is significantly larger than the Debye length14,15. Moreover, the decay length has been shown to increase strongly with ion
concentration when the ratio d / λD>1 16,17, where d is the ion diameter. This phenomenon was reported for different ionic liquids and concentrated chloride salts with d <¿ 0.5 nm.
Interestingly, the transition point d / λD≈ 1 , where the classical framework of uncorrelated electrolyte solutions was experimentally shown to fail, is close to the theoretical Kirkwood line d / λD≈
√
2 , at which the charge-charge correlation function transitions from monotonic to oscillatory16,18,19. Based on the observation that the decay length in ionic liquids and concentratedchloride salts increases similarly when d / λD>1 , Smith et al.16 hypothesized that bulk nanostructuring in ionic liquids or ion size and shape anisotropy do not affect the observed qualitative behavior. Since these observations have only been reported for small ions at high electrolyte concentrations (typically c >¿ 0.5 M), it remains unclear if this behavior is still
true in dilute electrolyte solutions of sufficiently large ions ensuring d / λD>1 .
Numerous theoretical studies have reported that finite ion size could significantly change the electrostatic interactions in the double layer due to steric interaction effects or by screening the ion charge10,11,20,21. In addition, the ion size has been found to influence ion distribution around
nucleic acids, critically affecting biological and physical processes such as RNA folding and molecular recognition22,23. Other experimental studies have also reported ion size-dependent
electrostatic interactions close to charged surfaces20,24,25.
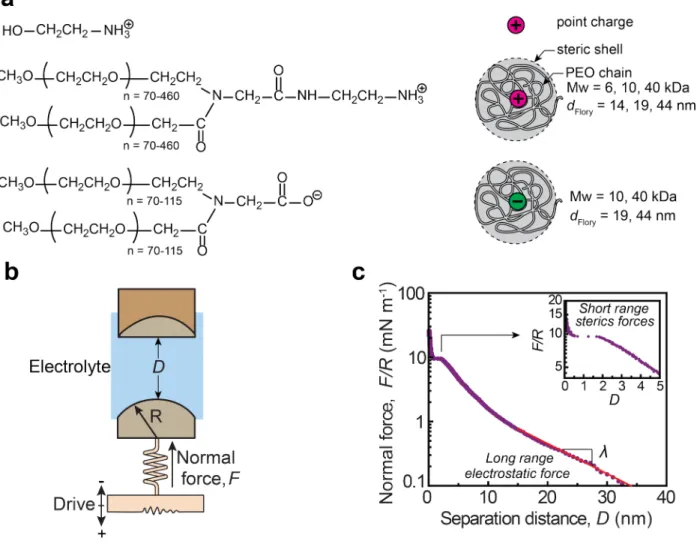
In this study, the electrostatic interactions are measured using the surface force apparatus (SFA) on mica surfaces, which are negatively charged in aqueous media. In addition to evaluating the concentration dependence of the electrostatic interactions in an organic-ion electrolyte with a small ion size, the electrostatic screening in electrolytes with anomalously large ion sizes are investigated. Protonated ethanolamine cations were chosen as small organic ions ( d=0.46
nm26). The large cations are constituted of a single ammonium group at the center of a neutral
polyethylene oxide (PEO) chain with molecular weights Mw = 6, 10 and 40 kDa (Flory diameters
of 14, 19, and 44 nm). In water, the PEO chain adopts a random coil conformation around the ammonium group, resulting in a structure that can be interpreted as a cation surrounded by a “soft” steric shell with a characteristic size determined by the Flory radius of the PEO chain. In aqueous media, the cation charge is activated in the presence of an equimolar concentration of a strong acid (HNO3 in this study). Surface interaction forces are measured across these electrolyte
solutions with various concentrations in the range 0.1-100 mM. The use of “soft” ions also allows to experimentally explore the role of cation/anion size asymmetry on the electrostatic interactions, which is previously unexplored. Here, electrolyte solutions with equimolar concentrations of soft cations (protonated polymers with molecular weight of 6 kDa or 10 kDa) and soft anions (PEO chains with molecular weights of 10 kDa or 40 kDa bearing a single deprotonated carboxylic acid group instead of ammonium group) were used. The chemical structure of these ions and their schematic ionic form are shown in Figure 1a. The SFA setup and an example of interaction force profiles for 10 kDa cation and NO3- anion are also shown in
Figure 1b and c, respectively. In the force profile shown in Figure 1c, the long range exponentially decaying interaction forces are associated to pure electrostatic forces with a characteristic decay length . The short range steric forces are associated to weakly adsorbed polymer layers on the mica surfaces, which can be squeezed out from the contact by a large enough force (see inset of Figure 1c).
Figure 1. (a) Chemical structure of the anion and cation used as soft ions with their
corresponding schematic representation (b) Schematic representation of the SFA setup used to assess electrostatic forces across soft ions solutions. The top and bottom surfaces are negatively charged mica pieces, and R=1 cm. (c) Typical interaction force profile across a solution of 10 kDa polymeric cation and NO3- anion at a concentration of 10 mM. The red solid line shows
the long-range electrostatic interaction exponential decay as F /R exp (−λ−1D) . The inset
highlights the short-range steric interactions.
As the polymer chains are mainly composed of ethylene oxide units, it is reasonable to use available empirical equations and parameters for polyethylene oxide to estimate the ion radius and polymeric interactions. Water is expected to be a good solvent for the polymers27. Therefore,
Flory radius, RF=l . n
0.6
l=0.365 nm is the length of a segment28, and number of segments n=M
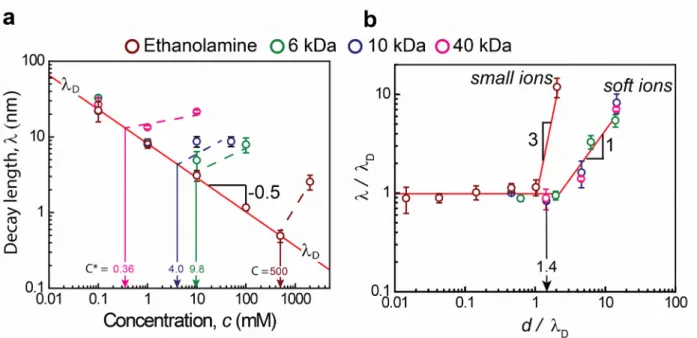
w/Mw 0, where Mw 0=44 Da is the molecular weight of the segment. The variation of λ for the electrolytes composed of organic ions with respect to the ion concentration, c, is shown in Figure 2a. For the small ethanolamine cations, the decay length is equal to the Debye length up to c = 0.5 M, whereas above this value, the experimental decay length is significantly larger than the Debye length ( λ /λD≈ 12 , at c=2 M). When the reduced decay length λ /λD is plotted against the ion correlation parameter d / λD (see Figure 2b), the limiting concentration of 0.5 M corresponds to d / λD≈ 1 , as was also observed by Smith et al.16 and Lee et al. for different point-charge salts.17 In addition, for ethanolamine, the deviation of from the Debye length at
d / λD>1 increases sharply as was reported previously16,17, and was consistent with the scaling
relation λ /λD
(
d /λD)
3
proposed by Lee et al.17 For the large soft ions, a different behavior
was observed leading to a different scaling relationship. First, the decay length across the soft electrolytes deviates from the Debye length only when these polymerc ions enter a correlated regime at a concentration where small electrolytes are still well within the semi-dilute polymer regime, i.e. when c is greater than the overlapping concentration c¿ (see Figure 2a). For
random coil polymer chains, the critical overlapping concentration is defined as 29
c¿
= Mw
4/3 π Rg3NA, where NA is the Avogadro number, and Rg=0.0215 Mw
0.583 , is the
gyration radius27. Secondly, when plotting the scaled decay length λ /λD versus d / λD , the
data from Figure 2a collapses onto a single master curve which is different from the master curve observed for small ions. For the large polymeric ions, at d / λD>1 , the measured scaling law is λ /λD d / λD , and the deviation begins at d / λD≈ 2 , when d = 2RF is used (see Figure 2b),
and at d / λD≈ 1 , when d = 2RG is used to estimate the ion diameters (see Figure S1). When
calculating the radius in the semi-dilute regime, a correction factor of c¿ c /¿ ¿ 8 1/¿ ¿ −¿ ¿ ¿ was applied to
account for the reduced blob size due to shielding of the polymer excluded volume30.
Figure 2. (a) The experimental decay length across the soft electrolytes versus the concentration.
The red line indicates the Debye length values. (b) The variation of scaled decay length versus the ion-correlation parameter. The diameters here are calculated according to the Flory radius. The Kirkwood limit d / λD≈
√
2 , is also indicated.Above c¿
, steric forces between ions are expected to affect the interaction forces between the mica surfaces. To evaluate if the steric forces can influence the force profile in the electrostatic
range, the experiments with d / λD>1 were repeated without the protonation of amine groups (effectively uncharged polymer solutions). In this case, the steric interactions were found to be short range compared to the electrostatic interactions. By increasing the polymer concentration,
the steric interaction range increased as d ¿ λD ¿ ¿ ¿ ¿ λD ¿ λs/¿
, where λs is the decay length of the steric
forces (see Figure S2 and S3). Therefore, the linear increase in the decay length with the ion diameter ( λ d at d / λD>1 ) can be attributed to the redistribution of ions in the electrical double layer due to electrostatic effects (see Figure S4 for the EDL expansion force profiles with increased polymer size). To explain the linear expansion of the double layer with respect to the ion diameter, it should be noted that the charged surface and the ion cloud around it form an electrically neutral system, requiring a fixed number of ions to neutralize the surface charge. By increasing the PEO chain length (increasing the polymeric ion blob diameter), the volume around the point charge ion that is excluded from other ions increases. Above c¿
, where polymer chains partially overlap, this causes an increase in the volume required to contain the ions in the double layer, and distributes counterions farther from the charged surface. The observed increase in the electrostatic decay length is attributed to such expansion of the double layer.
It is important to note that coions (here coions are the anions for the negatively charged mica) are also present in the double layer6. Therefore, their size could also affect the ion distribution in the
double layer. In this study, anion-cation size asymmetry is investigated by varying the molecular weights of both soft anions and cations. The use of soft ion allows to explore the effect of the size asymmetry on the double layer thickness and electrostatic forces by providing highly asymmetrical cation/anion pairs. Following Frosberg et al.11, the ion asymmetry factor is defined as d ¿ −¿ +¿+d−¿, d¿ 2¿ ¿ β=¿ (2)
where d is the ion diameter, with + and - superscripts referring to the cation and anion species, respectively. At the limiting condition that the anion is a point charge, β=2 .
The scaled electrostatic decay length across solutions of asymmetric electrolytes at a fixed concentration of 5 mM is plotted versus the asymmetry factor in Figure 3, keeping the cation size constant and increasing the anion size from a point charge (NO3-) to a significantly larger soft
anion ( −¿d=43.5 Flory
¿ nm). With decreasing the anion size, for a cation of size
+¿=¿ dFlory
¿ 18.9 nm,
the relative decay length appears to rapidly decrease for -1 < < 0 and then increase for > 0,
with a minimum value of λD=¿
λ /¿ 1.38 ± 0.02 at ~ 0. For smaller soft cation ( +¿
dFlory¿ = 13.9
nm), the results were similar except that the increase of λ /λD at > 0 did not occur. This could be explained by noting that among the six samples evaluated in Figure 3, only the 6 kDa cation - NO3- anion pair was in the dilute polymer regime (see Figure 2a for c¿ values).
Therefore, the decay length for this sample was very close to the Debye length. To explain the variation of the scaled decay length with the asymmetry factor, we refer to Frosberg et al.11 to describe the effective ion charge with respect to its counterion size. The presence of counterions around an ion screens the ion effective charge density. By increasing the counterion size, the number of the counterions that can sufficiently approach the ion to screen its charge decreases, causing an increase in the effective charge of the ion. This effect is manifested in Figure 3, where the decay length first decreases with increasing the anion size at 0<β <2 (from right to left in Figure 3). However, above c¿
, increasing the anion size, similarly to the cation size, expands the double layer due to the increase in the polymer excluded volume. Although this effect is less pronounced for anions than cations because of their lower number density in the double layer, it eventually causes the decay length to increase with further increasing the anion size when
β≲0 .
Figure 3. The scaled decay length versus the asymmetry factor defined in Equation (2). The
Flory diameter of cations are 13.9 nm (green data points) and 18.9 nm (blue data points), for 6 kDa and 10 kDa polymers, respectively. The anions from right to left are NO3- (assumed a point
charge), 10 kDa polymer ( −d¿=¿
Flory
¿ 18.9 nm) and 40 kDa polymer ( −d¿=¿
Flory
In summary, the present study showed that large soft polymeric ions can be used to investigate the effect of ion-ion correlation on electrostatic interaction forces between charged surfaces. Similarly to the experimental works of Smith et al.16 and Lee et al.17, which demonstrated that
the screened electrostatic interactions in concentrated electrolyte solutions increases with concentration, we demonstrated that, at significantly lower ion concentrations, the decay length of electrostatic interactions across soft ion solutions shows a similar increase due to steric interactions between ions. However, we also demonstrated that the relation between the scaled decay length and the ion correlation parameter is not universal and depends on the type of the correlation. For example, in concentrated electrolytes, the decay length has been proposed to
increase as d ¿ λD ¿ ¿ ¿ ¿ λD ¿ λ /¿
, due to the ion-ion correlations arising from the extreme proximity of the
ions to each other17. However, in solutions of point charges with anomalously large soft shells,
the steric and excluded volume effects lead to an increase in the decay length of the form λ /λD d / λD . Interestingly, this is not limited to the counterion size, since the coion size can also influence these ion-ion correlations in the double layer, and affect the interactions close to a charged surface.
Supporting Information available: including a description of the materials and methods, and
supplementary figures and force profiles.
Notes
The authors declare no competing financial interests.
ACKNOWLEDGMENT
This research was undertaken thanks, in part, to funding from the Canada First Research Excellence Fund through the TransMedTech Institute. V. A. and F. M. appreciates the financial support from the Fonds de Recherche du Québec (FRQ-NT). B.R.S. is grateful for the financial support from GRUM. XB acknowledges support from NSERC (Discovery) and the Canada Research Chair program.
REFERENCES
(1) Zhang, Q.; Uchaker, E.; Candelaria, S. L.; Cao, G. Nanomaterials for Energy Conversion and Storage. Chem. Soc. Rev. 2013, 42, 3127–3171.
(2) Watanabe, M.; Thomas, M. L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev.
2017, 117, 7190–7239.
(3) Wang, Y.; Xia, Y. Recent Progress in Supercapacitors: From Materials Design to System Construction. Adv. Mater. 2013, 25, 5336–5342.
H., Makino, K., Eds.; Elsevier, 2014.
(5) Moore, T. L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle Colloidal Stability in Cell Culture Media and Impact on Cellular Interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. (6) Hunter, R. J. Foundations of Colloid Science, Second Ed.; Oxford University Press, 2001. (7) Lee, B. P.; Fisher, M. E. Charge Oscillations in Debye-Hückel Theory. Europhys. Lett.
1997, 39, 611–616.
(8) Outhwaite, C. W.; Hutson, V. C. L. The Mean Spherical Model for Charged Hard Spheres. Mol. Phys. 1975, 29, 1521–1531.
(9) Levin, Y. Electrostatic Correlations : From Plasma to Biology. Reports Prog. Phys. 2002, 65, 1577–1632.
(10) Li, B.; Liu, P.; Xu, Z.; Zhou, S. Ionic Size Effects: Generalized Boltzmann Distributions, Counterion Stratification and Modified Debye Length. Nonlinearity 2013, 26, 2899–2922. (11) Forsberg, B.; Ulander, J.; Kjellander, R. Dressed Ion Theory of Size-Asymmetric
Electrolytes: Effective Ionic Charges and the Decay Length of Screened Coulomb Potential and Pair Correlations. J. Chem. Phys. 2005, 122, 064502.
(12) Leote de Carvalho, R. J. F.; Evans, R. The Decay of Correlations in Ionic Fluids. Mol. Phys. 1994, 83, 619–654.
(13) Naji, A.; Kanduč, M.; Forsman, J.; Podgornik, R. Perspective: Coulomb Fluids - Weak Coupling, Strong Coupling, in between and Beyond. J. Chem. Phys. 2013, 139, 150901. (14) Gebbie, M. A.; Valtiner, M.; Banquy, X.; Fox, E. T.; Henderson, W. A.; Israelachvili, J.
N. Ionic Liquids Behave as Dilute Electrolyte Solutions. Proc. Natl. Acad. Sci. 2013, 110, 9674–9679.
(15) Gebbie, M. A.; Dobbs, H. A.; Valtiner, M.; Israelachvili, J. N. Long-Range Electrostatic Screening in Ionic Liquids. Proc. Natl. Acad. Sci. 2015, 112, 7432–7437.
(16) Smith, A. M.; Lee, A. A.; Perkin, S. The Electrostatic Screening Length in Concentrated Electrolytes Increases with Concentration. J. Phys. Chem. Lett. 2016, 7, 2157–2163. (17) Lee, A. A.; Perez-Martinez, C. S.; Smith, A. M.; Perkin, S. Scaling Analysis of the
Screening Length in Concentrated Electrolytes. Phys. Rev. Lett. 2017, 119, 1–5.
(18) Kirkwood, J. G. On the Theory of Strong Electrolyte Solutions. J. Chem. Phys. 1934, 2, 767–781.
(19) Kirkwood, J. G. Statistical Mechanics of Liquid Solutions. Chem. Rev. 1936, 19, 275–307. (20) Gao, Y.; Liu, Y.; Chen, S. A Theoretical Consideration of Ion Size Effects on the Electric Double Layer and Voltammetry of Nanometer-Sized Disk Electrodes. Faraday Discuss.
2016, 193, 251–263.
(21) Gillespie, D. A Review of Steric Interactions of Ions: Why Some Theories Succeed and Others Fail to Account for Ion Size. Microfluid. Nanofluidics 2015, 18, 717–738.
(22) Bai, Y.; Greenfeld, M.; Travers, K. J.; Chu, V. B.; Lipfert, J.; Doniach, S.; Herschlag, D. Quantitative and Comprehensive Decomposition of the Ion Atmosphere around Nucleic Acids. J. Am. Chem. Soc. 2007, 129, 14981–14988.
(23) Lambert, D.; Leipply, D.; Shiman, R.; Draper, D. E. The Influence of Monovalent Cation Size on the Stability of RNA Tertiary Structures. J. Mol. Biol. 2009, 390, 791–804.
(24) Lockless, S. W.; Zhou, M.; MacKinnon, R. Structural and Thermodynamic Properties of Selective Ion Binding in a K+ Channel. PLoS Biol. 2007, 5, 1079–1088.
(25) Rajni; Oh, J. M.; Kang, I. S. Ion Size Effects on the Osmotic Pressure and Electrocapillarity in a Nanoslit: Symmetric and Asymmetric Ion Sizes. Phys. Rev. E 2016,
93, 1–11.
(26) Tinker, A. Probing the Structure of the Conduction Pathway of the Sheep Cardiac Sarcoplasmic Reticulum Calcium-Release Channel with Permeant and Impermeant Organic Cations. J. Gen. Physiol. 1993, 102, 1107–1129.
(27) Devanand, K.; Selser, J. C. Asymptotic Behavior and Long-Range Interactions in Aqueous Solutions of Poly(Ethylene Oxide). Macromolecules 1991, 24, 5943–5947. (28) Donaldson, S. H.; Utzig, T.; Gebbie, M. A.; Raman, S.; Shrestha, B. R.; Israelachvili, J.
N.; Valtiner, M. Electrochemical Control of Specific Adhesion between Amine-Functionalized Polymers and Noble Metal Electrode Interfaces. Mater. Corros. 2014, 65, 362–369.
(29) Kalwarczyk, T.; Ziȩbacz, N.; Bielejewska, A.; Zaboklicka, E.; Koynov, K.; Szymański, J.; Wilk, A.; Patkowski, A.; Gapiński, J.; Butt, H.-J.; Hołyst, R. Comparative Analysis of Viscosity of Complex Liquids and Cytoplasm of Mammalian Cells at the Nanoscale. Nano Lett. 2011, 11, 2157–2163.
(30) Teraoka, I. Polymer Solutions: An Introduction to Physical Properties; John Wiley & Sons, Inc., 2002.