BADJI MOKHTAR- ANNABA UNIVERSITY
ﺔﺑﺎﻨﻋ-رﺎﺘﺨﻣ ﻲﺟﺎﺑ ﺔﻌﻣﺎﺟ
UNIVERSITÉ BADJI MOKHTAR - ANNABA
Faculté Des Sciences Année 2016 Département De Chimie
THESE
Présentée en vue de l’obtention du diplôme de DOCTORAT
Synthèse des esters et des éthers du
D
-Xylose
amphiphiles. Evaluation physico-chimique de leurs
propriétés tensioactives.
Option: Chimie Organique Appliquée.
Par : M
elleKLAI Nadia.
Mme BIDJOU-HAIOUR Chahra Professeur Directrice de thèse U.B.M. Annaba
Devant le jury
Mr BOUKHARI Abbes Professeur Président U.B.M. Annaba
Mme BOUQUILLON Sandrine Professeur Examinateur U. Reims
Mme BENDJEDDOU Amel M.C.A Examinateur U. Souk-Ahras
Mr LIACHA Messaoud Professeur Examinateur U.B.M.Annaba
I Ce travail propose la synthèse et la caractérisation de molécules tensioactives originales, élaborées à partir du D-Xylose. Ces composés, connus pour leur biodégradabilité, sont employés couramment dans la formulation des détergents, des cosmétiques mais également dans certaines industries telles que la métallurgie, les peintures etc ...
L’estérification enzymatique du D-xylose est effectuée, par les acides gras portant des longueurs de chaînes variées : l’acide caproïque (C6), acide laurique (C12) et l’acide stéarique (C18) dans des solvants de diverses polarités: THF, t-BuOH et l’EMC à différentes températures, en présence de trois lipases : la lipase du Candida antractica B (CAL B) immobilisée sur résine (commerciale), la lipase Pancréatique du porc libre et immobilisée sur célite a l’échelle du laboratoire par la méthode d’adsorption. Nous avons rapporté dans ce travail une étude des paramètres qui influent sur la réactivité du sucre et l’activité enzymatique dans la réaction d’estérification du D-Xylose (agent acylant, rapport molaire Sub/AA, enzyme, solvant, température). De très bonnes performances ont été enregistrées avec la CAL B et la PPL Im. La réaction de glycosidation est également mise à profit pour la synthèse d’une série d’éthers de D-Xylose, portant diverses chaînes alkyles (C6, C8, C10, C12, C18). Ces composés tensioactifs sont par la suite utilisés dans la réaction de Heck comme agents de transfert de phases. A cet effet, nous avons étudié les paramètres qui influent sur la conversion du substrat de départ, à savoir la nature de la base, du catalyseur et de la quantité du tensioactif. Les meilleures conversions sont obtenues en présence de K2PdCl4 comme catalyseur, de la triéthylamine comme base et des tensioactifs avec des
longueurs de chaînes moyennes.
D’autre part, une étude se rapportant aux propriétés physicochimiques des esters et des éthers de D-Xylose synthétisés est entreprise. Pour compléter nos études, les conformations spatiales des alkyles xylosides purs synthétisées ont été déterminées par modélisation moléculaire.
Mots clés : tensioactif, estérification enzymatique, immobilisation, glycosidation, réaction de
II This work deals with the synthesis and the characterization of original surfactants, starting from carbohydrates. These attractive compounds, known for their biodegradability, are commonly employed in cleansing agents, cosmetics but also by few industries such as metallurgy, paintings...
Enzymatic esterification of D-Xylose was performed whith fatty acids which have variable
chain size: caproïc acid (C6), lauric acid (C12) and stéaric acide (C18) in different solvent:
THF, t-BuOH and EMK at different temperature. Three lipases were used, Candida
antractica B lipase (CAL B) immobilized on resin (commercial) and porcine pancreatic
lipase (PPL) free and immobilized on celite at the laboratory by the adsorption method. Influence of several parameters such as (fatty acids, ratio Sub/AA, solvents and temperature) was studied. The highest conversions and yields were obtained with CAL B and PPL Im. On an other hand, a range of D-Xylose ethers with alkylchain of different lengths (C6, C8, C10, C12, and C18) were easily synthetized with the glycosidation process. These compounds have been used as phase transfer agents in aqueous Heck reaction. Influence of the bases, the nature of the Pd salts, the nature of the catalysts and the amount of surfactants have been performed. The best results are achieved in the presence of soluble palladium K2PdCl4 as catalyst, NEt3
as a base and surfactants with medium alkylchain.
Critical micellar concentration and surface tension value of the synthetized amphiphilic compounds are determined. To complete this study, the determination of the spatial conformation of xylosides was realized by molecular modeling.
Key words: surfactant, enzymatic esterification, immobilization, glycosidation, the Heck
III ﺺﺋﺎﺼﺧ ﺔﺳارد و عﺎﻨﻄﺻإ ﻰﻠﻋ اﺬھ ﺎﻨﻠﻤﻋ ﻲﻓ ﺎﻧﺪﻤﺘﻋإ ﺔﯿﺤﻄﺴﻟا تاﺮﺛﺆﻤﻟا ﻟ ﻦﻣ ءاﺪﺘﺑإ ﺔﻤﮭﻣ ﺪﺟ تﺎﺒﻛﺮﻤ د -ز ا زﻮﻠﯾ . ﻲﺟﻮﻟﻮﯿﺒﻟا ﻞﻠﺤﺘﻟﺎﺑ ﺎھﺮﯿﻏ ﻦﻋ تﺎﺒﻛﺮﻤﻟا هﺬھ ﺰﯿﻤﺘﺗ , ﺔﻋﺎﻨﺻ ﻲﻓ ﻊﺳاو قﺎﻄﻧ ﻰﻠﻋ ةﺮﯿﺧﻷا هﺬھ مﺪﺨﺴﺗ ﺎﮭﻟﺎﻤﻌﺘﺳإ ﻰﻟإ ﺔﻓﺎﺿإ ﻞﯿﻤﺠﺘﻟا تاﺮﻀﺤﺘﺴﻣ و تﺎﻔﻈﻨﻤﻟا ﻞﺜﻣ ﺔﻋﺎﻨﺼﻟا لﺎﺠﻣ ﻲﻓ : تﺎﻧﺎھﺪﻟا و ندﺎﻌﻤﻟا . تاﺮﺘﺳأ ﻞﯿﻜﺸﺘﺑ ﺎﻨﻤﻗ ﺔﯾاﺪﺒﻟا ﻲﻓ ﺰﻟا ا زﻮﻠﯾ ﺘﺳﻷا ﻖﯾﺮط ﻦﻋ ﺮ ﺔﯿﻤﯾﺰﻧﻷا ة . ﺘﺳﻷا ﺖﻤﺗ ﺮ ﺰﻠﻟ ﺔﯿﻤﯾﺰﻧﻷا ة ا ﺔﻄﺳاﻮﺑ زﻮﻠﯾ لﻮﻄﻟا ﺔﻔﻠﺘﺨﻣ ﻞﺳﻼﺳ تاذ ﺔﯿﻨھذ ضﺎﻤﺣأ : ﻚﯾﻮﻧﺎﺴﻜﮭﻟا ﺾﻤﺣ ) 6 تاﻮﺑﺮﻛ ( , ﻚﯾرﻮﻠﻟا ﺾﻤﺣ ) 12 نﻮﺑﺮﻛ ( ﻚﯾرﺎﯿﺘﺴﻟا ﺾﻤﺣو ) 18 نﻮﺑﺮﻛ ( ﻞﯿﻟﺎﺤﻣ ةﺪﻋ ﻲﻓ : ﻲﻋﺎﺑر ناﺮﯿﻓورﺪﯿﮭﻟا , ﻲﺗﻼﺛ لﻮﻧﺎﺘﯿﺒﻟا , نﻮﺘﯿﻛ ﻞﯿﺜﯿﻣ ﻞﯿﺛإ . ﺎﻤﻛ ﺘﺳﻷا هﺬھ ﺖﻤﺗ ﺮ تﺎﻤﯾﺰﻧإ ﺔﺗﻼﺛ دﻮﺟو ﻲﻓ ة : لﺎﻛ زﺎﺒﯿﻠﻟا ب ) ﺞﻨﺗاﺮﻟا ﻰﻠﻋ ﺪﻤﺠﻣ يرﺎﺠﺗ زﺎﺒﯿﻟ ( سﺎﯾﺮﻜﻨﺑ زﺎﺒﯿﻟو ﺖﯿﻠﯿﺴﻟا ﻰﻠﻋ صﺎﺼﻣدﻹا ﺔﻘﯾﺮﻄﺑ ﺮﺒﺨﻤﻟا ﺪﯿﻌﺻ ﻰﻠﻋ ﺪﻤﺠﻤﻟا و ﺮﺤﻟا ﺮﯾﺰﻨﺨﻟا . ﻞﻤﻌﻟا اﺬھ لﻼﺧ ﺮﯿﺛﺄﺗ ﺔﺳارﺪﺑ ﺎﻨﻤﻗ ﺘﺳﻷا ﺔﯿﻠﻤﻋ ﻲﻓ ﺔﻤھﺎﺴﻤﻟا ﺮﯿﯾﺎﻌﻤﻟا ﻒﻠﺘﺨﻣ ﺮ ة ﺰﻠﻟ ا زﻮﻠﯾ ) ﻞﯿﺳﻷا تﺎﺤﻧﺎﻣ , ﻞﯿﻟﺎﺤﻤﻟا , ةراﺮﺤﻟا ﺔﺟرد ... ﺦﻟإ .( ﺞﺋﺎﺘﻨﻟا لﺎﻛزﺎﺒﯿﻟ ﻦﻣ ﻞﻜﻟ زﺎﺘﻤﻣ ءادأ ﺖﻠﺠﺳ -سﺎﯾﺮﻜﻨﺑزﺎﺒﯿﻟو ب ﺪﻤﺠﻤﻟاﺮﯾﺰﻨﺨﻟا . ﺮﻜﺴﻟا عﻮﻧ ﺲﻔﻧ ﻦﻣ ءاﺪﺘﺑإ , ﺮﻜﺴﻟا ﻦﻣ ﺔﻠﻜﺸﺘﻣ تﺎﺒﻛﺮﻤﻟا ﻦﻣ ﺔﻠﻤﺟ ﻊﯿﻨﺼﺘﺑ ﺎﻨﻤﻗ ﺔﻔﻠﺘﺨﻣ ﺔﯿﻧﻮﺑﺮﻛ ﻞﺳﻼﺳ و : 6 تﺎﻧﻮﺑﺮﻛ , 8 تﺎﻧﻮﺑﺮﻛ , 10 تﺎﻧﻮﺑﺮﻛ , 12 نﻮﺑﺮﻛ 18, نﻮﺑﺮﻛ . ﯾإ ﺔﻔﯿظﻮﺑ ﺔﻠﺼﺘﻣ ﺜ ﻖﯾﺮط ﻦﻋ ﺔﯾﺮﯿ " ﻮﯿﺳاﺪﯾزﻮﻜﯿﻠﻏ " ﻢﮭﺘﯿﻤھأ ﻦﯿﯿﺒﺘﻟ و , ﻞﻋﺎﻔﺗ ﻲﻓ ﺔﻠﻗﺎﻧ ةدﺎﻤﻛ ﺖﻠﻤﻌﺘﺳإ تﺎﺒﻛﺮﻤﻟا هﺬھ " ھ ﻚﯿ " ﻲﺋﺎﻣ ﻂﺳو ﻲﻓ . ﮫﯿﻠﻋ و ﺎﮭﺘﺳارد ﺖﻤﺗ ﻞﻋﺎﻔﺘﻟا ﻰﻠﻋ ةﺮﺛﺆﻣ ﺮﯿﯾﺎﻌﻣ ةﺪﻋ : ةﺪﻋﺎﻘﻟا عﻮﻧ , إ ﺔﯿﻤﻛ و ﺰﻔﺤﻤﻟا عﻮﻧ زﻮﻠﯾاﺰﻟا تاﺮﯿﺜﯾ . ﺞﺋﺎﺘﻨﻟا ﻦﺴﺣأ ةﺪﻋﺎﻘﻛ ﻦﯿﻣأ ﻞﯿﺘﯾﻻا ﻲﺗﻼﺛ دﻮﺟو ﻲﻓ ﺎﮭﯿﻠﻋ لﻮﺼﺤﻟا ﻢﺗ , K2PdCl4 و ﺰﻔﺤﻤﻛ إ زﻮﻠﯾاﺰﻟا تاﺮﯿﺜﯾ تاذ ﻞﺳﻼﺳ ﺔﯿﻧﻮﺑﺮﻛ لﻮﻄﻟا ﺔﻄﺳﻮﺘﻣ . ﺰﻤﺘﺗ إ تاﺮﯿﺜﯾ و تاﺮﺘﺳأ ﺑ ﺎﮭﻌﯿﻨﺼﺗ ﻢﺗ ﻲﺘﻟا زﻮﻠﯾاﺰﻟا ﺮﯿﻐﻟا ﻲﺤﻄﺴﻟا ﺮﺛﺆﻤﻟا ﺔﯿﺻﺎﺨ أ ﻲﻧﻮﯾ , ﺺﺋﺎﺼﺨﻟا ﮫﯿﻠﻋ و ھ ﻰﻠﻋ ﺎﮭﺘﺳارد ﻢﺗ ﺔﯿﺋﺎﯿﻤﯿﻛﻮﯾﺰﯿﻔﻟا ﺬ تﺎﺒﻛﺮﻤﻟا ه . تﺎﺌﯾﺰﺠﻟ ﻲﻧﺎﻜﻤﻟا ﻞﻜﺸﺘﻟا ﺪﯾﺪﺤﺗ ﻢﺗ ﺎﻤﻛ إ ﻦﻋ زﻮﻠﯾاﺰﻟا تاﺮﯿﺜﯾ ﻰﺌﯾﺰﺠﻟا جذﻮﻤﻨﻟا ﻖﯾﺮط . ﺔ ﯿﺣﺎﺘﻔﻣ تﺎ ﻤﻠﻛ : ﻲﺤﻄﺴﻟا ﺮﺛﺆﻤﻟا , ﺘﺳﻷا ﺮ ة ﺔﯿﻤﯾﺰﻧﻷا , ﺪﯿﻤﺠﺗ , ﻮﯿﺳاﺪﯾزﻮﻜﯿﻠﻏ , ھ ﻞﻋﺎﻔﺗ ﻚﯿ , ﻲﺤﻄﺴﻟا ﺮﺗﻮﺘﻟا , ﻰﺌﯾﺰﺠﻟا جذﻮﻤﻨﻟا .
IV
Je dédie le fruit de ce travail
A
La mémoire de mon père. Ma mère.
Mes sœurs et frères.
V
R
emercier c’est exprimer à quelqu’un de la gratitude, de la reconnaissance pour ce qu’il a fait. Alors, à vous tous merci!L
es travaux présentés dans cette thèse ont été réalisés au Laboratoire de synthèse Organique, Modélisation et Optimisation des Procédés chimiques (LOMOP) à l’université Badji-Mokhtar de Annaba. En tant que directrice de thèse, le Professeur ChahraBIDJOU-HAIOUR ma accueillie au sein de son équipe. Elle a su me guider habilement dans un sujet
riche tout en m’accordant sa confiance. Ses compétences et son enthousiasme scientifiques ont été des sources permanentes de soutient et de motivation. Un grand merci pour ta disponibilité et ton dévouement.
L
es résultats présentés dans ce manuscrit sont également le fruit d’un travail au sein du Groupe de Chimie de Coordination de l’UMR 6229 - Université de Reims Champagne-Ardenne en France. J’aimerais remercier sincèrement le Professeur SandrineBOUQUILLON pour m’avoir accueillie et donnée l'opportunité de réaliser ces travaux, dans
le cadre des stages de courte durée. Je la remercie également de m’avoir accordée une très grande liberté dans mes travaux ainsi que pour sa disponibilité, ses qualités humaines et scientifiques. Son accueil, sa confiance et son enthousiasme ont rendu mes séjours agréables au sein de son équipe.
J
e tiens à exprimer ma gratitude au Professeur Abbes BOUKHARI de l’université d’Annaba, pour l'honneur qu'il me fait en acceptant de présider ce jury.J
e tiens aussi à remercier le Docteur Amel BENDJEDDOU,Maître de conférences A à l’université de Souk-Ahras, de bien vouloir participer à ce jury et argumenter ce travail.J
e suis profondément reconnaissante envers le Professeur Messaoud LIACHA de l’université d’Annaba, pour l’intérêt manifesté vis-à-vis de ce travail en acceptant de participer au jury de cette thèse.VI conférences A à l’université de Constantine de bien vouloir accepter de critiquer ce travail, je l’en remercie infiniment.
M
es remerciements s’adressent également au service analytique de l’université de Constantine pour les spectres RMN ainsi que tous les membres de la plateforme analytique de l’université de Reims Champagne-Ardenne.M
es remerciements vont également à madame Ouassila ATTOUI, Maître de conférences B à l’université d’Annaba, pour la modélisation moléculaire.C
ette thèse a pu être réalisée grâce au soutien financier de l’université de M’sila à laquelle je souhaite exprimer ma gratitude.Pour terminer, j’ai le plaisir de remercier tous les membres du laboratoire qui par leur
gentillesse et leur disponibilité, ont rendu le quotidien de ces années de thèse agréable et chaleureux : Docteur Sylvain Gatard, Docteur Fabien Accadbled, Nadège Ferlin post-doc ainsi qu’a Nadia Bouzaouit et Nacer Rezgui bientôt docteurs. Un grand merci à mes amies et collègues de travail à l’université de M’sila : Ouarda, Fatiha, Amel, Rima, Souad,
Abdelkrim et Mounir pour leur soutient et encouragement.
VII
Abréviation / Symbole
Signification
Å Angström
AA Agent acylant
AcOEt Acétate d’éthyle
AcONa Acétate de sodium
Ac2O Anhydride acétique
Ae Activité de l’eau
AL Acide laurique
AL-89 Protéase alcaline lypophilisée
APTS Acide para toluene sulfonique
Boc Tert-butoxycarbonyle
Bmim BF4 1-Butyl-3-methylimidazolium tetrafluoroborate
Cat Catalyseur
CAL B Lipase du Candida Antractica B
CCM Chromatographie sur couche mince
CD Cyclodextrine
CDCl3 Chloroforme deutéré
CD3OD Méthanol deutéré
CMC Concentration Micellaire Critique
Conv Conversion
CPG Chromatographie en phase gazeuse
DCM dichlorométhane
DIAD Diisopropyl azodicarboxylate
DMAc N, N-Diméthylacetamide DMAP N, N-Diméthylamino-pyridine DMF N, N-Diméthylformamide DMSO Dimethylsulfoxide EP Ether de pétrole éq Equivalent
VIII h Heure HLB Balance hydrophile/hydrophobe HMDS Hexaméthyldisilazane i-PrOH Isopropanol IR KDa Infrarouge Kilodalton min Minute NEt3 Triéthylamine
PEG Polyéthylène glycol
POE Poly(oxyde d’éthylène).
PPL Lipase Pancréatique du porc
PPL im Lipase Pancréatique du porc immobilisé
PS Polystyrène
PSS Polyéthylène glycol-β-sitosteryl sébacate
PTS Polyéthylène glycol-α-tocopheryl sébacate
Pyr Pyridine
Rdt Rendement
Rf Rapport frontal
RMN Résonance magnétique nucléaire
SDS Dodécylsulfate de sodium
SM Spectrométrie de masse
T.A Température ambiante
t-BuOH Tertiobutanol
THF Tétrahydrofurane
TM Tamis moléculaire
TMS Triméthylsilyl
TPGS-750-M DL-α-tocophérol méthoxypolyéthylène glycol
IX
---2
èmePartie---
Chapitre-I-
I-1 Rendements de la glycosidation du D-Xylose avec différents alcools. 52 Chapitre-II-
II-1 Influence de catalyseur et la nature de la base. 70
II-2 Influence de la longueur de la chaîne hydrophobe. 72
II-3 Influence de la concentration du tensioactif. 73
---3
èmePartie---
Chapitre-I-
I-1 Consommations mondiales par principales régions en 2006. 77 I-2 Les différents types de tensioactifs. 78 I-3 Bilan des tensioactifs non ioniques utilisés en cosmétique. 81 I-4 Prédiction du type d’agrégation en fonction du paramètre d’empilement
des tensioactifs.
94
I-5 Incréments des principaux groupements nécessaires au calcul de la HLB. 96 I-6 Classification des tensioactifs selon la valeur de la HLB. 96
Chapitre-II-
II-1 HLB et propriétés des tensioactifs synthétisés. 101
II-2 Récapitulatif des grandeurs (CMC, γ) relatives aux esters du D-xylose. 102 II-3 Aire occupée par les tensioactifs II-1, II-2, II-3 et leurs empilements. 103 II-4 Données physicochimiques des alkyl-xylosides. 105 II-5 Caractéristiques physicochimiques d’alkyl-glycosides pure. 106 II-6 Influence de la stéréochimie du sucre sur l’aire occupée par les
tensioactifs I-4b, I-4b, I-5a et I-5b.
108
X
LISTE DES FIGURES
---Introduction générale---
1 Consommation mondiale des produits dérivés de sucres. 2
---1
èmePartie---
Introduction de la première partie
1 Esters de saccharose. 4 Chapitre-I-
I-1 Principe de la catalyse. 5 I-2 Représentation schématique de la structure primaire d'une protéine. 6 I-3 Représentation schématique de la structure secondaire d'une protéine. 7 I-4 Représentation schématique de la structure tertiaire d'une protéine. 7 I-5 Représentation schématique de la structure quaternaire d'une protéine. 7 I-6 Modèle de simple complémentarité stérique. 8 I-7 Modèle de l’ajustement induit. 8 I-8 Mode d’action des enzymes. 9 I-9 Fréquence d’utilisations des enzymes en synthèse organique. 12
I-10 Fréquence de l'utilisation des enzymes hydrolytique dans les
Biotransformations.
12
I-11 Structure tridimensionnelle de la lipase de Candida antarctica B. 15
I-12 Représentation schématique du volet amphiphile et du phénomène
d'activation interfaciale en milieu organique.
16
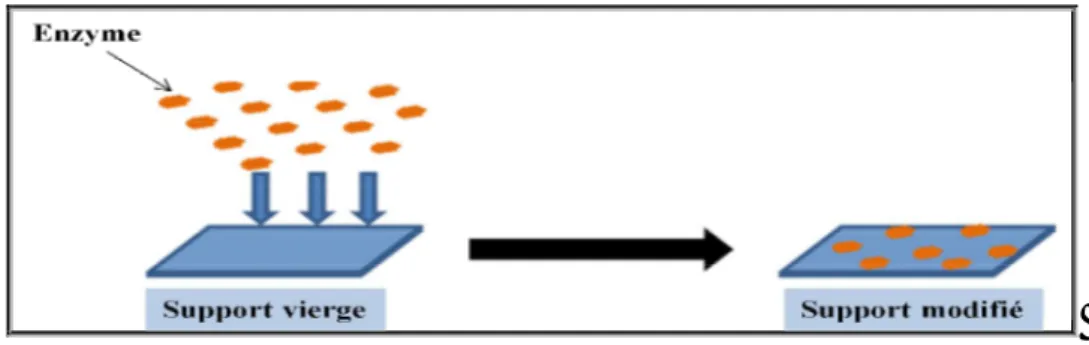
I-13 Adsorption des enzymes sur un support. 19
Chapitre-II-
II-14 Effet de la température sur la stabilité de la PPL libre et immobilisée. 35
XI
II-18 Effet de la longueur de la chaîne de l’agent acylant. 40
II-19 Effet du rapport D-Xylose/acide laurique. 41
---2
èmePartie---
Chapitre-II-
II-1 Exemple de molécules d’intérêt synthétisées via un couplage de Heck. 59
II-2 Phénomène observé pour les réactions catalytiques en milieu aqueux en présence detensioactif.
63
II-3 PS-PEG-Pd. 65
II-4 Classement des tensioactifs selon les valeurs de la HLB. 67
II-5 Structure moléculaire du PEG-α-tocophéryl sebacate (PTS-600, n= 13) et
sa 2ème génération le DL-α-tocophérol méthoxypolyéthylène glycol (TPGS-750-M, n=16).
68
II-6 Suivi de la réaction de Heck dans le K2PdCl4. 72
---3
èmePartie---
Chapitre-I-
I-1 Représentation schématique d’un monomère de tensioactif. 75 I-2 Volume mondial par domaines d’application en 2003. 76 I-3 Schématisation des forces intermoléculaires s'exerçant au sein d'un
liquide et à sa surface.
82
I-4 Montage pour les mesures de tension de surface par la goutte pendante. 84 I-5 goutte pendante à l’équilibre. 84 I-6 Schéma illustrant la mesure de la tension superficielle selon la méthode
de Wilhelmy. Illustration des forces s’exerçant sur la lame de platine lors d’une mesure.
85
I-7 Courbe de tension de surface en fonction de log c d’un tensioactif dans l’eau.
86
I-8 Détermination de la CMC à partir de la courbe de tension de surface en fonction du log c d’un tensioactif.
XII
I-10 Diagramme de phase d’un tensioactif soluble dans l’eau. 90
I-11 Représentation d’une micelle sphérique en trois dimensions. 92
I-12 Représentation schématique d’un tensioactif et de l’espace qu’il occuperait dans un agrégat.
93
I-13 Schéma de la formation d’une mousse. 97
I-14 Schémas du mouillage d'une surface par des gouttes de différentes solutions.
98
I-15 Structure microscopique d’émulsions E/H et H/E. 98
I-16 Solubilisation d’un substrat lipophile dans une micelle. 99
Chapitre-II-
II-1 Représentation schématique illustrant les configurations α et β du carbone anomérique du xylose.
107
II-2 Schéma illustrant les angles des liaisons anomériques α et β. 107
II-3 L’organisation des alkylglycosides α et β à l’interface air/liq. 107
II-4 Schéma illustrant les angles des liaisons anomériques α et β susceptible d'influencer l'aire occupée par une molécule de tensioactif.
109
XIII
LISTE DES SCHEMAS
---Introduction générale---
1 Exemples de glycosides. 1
---1
èmePartie---
Chapitre-I-
I-1 Différentes réactions possibles par utilisation de lipases. 13 I-2 Mécanisme réactionnel de la triade catalytique. Nu : H2O, H2NR,
ROH.
14
Chapitre-II-
II-1 Estérification par un chlorure d’acide. 21
II-2 Estérification de la cellulose par activation micro-ondes. 22
II-3 Estérification du saccharose dans l’eau. 22
II-4 Estérification par la méthode de Köenigs-Knorr à partir du D-Xylose. 23
II-5 Transestérification du D-glucose par la méthode « A.A.E ». 23
II-6 Estérification à l’aide du n-BuLi. 24
II-7 Estérification du saccharose dans les conditions de Mitsunobu. 24
II-8 Transestérification du D-glucose par un dérivé de métronidazole. 26
II-9 Estérification du saccharose catalysée par la Protéinase. 26
II-10 Synthèse de 6-O-monoesters de galactose. 27
II-11 Estérification enzymatique du saccharose. 27
II-12 Estérification enzymatique du Lactose. 28
II-13 Influence de l’activité de l’eau. 29
II-14 Influence de la nature du solvant. 29
II-15 Influence de la longueur de la chaîne de l’agent acylant. 30
II-16 Influence de la nature de la lipase. 31
II-17 Influence de la température. 31
II-18 Influence du rapport molaire entre les substrats. 32
XIV
---2
èmePartie---
Chapitre-I-
I-1 Transmannolysation du mannobiose. 44 I-2 Synthèse enzymatique d’octyl β-D-glucoside. 45 I-3 Synthèse enzymatique des alkyles arabinosides. 45 I-4 La réaction de transglycosylation de xylans par la xylanase. 46 I-5 Réaction de glycosylation. 46 I-6 Synthèse des glycosides par voie chimique. 47 I-7 Ethérification du saccharose en présence de résine A26. 48 I-8 Méthylation du D-Xylose en présence d’une résine acide. 49 I-9 1-O-benzylation du D-Xylose. 50
I-10 Glycosidation du glucose en présence d’APTS. 50
I-11 Schéma général de synthèse des alkyl-xylosides par glycosidation. 51
I-12 Mécanisme de la glycosidation de Fischer. 51
I-13 Schéma général de la purification des alkyl-xylopyranosides. 53
I-14 Acétylation des alkyles α,β-D-xylopyranosides. 53
I-15 Déprotection des alkyles α-β-D-xylopyranosides acétylés. 54
Chapitre-II-
II-1 Les principales réactions de couplage carbone-carbone catalysées par le palladium.
57
II-2 Réaction de couplage de Heck. 58
II-3 Synthèse de Prosulforon. 59
II-4 Cycle catalytique proposé pour le couplage de Heck. 60
II-5 Réaction chimique par greffage d’un groupement hydrophile. 62
II-6 Réaction de Heck en milieu aqueux avec des co-solvants. 64
II-7 Couplage de l’iodobenzène avec l’acide acrylique. 65
II-8 Réaction de Heck utilisant des nanoparticules de Pd-α-HPCD. 66
II-9 Couplage de Heck de 4-bromoacétophenone avec le styrène. 66
XV
II-12 Couplage de Heck en présence de PTS et sa génération (TPGS-750-M).
69
II-13 Couplage de Heck entre l’iodobenzéne et le t-butylacrylate. 69
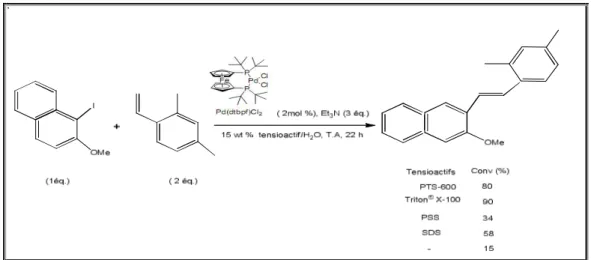
II-14 Réaction modèle pour l’étude méthodologique. 70
---3
èmePartie---
Chapitre-I-
I-1 Tensioactifs anioniques. 79 I-2 Tensioactifs cationiques. 80 I-3 Tensioactifs zwitterioniques. 80
--- Partie Expérimentale---
XVI
Sommaire
Introduction générale
11
èrePartie : Synthèse des esters du D-Xylose
Introduction de la première partie 4
Chapitre -I- : Revue bibliographique sur les enzymes
I.A. Introduction 5
I.B. Marché des enzymes 5
I.C. Structure des enzymes 6
I.D. Phénomène du cycle catalytique enzymatique 8
I.E. Cinétique enzymatique (équation de Michaelis-Menten) 9
I.F. Classification des enzymes 11
I.F.1. Les oxydo-réductases 11
I.F.2. Les transférases 11
I.F.3. Les hydrolases 11
I.F.4. Les lyases 11
I.F.5. Les isomérases 11
I.F.6. Les ligases (synthétases) 11
I.G. Les Lipases 12
I.G.1. Généralités 12
I.G.2. Réactions réalisables par les lipases 13
I.G.3. Mécanisme d’action des lipases 14
I.G.4. La structure des lipases 15
I.G.4.a. L’activation interfaciale 16
I.G.5. Spécificité des lipases 17
I.G.5.a. La régiosélectivité 17
I.G.5.b. La typosélectivité 17
I.G.5.c. La stéréosélectivité 17
I.H. Immobilisation des enzymes 17
XVII I.H.2. Immobilisation par réticulation et co-réticulation 18
I.H.3. Immobilisation par inclusion ou piégeage 18
I.H.4. Immobilisation par adsorption 18
I.I. Conclusion 19
Chapitre -II- : Synthèse enzymatique des esters du D-Xylose
II.A. Introduction 21
II.B. Différentes modes d’accès aux esters de sucres 21
II.B.1. Méthodes par voie chimique 21
II.B.1.a. Estérification à l’aide d’un chlorure d’acide 21
II.B.1.b. Réaction de type Köenigs-Knorr 22
II.B.1.c. Les méthodes dites de « condensation » 23
II.B.1.c.i. Transestérification par la méthode Accélérative Active Ester « A.A.E » 23
II.B.1.c.ii. Utilisation d’esters activés 24
II.B.1.c.iii. Conditions de Mitsunobu 24
II.B.2. Méthodes par voie enzymatique 25
II.B.2.a. Transestérification à l’aide d’une protéase 25
II.B.2.b. Estérification par une protéinase 26
II.B.2.c. Estérification catalysée par une lipase 26
II.C. Influence des paramètres de la réaction 28
II.C.1. Influence de l’eau 28
II.C.2. Influence de solvant organique 29
II.C.3. Influence de la longueur de la chaîne de l’agent acylant 30
II.C.4. Influence de la nature de la lipase 30
II.C.5. Influence de la température 31
II.C.6. Influence du rapport molaire entre les substrats 31
II.D. Estérification enzymatique du D-Xylose 32
II.D.1. Résultats et discussion 34
II.D.1.a. Influence des enzymes 34
II.D.1.a.i. Effet de la température sur la stabilité de la PPL libre et immobilisée 34
II.D.1.a.ii. Effet de la nature de la lipase 36
XVIII II.D.1.b. Influence du solvant organique 38
II.D.1.c. Effet de la longueur de la chaîne de l’agent acylant 39
II.D.1.d. Effet du rapport D-Xylose / acide laurique 40
II.E. Conclusion 41
2
èmePartie : Synthèse des éthers du D-Xylose. Etude de leurs effets dans la
réaction de Heck.
Introduction de la deuxième partie 43
Chapitre -I- : Synthèse des éthers du D-Xylose
I.A. Introduction 44
I.B. Différentes mode d’accès aux éthers de sucres 44
I.B.1. Méthodes par voie enzymatique 44
I.B.2. Principales méthodes de synthèse des alkyl-glycosides par voie chimique 46
I.B.2.a. Activation par groupement partant 47
I.B.2.a.i. Activation basique 48
I.B.2.a.ii. Activation acide 48
I.B.2.a.iii. Utilisation de résines 49
I.B.2.a.iv. Utilisation de chlorure d’acétyle 49
I.B.2.a.v. Utilisation d’acide sulfonique 50
I.C. Résultats et discussion
I.C.1. Séparation des anomères α, β des composés II-4 et II-5
51 53
I.C.1. a. Acétylation 53
I.C.1. b. Désacétylation 54
I.D. Conclusion 55
Chapitre-II- : Effets des éthers du D-Xylose dans la réaction de Heck.
II.A. Introduction 56
II.B. Le principe de la réaction de Heck 57
II.C. Les domaines d’utilisation de la réaction de Heck 58
XIX II.E. Les problèmes liés à la catalyse dans l’eau 61
II.E.1. Ajout d’un cosolvant dans le milieu 61
II.E.2. Contrôle du pH 61
II.E.3. Greffage de groupements hydrophiles 61
II.E.4. Utilisation de tensioactifs 62
II.E. Bibliographie sur La réaction de couplage de Heck en milieu aqueux 63
II.E.1. Utilisation des co-solvants 64
II.E.2. La réaction en milieu micellaire 64
II.E.2.i. Utilisation des catalyseurs amphiphiles 65
II.E.2.ii. Utilisation des additifs amphiphiles 67
II.F. Travaux antérieurs 69
II.G. Résultats et discussion 70
II.G.1. Influence du catalyseur et la nature de la base 70
II.G.2. Influence de la longueur de la chaîne hydrophobe 72
II.G.3. Influence de la concentration du tensioactif 72
II.H. Conclusion 73
3
èmePartie : Propriétés physico-chimiques des esters et des éthers du
D-Xylose synthétisés.Introduction de la troisième partie 74
Chapitre-I- : Aperçu bibliographique sur les tensioactifs.
I.A. Introduction 75
I.B. Domaines d’application des tensioactifs 75
I.C. Etat du marché mondial des tensioactifs 76
I.D. Structures des tensioactifs 77
I.E. Classification 78
I.E.1. Les tensioactifs anioniques 78
I.E.2. Les tensioactifs cationiques 79
XX I.E.4. Les tensioactifs non ioniques 81
I.E.5. Etat du marché par classes de tensioactifs 81
I.F. Les propriétés physico-chimiques des tensioactifs 82
I.F.1. La tension de surface (γ) 82
I.F.1.a. Mesure de la tension de surface 83
I.F.1.a.i. Méthode de la goutte pendante 83
I.F.1.a.ii. Méthode de Wilhelmy 85
I.F.2. Concentration Micellaire Critique (CMC) 87
I.F.2.a. Facteurs influençant la Concentration Micellaire Critique 88
I.F.2.a.i. La structure du tensioactif 88
I.F.2.a.ii. La température de la solution 90
I.F.3. L’excès superficiel par unité de surface (Γ) 91
I.F.4. Aire de la molécule à l’interface (A) 92
I.F.5. Paramètre d’empilement (P) 92
I.F.6. Notion de la HLB 94
I.G. Propriétés fonctionnelles des tensioactifs 96
I.G.1. Le pouvoir moussant 96
I.G.2. Le pouvoir mouillant 97
I.G.3. Pouvoir émulsifiant 98
I.G.4. Pouvoir solubilisant 99
I.H. Conclusion
Chapitre-II- : Résultats et Discussion
99
II.A. les propriétés des tensioactifs en fonction de la HLB 101
II.B. Les propriétés de surface des tensioactifs 101
II.B.1. Les esters du D-xylose 101
II.B.1.a. Mesure de la CMC et la tension de surface 102
II.B.1.a.i. Discussion 103
II.B.1.b. Mesure de l’aire occupée par molécule de tensioactif 103
II.B.1.b.i. Discussion 104
II.B.2. Les éthers du D-Xylose 104
XXI II.B.2.a.i. Discussion 106
II.B.2.b. Aire occupée par une molécule des alkyles-xyloside pure 108
II.C. Détermination de la conformation spatiale des alkyl-xyloside I-4 et I-5 109
II.C.1. Interprétation des résultants 110
II.D. Conclusion 111
Conclusion générale
112Protocoles expérimentaux
114Références bibliographiques
1331
Introduction générale
Actuellement, de nombreux facteurs économiques (instabilité du prix du baril de pétrole, développement des pays émergeants d’Asie), réglementaires (mise en place de la directive 1907/2006/CE (REACH), interdiction des alkylphénols polyéthoxylés) et l’évolution de l’opinion publique (syndrome de l’Encéphalopathie Spongiforme Bovine (ESB), prise en compte de l’impact environnemental), concourent à favoriser la croissance des composés obtenus à partir de ressources renouvelables. Pour faire face à ces changements, on assiste depuis plusieurs années à l’apparition de produits issus de ressources végétales comme les glycosides, citons : les esters de sorbitan,1 les esters de saccharose,2 les glucamines3 et les PolyGlycosides d’Alkyle (APGly)4, β-xylosides5 (Schéma -1-).
Schéma -1- : Exemples de glycosides.
Ces dernières années, le volume mondial de consommation de ce type de produit était de l’ordre de 200,000 tonnes/an. La Figure-1- englobe la répartition de leur consommation dans le monde.
1
Y. Queneau, S. Chambert, C. Besset, R. Cheaib, Carbohydr. Res. 2008, 343, 1999.
2
S. Piccicuto, C. Blecker, J. C. Brohée, A. Mbampara, G. Lognay, C. Deroanne, M. Paquot, M. Marlier,
Biotechnol. Agron. Soc. Environ. 2001, 5, 209.
3
J. Guilbot, Oléagineux, corps gras, lipides. 2006, 13, 178.
4
a.W. von Rybinski, K. Hill, G. Stoll, Alkyl Polyglucosides. Technology, Properties and Applications. Verlag
Chemie, Weinheim. 1996.
b. D. Balzer, H. Lüders, Nonionic surfactants Alkyl Polyglucosides, vol. 91, Surfactant Science Series, Marcel Dekker. 2000.
5
2
Figure -1- : Consommation mondiale des produits dérivés de sucres.
L’intérêt des sucres de faible poids moléculaire en tant que substrats pour l’industrie est basé sur leur disponibilité en quantités importantes, leur faible coût de production et leur pureté énantiomérique.6 Les glycosides dont le squelette amphiphile laisse apparaître des propriétés de surface spécifiques ont connu un développement important en raison de leur caractère non ionique impliquant de vastes utilisations dans des domaines aussi variés que la détergence,7 la pharmacie,8 la cosmétologie9 et la biologie.10 Parallèlement à leur utilisation dans des applications fines, telles que l’extraction des protéines membranaires,11 certains tensioactifs ont acquis depuis quelques années une réelle importance dans le domaine de la catalyse. Les tensioactifs sont des molécules aux propriétés spécifiques (détergente, mouillante, solubilisante, etc…) grâce à leur structure amphiphile, ils réagissent de manière à créer à l’intérieur du solvant, des cavités hydrophobes à l’intérieur desquelles les molécules organiques vont pouvoir s’insérer et éventuellement réagir. Les tensioactifs sont des composés clés dans les réactions organométalliques en milieu aqueux, dans lesquels ils interviennent comme agent de transfert de phase. Parmi les réactions organométalliques les réactions de couplage carbone-carbone. Nous pouvons citer, les réactions de Suzuki, Negishi, Sonogashira et Heck.
Dans ce contexte, l’utilisation d’hydrates de carbone, composés polyhydroxylés abondants comme matières premières et ayant une hydrophilie importante, présente un intérêt certain.12 Leur transformation en tensioactifs biodégradables augmente les possibilités de leur
6
F. N. Lichtenthaler (ed.), Carbohydrates as Organic Raw Materials, VCH, Weinheim, Germany, 1991.
7
K. Schmid, H. Tesmann, Surfactant Science Series. 2001, 98, 1.
8
T. Ushibori, K. Kawada, S. Matsumura Jpn Kokai Tokkyo Koho. 1990, JP 0230696; Chem. Abstr. 1991, 115, 44228.
9
H. Kiwada, H. Niimura, Y. Fujisaki, S. Yamada, Y. Kato, Chem. Pharm. Bull. 1985.
10
A. Shioi, M. Harada, H. Takahashi, M. Adachi, Langmuir. 1997, 15, 609.
11
M. Prasad, S. P. Moulik, A. Wardian, S. Moore, R. Palepu, Coll. Poly. Sci. 2005, 238, 887.
12
F. W. Lichtenthaler, S. Peters, C. R. Chimie. 2004, 7, 65.
40% USA 25% Asie 20% Europe 15% Autres
3 valorisation. Notre équipe avait axé la valorisation d’agroressources essentiellement le xylose et le glucose pour la préparation de tensioactifs non ioniques.13Leur synthèse est réalisée soit par voie chimique ou bien par voie enzymatique. Dans le présent travail, nous nous sommes
intéressés à l'élaboration en une seule étape, de tensioactifs non ioniques à partir du
D-Xylose : de type esters par une estérification enzymatique en présence d’un acide gras et de type éthers par glycosidation en présence d’un alcool gras. Ce pentose est peu employé dans la synthèse des glycosides contrairement au glucose (sucre alimentaire), il est donc très intéressant de développer ces voies de synthèse.
Les résultats de ce travail sont regroupés dans trois parties distinctes.
La première partie introduit des généralités sur les enzymes et la catalyse enzymatique ce, dans un premier chapitre. Dans un deuxième chapitre, nous étudions la synthèse enzymatique des esters de xylose, mettant l’accent sur l’influence de certains paramètres qui influent sur l’évolution des réactions mises en œuvre.
La deuxième partie de la thèse sera consacrée à la réalisation d’une catalyse en milieu micellaire dans l’eau. Dans le premier chapitre, nous décrirons l’élaboration des éthers de xylose. Le deuxième chapitre a pour objectif d’étudier leurs effets dans le couplage de Heck comme réaction modèle en milieu aqueux.
La troisième partie est divisée en deux chapitres : le premier chapitre concerne un aperçu bibliographique sur les tensioactifs. Dans le chapitre qui suit, nous avons évalué les propriétés tensioactives des composés synthétisés au cours de ce travail.
Le manuscrit sera terminé par une conclusion générale et la présentation des perspectives envisagées. Les références bibliographiques sont notées en bas de page puis regroupés à la fin du présent mémoire. Toutes les informations concernant l’ensemble des synthèses effectuées sont rassemblées dans la partie expérimentale. Les travaux publiés dans cette thèse sont mis en annexe.
13
a. D. Hallal. Optimisation de la réactivité dans la réaction d’estérification enzymatique du D-glucose. Thèse de magister d’université d’Annaba. Algérie. 2012.
b. N. Bouzaouit, C. Bidjou-Haiour, Der Pharma Chemica. 2015, 7, 261.
c. L. Lins, K. Nott,C. Flore, R. Thomas, M. Paquot, S. Bouquillon, C. R. Chimie. 2012, 12, 68. d. S. Gatard, M. Nail Nasir, M. Deleu, N. Klai, V. Legrand, S. Bouquillon. Molecules . 2013, 18, 6101.
1
èrepartie
4
Introduction de la première partie
Les esters gras de sucres sont des tensioactifs non ioniques intéressants en raison de leur grande biodégradabilité, leur faible toxicité et leur grande tolérance vis-à-vis des yeux et de la peau.14 Ils occupent un vaste marché industriel, et rien n’échappe à leur utilisation, en passant par l’industrie alimentaire,15 cosmétique, détergente et pharmaceutique. Des propriétés antibiotiques,16 antitumorales17 et insecticides18 ont été mises en évidence.
Les premiers esters de sucres commercialisés sont les esters de saccharose (Figure -1-). Tout d’abord autorisé en 1959 au Japon, ces composés ont ensuite été agrées par la Food and Drug Administration (USA) en 1983.19 O O H O H O H O O OH O H OH O H O O (CH2)nCH3
Figure -1- : Esters de saccharose.
Le nombre de publications relatives aux esters de sucres a considérablement augmenté depuis dix ans. D’un point de vue général, ce type des molécules est obtenu par estérification ou par transestérification, chimique ou enzymatique, selon différentes méthodes. Une méthode de synthèse a été la plus étudiée au cours de cette dernière décennie : la voie biotechnologique exploitant les capacités de synthèse des enzymes.
Les travaux présentés dans cette première partie concernent une revue bibliographique sur les enzymes et les résultats obtenus pour la synthèse des esters du D-Xylose par voie enzymatique.
14
I. J. A. Baker, B. Matthews, H. Suares, I. Krodkiewska, D. N. Furlong, F. Grieser, C. J. Drummond, J. Surf.
Deterg. 2000, 3, 1.
15
a. S. Nakamura, Oleochemicals.1997, 8, 866.
b. G. Sekeroglu, S. Fadiloglu, E. Ibanoglu, J. Sci. Food. Agric. 2002, 82, 1516. c. S. Sabeder, M. Habulin, Z. Knez, J. Food. Eng. 2006, 77, 880.
16
D. L. Marshall, L. B. Bullerman. In: Akoh CC, Swanson BG, editors. “Carbohydrate polyesters as fat substitutes”. Marcel Dekker: New York, 1994. 149-167.
17
S. Okabe, M. Saganuma, Y. Tada, Y. Ochiai, E. Sueoka, H. Kohya, A. Shibata, M. Takahashi, M. Mizutani, T. Matsuzaki, H. Fujiki, Jpn. J. Cancer. Res. 1999, 90, 669.
18
A. Smith, P. Nobmann, J. Dunne, Carbohydr. Res. 2008, 343, 2557.
19
1
èrepartie
Chapitre -I-
Revue bibliographique sur les
enzymes.
5
I.A. Introduction
Les enzymes sont des catalyseurs naturelles qui émergent du monde vivant, elles sont des macromolécules biologiques de nature protéique20 qui possèdent des propriétés catalytiques remarquables. Nettement plus efficaces que les catalyseurs chimiques, elles sont capables de multiplier la vitesse d’une réaction par des facteurs très élevés21 (107 – 1019) diminuant son énergie libre d’activation (Figure I-1).
Figure I-1 : Principe de la catalyse.
De plus, les enzymes permettent d’opérer dans des conditions douces de pH, de température et de pression contrairement aux catalyseurs chimiques. Le procédé biologique est dans certains cas plus rentable, d’une part, avec une meilleure productivité et d’autre part, avec un bilan énergétique et environnemental amélioré.
I.B. Marché des enzymes
Le marché des enzymes industrielles, quant à lui, est en pleine croissance grâce notamment à l’apparition de nouveaux domaines d’application. En 2007, le marché mondial des enzymes industrielles a été estimé à 2,3 milliards de dollars. Avec un taux d’utilisation d’enzymes industrielles de 41%, le domaine pharmaceutique se place en tête du classement suivi par l’industrie des détergents (17%), l’industrie agroalimentaire (17%), la papeterie (17%) et le
20
O. H. Housse, B. M. Trost, J. Org. Chem. 1964, 30, 2502.
21
6 textile (8%).22 Par conséquent, la connaissance des enzymes, de leur nature et de leurs propriétés est fondamentale.
I.C. Structure des enzymes
Elles sont constituées de plusieurs acides α-aminés de la série L unis entre eux par une liaison formée par condensation entre le groupement carboxyle d’un acide aminé et le groupement amine d’un autre acide aminé afin de former une liaison amide. Les enzymes sont donc des polypeptides de masses moléculaires élevées entre 10 à 1000 kDa. L’ordre, dans lequel sont arrangés les acides aminés, constitue ce que l’on appelle la structure primaire des enzymes (Figue I-2).
Figure I-2 : Représentation schématique de la structure primaire d'une protéine.
Ces protéines vont avoir tendance à se replier sur elles-mêmes afin de former des arrangements secondaires principalement en hélices α et en feuillets β (Figure I-3) ; cette structure est stabilisée grâce à la génération de liaisons hydrogènes.
22
A. K. Chandel, R. Rudravaram, L. V. Rao, P. Ravindr, M. L. Narasu, J. Commercial Biotechnology. 2007, 13, 283.
7
Figure I-3 : Représentation schématique de la structure secondaire d'une protéine
.
L’arrangement de ces structures secondaires les unes par rapport aux autres forme une structure tertiaire qui, elle, sera stabilisée par des ponts disulfures (Figure I-4).
Figure I-4 : Représentation schématique de la structure tertiaire d'une protéine.
Une structure quaternaire peut même être décrite pour les très grosses enzymes (Figure I-5). Cette structure tridimensionnelle de l’enzyme lui donnera sa spécificité permettant à celle-ci de reconnaitre un substrat en particulier via une région distincte de l’enzyme, appelée le site actif.
8
I.D. Phénomène du cycle catalytique enzymatique
Les enzymes assurent une fonction de catalyseurs. En présence d'une enzyme donnée, plusieurs molécules sont amenées à interagir et à subir des transformations chimiques déterminées. L’enzyme se lie au substrat. Cette liaison s'établit avec une région de l'enzyme, appelée site actif, dont la configuration spatiale est complémentaire de celle des molécules de substrats. On appelle complexe enzyme-substrats l'ensemble formé par la molécule d'enzyme et les molécules de substrats spécifiques fixées sur elle. La formation du complexe E-S peut s’expliquer soit par le modèle de simple complémentarité stérique (Figure I-6), soit par celui de l’ajustement induit (Figure I-7).
Figure I-6 : Modèle de simple complémentarité stérique.
Figure I-7 : Modèle de l’ajustement induit.
Récemment, une nouvelle suggestion sur la relation du substrat envers le site actif de l’enzyme a été faite par Hames qui suggéra que les enzymes partagent des caractéristiques de ces deux modèles : une dose de complémentarité et une autre de changement conformationnel induit.
L’enzyme et son substrat sont associés par diverses forces d’interaction. Ils constituent alors une ou plusieurs interactions réactionnelles instables qui permettent d’abaisser la barrière d’énergie de la réaction et d’aller vers la formation du produit. C’est la reconnaissance Enzyme-Substrat qui permet le rapprochement de certains groupes fonctionnels de l’enzyme
9 et du substrat et facilite la rupture et la formation de certaines liaisons du substrat. Une fois l’étape catalytique proprement dite effectuée, le produit formé reste transitoirement fixé à l’enzyme par des interactions de faible énergie. Il est ensuite libéré avec une vitesse variable selon la réaction (Figure I-8).
Figure I-8: Mode d’action des enzymes.
E : Enzyme, S : Substrat, P : Produit, ES : Complexe enzyme-substrat.
I.E. Cinétique enzymatique (équation de Michaelis-Menten)
Comme dans tout procédé catalytique, il est important, avant d’utiliser une enzyme dans une synthèse ou une analyse, d’en connaître le comportement cinétique. De façon simplifiée, nous allons admettre pour la réaction enzymatique suivante :
Soient [E], [S], [ES] et [P] les concentrations des différentes espèces au temps t. Si l’on admet que la concentration en enzyme est beaucoup plus petite que celle du substrat et que le taux d’avancement de la réaction est faible de façon à pouvoir négliger la variation de la concentration en substrat, la vitesse de la réaction est :
L’état stationnaire implique que la concentration de ES reste constante pendant la mesure de la vitesse. Donc :
10 Or la quantité totale d’enzyme [E]0 ne varie pas . Donc :
Et la vitesse devient :
Ainsil’étude de la vitesse en fonction de [E]0 et de [S] permet de déterminer k2 et (k-1+ k2)/ k1
mais pas k1 et k-1. Le coefficient (k-1+ k2)/k1 est appelé constante de Michaelis (km) et
l’expression de la vitesse devient :
Si la formation et la dissociation du complexe ES sont rapides par rapport à la transformation écrit par k2, la constante de vitesse est voisine de la constante de dissociation du complexe
enzyme-substrat ES : ks = k-1/k1. L’étude de l’influence de la concentration du substrat sur la
vitesse montre qu’à concentration élevée de S par rapport à km, la vitesse est constante. En
effet, si [S] est très grand par rapport à km, la vitesse devient : v = k2[E]0 = vmax. La moitié de
cette vitesse maximale est atteinte quand [S] est égale à km. L’équation de Michaelis devient :
(4) (5) (6) (Equation de Michaelis-Menten) (7) (2) (3)
11
I.F. Classification des enzymes
En 1961,23 l’Union Internationale de la Biochimie (U.I.B) donne la classification suivante des enzymes, qui sont divisées en six groupes selon leurs activités catalytiques, ceci étant le caractère essentiel de toute enzyme sachant, qu’en général, qu’une enzyme ne peut catalyser qu’un seul type de réaction.
I.F.1. Les oxydo-réductases : les enzymes de ce groupe catalysent les réactions
d’oxydoréduction et englobent les réactions d’oxygénation, comme par exemple le passage de C-H à C-OH, ainsi que l’addition ou l’élimination d’atomes d’hydrogène, comme par exemple CH(OH) à C=O et CH-CH à C=C.
I.F.2. Les transférases : ces enzymes transfèrent des radicaux (méthyle, éthyle,…) ou des
groupements d’atomes (l’hydroxyméthyl, carbonyl et les groupements carbonés des fonctions aldéhydes ou cétones,…) d’une molécule (substrat donneur) à une autre (substrat accepteur).
I.F.3. Les hydrolases : Cette classe permet l’hydrolyse d’esters, d’anhydrides, de glycosides,
d’amides et permet aussi la transestérification des alcools.
I.F.4. Les lyases : ce type de catalyseurs favorise les additions de HX sur les doubles liaisons
comme C=C, C=N, C=O et leur processus inverse.
I.F.5. Les isomérases : elles catalysent aussi bien les migrations intramoléculaires des
doubles liaisons C=C que l’isomérisation cis – trans et peuvent effectuer les racémisations désirées.
I.F.6. Les ligases (synthétases) : elles permettent la formation de divers types de liaisons
telles que C-O, C-C, C-S, C-N.
La Figure I-9 illustre la fréquence d’utilisation des enzymes en synthèse organique.24
23
Internationnal Union of Biochemistry Enzyme Nomenclature Academic Press:Orlando, 1984.
24 K. Faber. “Biotransformations in organic chemistry”, 5th
12
Figure I-9 : Fréquence d’utilisations des enzymes en synthèse organique.
Les hydrolases25 (lipases, estérases, protéases, acylases,….) et plus précisément les lipases (Figure I-10), sont les enzymes les plus utilisées par le chimiste organicien. Les enzymes ne nécessitent pas de co-enzyme pour leur fonctionnement.
Figure I-10 : Fréquence de l'utilisation des enzymes hydrolytique dans les Biotransformations.26
I.G. Les Lipases I.G.1. Généralités
Les lipases ou les triacylglycéroacyl-hydrolases, sont des enzymes atypiques de par leur mécanisme d’action et leur spécificité de substrats. Elles peuvent agir en tant qu’hydrolases en milieu aqueux ou comme catalyseurs en synthèse organique. En tant qu’hydrolases, elles sont responsables du catabolisme des triglycérides, leurs substrats préférentiels, en acide gras et en glycérol. En milieu solvant, elles peuvent catalyser un bon nombre de réactions allant de
25
a. B. Danieli, S. Riva, Pure & Appl. Chem. 1994, 66, 2215.
b. A. A. Assamoi, J. Destain, P. Thonart, Biotechnol. Agron. Soc. Environ. 2009, 13, 281.
26
K. Faber, Pure & Appl. Chem. 1997, 69, 1613.
5% 2%
60% 25%
13 l’estérification à l’acidolyse ou l’alcoolyse tout en présentant une certaine énantio-régio- et chimio-sélectivité. Les lipases sont largement répandues dans la nature où elles ont un rôle physiologique important dans le métabolisme des graisses. On les retrouve aussi bien dans le règne végétal, animaux,27 bactéries Gram+28 ou Gram-29 mais également chez de nombreux microorganismes, principalement sous forme de protéines extracellulaires.
I.G.2. Réactions réalisables par les lipases
Les lipases peuvent être employées en synthèse organique en tant que catalyseurs de choix. En effet, elles présentent l’avantage de réaliser une multitude de réactions chimiques, allant de l’estérification (1),30 à l’acidolyse (2),31 en passant par l’amidification (3),32 l’hydrolyse (4) 33 ou la transestérification (5) 34 (Schéma I-1). En fonction de l'environnement de l’enzyme, elles peuvent agir en tant qu’hydrolases en milieu aqueux ou comme catalyseurs en synthèse organique.
Schéma I-1 : Différentes réactions possibles par utilisation de lipases.
27
S. Cherif, Y. Gargouri, Food Chemistry. 2009, 116, 82.
28
M. Guncheva, D. Zhiryakova, J. Mol. Catal. B: Enzym. 2011, 68, 1.21.
29
R. Gaur, A. Gupta, S. K. Khare, Process Biochemistry. 2008, 43, 1040.
30
P. Vidya, A. Chadha, J. Mol. Catal. B: Enzym. 2010, 65, 68.
31
M. L. Foresti, M. L. Ferreira, Enzyme Microb. Technol. 2010, 46, 419.
32
A. Prasad, M. Husain, B. Singh, R. Gupta, V. Manchanda, C. Olsen, V. Parmar, Tetrahedron Lett. 2005, 46, 4511.
33
Y. Zhang, J. Liu, J. Biochem. Eng. 2011, 54, 40.
34
14 Les lipases offrent tout un panel de réactions possibles, directement utilisables en synthèse organique.
I.G.3. Mécanisme d’action des lipases
La caractéristique commune de toutes les lipases est que leurs sites actifs sont constitués de trois acides aminés, appelés la triade catalytique : la sérine, l'acide aspartique et l'histidine.35 Cette triade d’acides aminés, ajoutée à quelques autres résidus, constitue le site actif de l’enzyme. Celui-ci est protégé par un « couvercle » (en fait un segment hélicoïdal hydrophobe) qui en bloque l’accès et ne s’ouvre qu’au contact d’une phase hydrophobe pour permettre la complexation du substrat, lipide hydrophobe, et faciliter ainsi la catalyse de la réaction.
Le mécanisme d’intervention de la triade (Ser, Hist et Asp) lors de l’hydrolyse d’un ester est donné par le schéma suivant:
Schéma I-2 : Mécanisme réactionnel de la triade catalytique. Nu : H2O, H2NR, ROH,…
Comme le montre le schéma ci-dessus, la disposition particulière des 3 résidus formant la triade permet, dans une première étape, l’attaque nucléophile du substrat R1CO2R2 pour
donner un complexe appelé acyl-enzyme avec libération de l’alcool R2OH. Dans une
deuxième étape, si par exemple le nucléophile est l’eau, ce dernier attaque le complexe intermédiaire acyl-enzyme générant ainsi l’enzyme et l’acide carboxylique R1COOH.
35
15 Selon le milieu utilisé (aqueux ou organique), d’autres nucléophiles peuvent intervenir, ce qui conduit à plusieurs transformations possibles. Pour bien comprendre le mécanisme d'action des lipases, il est nécessaire de connaître leur structure.
I.G.4. La structure des lipases
Des études plus détaillées ont été réalisées par la suite et ont montré que, quelle que soit leur origine, les lipases présentent de nombreuses caractéristiques structurales communes. li s'agit du repliement α/β, de la triade catalytique et du volet amphiphile.36 A titre d’exemple, la lipase de Candida antarctica B une protéine de 33kDa, constituée de 317 acides aminés, se caractérise, comme chez les estérases, par la présence d’une structure α/β (Figure I-11).
Figure I-11 : Structure tridimensionnelle de la lipase de Candida antarctica B.
Le site actif d’une lipase est constitué d’une triade catalytique composée de :
• la serine 105, agissant comme nucléophile lors de la formation du premier intermédiaire tétraédrique.
• de l’histidine 224.
• l’acide aspartique 187 qui stabilise ce dernier.
Lorsque celle-ci est fermée, l’enzyme est dans sa conformation inactive. Cette conformation ne permet donc pas au substrat d’être en contact avec la triade catalytique se trouvant à l’intérieur du site actif. Dans la forme ouverte ou active, l’hélice α et sa face hydrophobe se
36
M. F. Egloff . S. Ransac, F. Marguet, E. Rogalska, H. Van Tilbeurgh, G. Buono. C. Cambillau, R. Verger, OCL. 1995, 2, 52.
16 met en contact avec la phase lipide de l’interface eau/lipide, le site actif devenant accessible au substrat : c’est l’activation interfaciale des lipases.
I.G.4.a. L’activation interfaciale
La majorité des lipases sont caractérisées par le phénomène d’activation interfaciale37 : lorsque la concentration micellaire critique est atteinte, les lipases situées à l’interface de la phase eau/lipide voient leur activité d’hydrolyse des acylglycérols fortement augmenter. En milieu organique, le site actif des lipases n'est généralement pas accessible au solvant. En effet, il est recouvert par une boucle amphiphile appelée volet amphiphile ou "lid" ou "flap". Il s'agit de la forme fermée ou inactive de l'enzyme (Figure I-12). Certains auteurs ont mis en évidence l'existence d'une forme ouverte active de l'enzyme. Dans ce cas, la face hydrophobe du volet, orienté auparavant vers l'intérieur du site actif, s'expose alors au solvant, créant une surface hydrophobe supposée interagir avec l'interface lipide-eau.38 Du fait de sa proximité avec le site actif, des études ont montré que l’on ne retrouve pas ce phénomène chez cet enzyme.39 Les travaux d’Uppenberg et coll.40 sur la structure de l’enzyme concluent à l’absence de volet amphiphile. Des études plus récentes ont montré l'existence du "lid" pour la CAL B en présence se substrats volumineux. 41
Figure I-12: Représentation schématique du volet amphiphile et du phénomène d'activation interfaciale en milieu organique.
37
R. D. Schmid, R. Verger. Angew. Chem. Int. Ed. 1998, 37, 1608.
38
a. Y. Gargouri, S. Ransac, R. Verger, Biochim. Biophys. Acta. 1997, 1344, 6.
b. T. Maruyama, M. Nakajima, S. lchikama, H. Nabetani, S. Furusaki, M. Seki, J. Am. Oit Chem. Soc. 2000,
77, 1121.
39
a. P. L. A. Overbeeke, G. C, N. Khalaf, J. A. Jongejan, J. J. Heijnen, J. Mol. Catal. B : Enzym. 2000, 10, 385.
b. M. Martinelle, M. Holmquist, K. Hult, Biochim. Biophys. Acta. 1995. 1258, 272.
40
J. Uppenberg, N. Oehrner, M. Norin, K. Hult, G. J. Kleywegt, S. Patkar, V. Waagen, T. Anthonsen, J. T.
Alwyn, Biochem. 1995, 34, 16838.
41
T. Zisis, P. L. Freddolino , P. Turunen, M. C. van Teeseling, A. E. Rowan, K. G. Blank , Biochem. 2015, 54, 5969.
17
I.G.5. Spécificité des lipases
Les lipases catalysent le même type de réaction, mais leur action vis-à-vis des substrats en terme de chimio-, régio- et énantio- sélectivité change considérablement. Les conditions opératoires optimales sont différentes d'une lipase à l'autre, rendant un procédé général approprié à toutes les lipases difficile ou même impossible à être développé. Les lipases présentent plusieurs spécificités liées à la structure du site actif.20
I.G.5.a. La régiosélectivité : pouvoir d'hydrolyser préférentiellement les liaisons ester
carboxylique en position externe sn-1 et sn-3 (esters primaires) vis-à-vis de la position interne sn-2 (esters secondaires).
I.G.5.b. La typosélectivité : pouvoir de distinguer plusieurs types de donneurs d'acyle.
I.G.5.c. La stéréosélectivité : capacité de discrimination entre deux énantiomères dans le cas
d'un substrat racémique et la capacité de discrimination entre deux groupements stéréohétérotopiques mais homomorphiques (énantiotopiques) dans le cas des acylglycérols prochiraux (position sn-1 contre sn-3). L'aptitude des enzymes à distinguer les énantiomères se fait grâce aux différentes interactions diastéréoisomériques intervenant entre les énantiomères et le site actif de l'enzyme.
I.H. Immobilisation des enzymes
Les enzymes présentent un fort intérêt dans le domaine de la biocatalyse. Cependant, comme nous l’avons évoqué, leur coût et leur stabilité limitée dans le temps sont des facteurs limitant leur utilisation industrielle. Afin de palier à ces inconvénients, une stratégie fut proposée : l’immobilisation des enzymes qui permet de stabiliser celles-ci au cours de leur utilisation, de pouvoir les réutiliser et de séparer l’enzyme des produits de la réaction enzymatique. Les biocatalyseurs immobilisés présentent une stabilité augmentée, mais des changements dans l’activité enzymatique, le pH optimum et l’affinité pour le substrat ont été observés.42 Ces changements dépendent de la source de l’enzyme, du type de support et de la méthode
42
a. V. M. Balco, A. L. Paiva, F. X. Malcata, Enzyme Microb. Technol. 1996, 18, 392. b. A. E. Ivanov, M. P. Schneider, J. Mol. Catal. B: Enzym. 1997, 3, 303.
18 d’immobilisation.43 En 1916, Nelson et Griffin seront les premiers à démontrer qu’une enzyme, en l’occurrence l’invertase, conserve son activité catalytique et ce même après avoir été immobilisée par adsorption sur du charbon actif. Cette technique connaîtra un véritable essor à partir des années 1950 avec les premières applications dans divers domaines. Il existe différentes techniques d’immobilisation pouvant être aussi bien chimiques que physiques. On peut notamment citer quatre méthodes, couramment utilisées :44
I.H.1. Immobilisation par liaison covalente
L’immobilisation par liaison covalente45 conduit à la formation des liaisons très solides entre enzyme et support. Le couplage covalent d’enzyme sur support nécessite la présence d’un groupe fonctionnel sur la surface de celui-ci. Ces groupements sont en général –COOH, -NH2, -OH, ou –SH ; ils sont peu réactifs chimiquement et il convient de les activer pour
qu’ils réagissent dans des conditions douces avec des groupements fonctionnels de l’enzyme.
I.H.2. Immobilisation par réticulation et co-réticulation
Le principe est ici de ponter les molécules d’enzymes respectivement entre elles, ou avec d’autres protéines, à l’aide d’un réactif polyfonctionnel tel que le glutaraldéhyde, afin d’obtenir des macrostructures insolubles.
I.H.3. Immobilisation par inclusion ou piégeage
La technique revient à emprisonner l’enzyme dans les mailles du réseau d’une matrice polymérique ou inorganique.46 La maille de la matrice assure de manière purement physique la rétention de l’enzyme tout en permettant la diffusion du substrat jusqu’au site actif de l’enzyme grâce à une porosité du gel suffisante.
43
a.W. A. M. Alloue, M. Aguedo, J. Destain, H. Ghalfi, Biotechnol, Agon, Soc, Environ. 2008, 12, 57. b. Z. D. Knezevic, S. S. Siler. Marinkovic, L. V. Mojović,APTEFF. 2004, 35, 151.
44
P. Worsfold, J. Enzymes, Pure & Appl. Chem. 1995, 67, 597.
45
Y. X. Bai, Y. F. Li, Y. Y. Liu-Xiang Yi, J. Biotechnol. 2006, 125, 574.
46