N° d’ordre : 2877
THÈSE DE DOCTORAT D’ETAT
Présentée par
Loubna GUENNOUN
Discipline : Chimie
Spécialité : Chimie Physique (Option : Spectroscopie)
ETUDE THEORIQUE ET EXPERIMENTALE DES
PROPRIETES STRUCTURALES ET VIBRATIONNELLES DU
3,5-DIAMINO-1,2,4-TRIAZOLE ET DU DICATION
GUANAZOLIUM
Soutenue le 01 Juin 2016
Devant le jury
Président :
Oum Keltoum KABBAJ PES, Faculté des Sciences, Rabat
Examinateurs :
Fouzia GUEDIRA PES, Faculté des Sciences, Rabat
Mohamed HAMAD PES, Faculté des Sciences, Rabat
Khadija MARAKCHI PES, Faculté des Sciences, Rabat
Lotfi RGHIOUI PES, Faculté des Sciences, Meknès
Hamid TABYAOUI PES, Faculté des Sciences, Rabat
Souad ZAYDOUN PES, Faculté des Sciences, Rabat
Faculté des Sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat – Maroc Tel +212 (05) 37 77 18 34/35/38, Fax : +212 (05) 37 77 42 61, http://www.fsr.ac.ma
Je dédie ce travail
A la mémoire de mon père
A ma mère que Dieu lui accorde longue vie
A mon mari pour son soutien constant
A mes enfants
A mes frères et à ma sœur
A ma famille
A ma belle famille
AVANT-PROPOS
Ce travail a été initialement réalisé au Laboratoire de Spectroscopie Infrarouge qui depuis 2012 fait partie du Laboratoire de Spectroscopie, Modélisation Moléculaire, Matériaux et Environnement (LS3ME) de la Faculté des Sciences de Rabat, sous la direction de Mesdames les professeurs Fouzia GUEDIRA et Souad ZAYDOUN.
Mes premières pensées vont à mes directrices de thèse. Je tiens à leur exprimer toute ma reconnaissance et mes plus chaleureux remerciements pour avoir suivi mon travail avec beaucoup d'intérêt, pour leur disponibilité, leurs conseils avisés ainsi que pour l'inaltérable confiance qu'elles n'ont eu de cesse de m'accorder. Leurs qualités personnelles et professionnelles resteront pour moi une immense source d'enrichissement.
Je suis très sensible à l’honneur qu’a bien voulu me faire Madame Oum Keltoum KABBAJ, Professeur à la Faculté des Sciences de Rabat et Directrice du LS3ME, en acceptant de présider ce jury de thèse. Qu'elle trouve ici l'expression de toute ma gratitude.
Je voudrais également exprimer ma reconnaissance à Monsieur Mohamed HAMAD, Professeur à la Faculté des Sciences de Rabat, d'avoir accepté de juger le contenu de ma thèse et d'en être l'un des rapporteurs.
Qu’il me soit permis d'exprimer ma profonde reconnaissance à Madame Khadija MARAKCHI, Professeur à la Faculté des Sciences de Rabat, pour sa collaboration efficace et le temps qu'elle a consacré à m'initier aux calculs théoriques. Qu’elle soit vivement remerciée pour l’intérêt qu’elle a porté à ce travail et pour avoir accepté de faire partie de ce jury de thèse.
Je sais gré à Monsieur Hamid TABYAOUI, Professeur et Vice-Doyen aux affaires Pédagogiques à la Faculté des Sciences de Rabat, qui a bien voulu se joindre au jury de cette thèse en délaissant momentanément ses nombreuses responsabilités.
Monsieur Lotfi RGHIOUI, Professeur à la Faculté des Sciences de Meknès, a bien voulu, avec une patience pleine d’amabilité, examiner ce mémoire et en être rapporteur. Qu’il
soit assuré de mes sincères remerciements pour le temps qu'il a consacré à ce travail et pour les nombreuses discussions que nous avons eues ensemble.
Les mots ne sauraient suffire pour remercier Madame Afaf EL HAJJI, Professeur à la Faculté des Sciences de Rabat, pour sa disponibilité à mon égard, son soutien et pour ses remarques judicieuses et nos discussions fructueuses. Son amitié m'a été précieuse dans les moments difficiles.
Je souhaite aussi manifester ma très grande gratitude à tous les membres de l'équipe de Spectroscopie pour l’accueil qu’ils m’ont réservé quand j'ai intégré le laboratoire. Leur enthousiasme communicatif, leurs inlassables encouragements et leurs qualités humaines ont rendu mon travail plus agréable.
Mes remerciements vont également aux autres membres du LS3ME : Mesdames Najia KOMIHA, Hassna ABOU EL MAKARIM, Souad EL HAJJAJI, Malika SERGHINI IDRISSI et Saloua SEBBAHI pour le soutien que j'ai trouvé auprès d'elles et pour l'ambiance amicale au sein du laboratoire.
Un grand merci à mon amie, Madame Jamila EL JASTIMI, qui a toujours su m'encourager et, surtout, être là lorsque j’en avais besoin.
Enfin, je voudrais remercier tous ceux qui ont contribué, de près ou de loin, à la réalisation de ce travail.
RESUME
Nous avons effectué une étude de la structure et des vibrations moléculaires du 3,5-diamino-1,2,4-triazole (guanazole) et de sa forme diprotonée par les méthodes ab-initio et les spectroscopies infrarouge et Raman.
Le calcul énergétique pour le guanazole montre que la forme tautomère 1H est la plus stable à l’état gazeux et en solution. Le dication guanazolium le plus stable est protoné sur les azotes du cycle.
Concernant le calcul des paramètres géométriques, les valeurs restent voisines pour le guanazole dans tous les états physiques; le noyau triazolique est plan avec les groupements amino inclinés par rapport au plan. La protonation induit une symétrie de la molécule et une planéité totale du noyau et des NH2.
La densité électronique calculée montre clairement des variations de charge considérables lors de la protonation.
Le calcul des fréquences de vibration et de la DEP ont permis de souligner les différents couplages mis en jeu. Les nombres d'onde expérimentaux restent proches de ceux calculés. L'effet de la protonation se traduit par des perturbations de la plupart des vibrations explicables surtout par les effets électroniques et les variations des paramètres géométriques.
Dans le guanazole, il existe des liaisons hydrogène de forces moyenne et faible respectivement pour le NH du cycle et les groupes NH2 (NH…N′, NH2...N′ et NH2…NH2).
Leurs forces augmentent dans le sel avec des interactions de type NH…Cl- et NH…N pour les
NH des NH2 et N4H…NH2, N1H….Cl- et N2….Cl- pour les NH triazoliques.
MOTS-CLEFS (5) : 3,5-diamino-1,2,4-triazole, calculs DFT, spectroscopie vibrationnelle, géométrie moléculaire, sites de protonation
ABSTRACT
We conducted a study of the structure and molecular vibrations of 3,5- diamino-1,2,4-triazole (guanazole) and its diprotonated form by ab initio methods and infrared and Raman spectroscopy.
Energy calculation for guanazole shows that the tautomeric 1H form is the most stable in the gaseous state and in solution. The dication guanazolium protonated on the two nitrogen cycle is the most stable.
Regarding the calculation of geometric parameters, the values remain close those for the guanazole in all physical states; the geometry of triazole ring is plane with the amino groups inclined towards the plane. Protonation induces symmetry of the molecule and total flatness of the cycle and NH2 groups.
The calculated electron density clearly shows considerable load variations during the protonation.
The calculated vibration frequencies and DEP have highlighted the different couplings involved. The experimental wave numbers are close to those calculated. The effect of the protonation on vibrations results in important variations which are explainable especially by electronic effects and geometric parameters variations.
In guanazole, there are hydrogen bonds of medium and low strengths involving respectively the ring NH and NH2 groups (NH ... N', NH2 ... N' and NH2... NH2). Their
forces increase in the salt; the interactions are NH…Cl- and NH…N for NH2 groups and
N4H…NH2,N1H….Cl- and N2….Cl-for triazolic NH.
KEYWORDS (5) : 3,5- diamino- 1,2,4-triazole, DFT calculations, vibrational spectroscopy, molecular geometry, protonation sites
SOMMAIRE
LISTE DES ABREVIATIONS LISTE DES TABLEAUX LISTE DES ILLUSTRATIONS
INTRODUCTION GENERALE ... 1
BIBLIOGRAPHIE ... 3
CHAPITRE I : REVUE BIBLIOGRAPHIQUE ... 7
INTRODUCTION ... 7
I. MOLECULE DE GUANAZOLE ... 8
I.1. Synthèse ... 8
I.2. Etude par spectroscopie UV-visible ... 8
I.3. Etude par spectroscopie RMN ... 8
I.4. Etude par diffraction des rayons X ... 9
I.5. Etude théorique ... 11
I.6. Etude vibrationnelle ... 11
I.7. Etude électrochimique ... 12
II. SELS DE GUANAZOLIUM ... 12
II.1. Etude par spectroscopie UV-visible ... 12
II.2. Etude par spectroscopie RMN ... 12
II.3. Etude par diffraction des rayons X ... 13
II.4. Etude vibrationnelle ... 17
CONCLUSION ... 17
BIBLIOGRAPHIE ... 18
CHAPITRE II : ETUDE THEORIQUE PAR LES METHODES AB-INITIO DES PROPRIETES ENERGETIQUES, GEOMETRIQUES, ELECTRONIQUES ET VIBRATIONNELLES DU 3,5-DIAMINO-1,2,4-TRIAZOLE ... 20
INTRODUCTION ... 20
I. MÉTHODES DE CALCUL ... 21
I.1. Structure et symétrie de la molécule ... 21
I.2. Calcul des charges atomiques ... 22
I.3. Choix des coordonnées internes ... 23
I.4. Calcul des fréquences vibrationnelles et de la distribution d’énergie potentielle DEP ... 23
I.5. Facteur d’échelle empirique ... 24
II. RESULTATS ET DISCUSSION ... 24
II.1. Analyse énergétique ... 25
II.3. Analyse des données géométriques ... 27
II.3.1. Longueurs de liaison ... 27
II.3.2. Angles valentiels ... 29
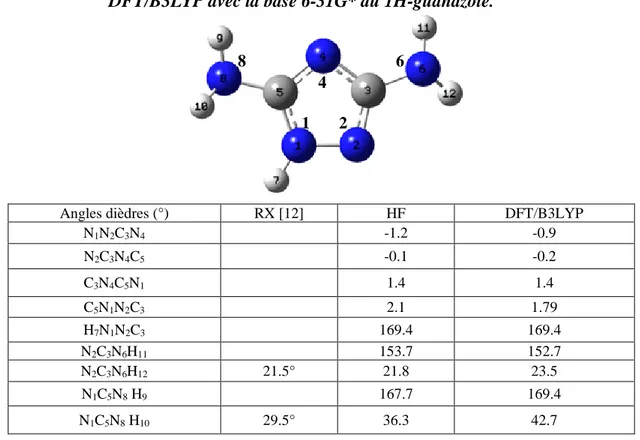
II.3.3. Angles dièdres ... 30
II.4. Etude vibrationnelle ... 31
II.4.1. Dénombrement des modes normaux de vibration ... 31
II.4.2. Présentation des résultats ... 33
II.4.3. Attributions et distribution d’énergie potentielle ... 38
II.5. Effet de solvant ... 45
II.5.1. Energies ... 45
II.5.2. Moments dipolaires et charges atomiques ... 46
II.5.3. Paramètres géométriques ... 48
II.5.4. Nombres d’onde de vibration et DEP ... 51
II.5.4.a. Vibrations des groupements NH2 ... 54
II.5.4.b. Vibrations du groupement NH ... 55
II.5.4.c. Vibrations des jonctions C-NH2 ... 56
II.5.4.d. Vibrations du cycle triazolique ... 57
CONCLUSION ... 58
BIBLIOGRAPHIE ... 60
CHAPITRE III : ETUDE EXPERIMENTALE PAR SPECTROSCOPIE DE VIBRATION DU 3,5-DIAMINO-1,2,4-TRIAZOLE... 63
INTRODUCTION ... 63
I. DÉNOMBREMENT ET CLASSEMENT DES VIBRATIONS ... 63
II. CRITERES D’ATTRIBUTION ... 64
III. ETUDE DES SPECTRES DE VIBRATION ... 65
III.1. Composés de départ et conditions expérimentales ... 65
III.1.1. Composés de départ ... 65
III.1.2. Techniques expérimentales ... 65
III.1.2.1. Spectroscopie d’absorption dans l’infrarouge ... 65
III.1.2.2. Spectroscopie de diffusion Raman... 66
III.2. Présentation des résultats ... 66
III.3. Analyse des spectres ... 66
III.3.1. Vibrations des groupements NH2 ... 70
III.3.2. Vibrations des jonctions C-NH2 ... 74
III.3.3. Vibrations du groupement NH "triazolique" ... 76
IV. EFFET DE LA C-SUBSTITUTION PAR LES GROUPEMENTS AMINO SUR LES
VIBRATIONS DU SQUELETTE TRIAZOLIQUE ... 80
IV.1. Effet sur la vibration NH ... 80
IV.2. Effet sur les vibrations cycliques ... 83
CONCLUSION ... 85
BIBLIOGRAPHIE ... 86
CHAPITRE IV : ETUDE THEORIQUE PAR LA METHODE DFT DES PROPRIETES ENERGETIQUES, GEOMETRIQUES, ELECTRONIQUES ET VIBRATIONNELLES DU DICATION 3,5-DIAMINO-1,2,4-TRIAZOLIUM ... 88
INTRODUCTION ... 88
I. METHODES DE CALCUL ... 88
I.1. Structure et symétrie de la molécule ... 89
I.2. Choix des coordonnées internes ... 89
II. RESULTATS ET DISCUSSION ... 89
II.1. Analyse énergétique ... 90
II.2. Analyse de charge ... 91
II.3. Analyse des données géométriques ... 94
II.3.1. Longueurs de liaison ... 94
II.3.2. Angles valentiels ... 96
II.3.3. Angles dièdres ... 98
II.4. Etude vibrationnelle ... 99
II.4.1. Dénombrement des modes normaux de vibration ... 99
II.4.2. Présentation des résultats ... 101
II.4.3. Attributions et analyse de la distribution d’énergie potentielle ... 105
II.4.3.1. Vibrations dans le plan de types A1 et B2 ... 108
II.4.3.2. Vibrations hors du plan de types A2 et B1 ... 114
II.4.4. Effet de la protonation sur les vibrations ... 116
CONCLUSION ... 119
BIBLIOGRAPHIE ... 121
CHAPITRE V : ETUDE EXPERIMENTALE DES SPECTRES DE VIBRATION DU CHLORHYDRATE DU 3,5-DIAMINO-1,2,4-TRIAZOLIUM ... 123
INTRODUCTION ... 123
I. DENOMBREMENT ET CLASSEMENT DES VIBRATIONS - CRITERES D’ATTRIBUTION ... 124
I.1. Dénombrement et classement des vibrations ... 124
I.2. Critères d’attribution ... 124
II. ANALYSE VIBRATIONNELLE DU CHLORHYDRATE DE GUANAZOLIUM (NH2)2TA.2HCL 125 II.1. Partie expérimentale ... 125
II.1.1.a. Chlorhydrate et bromhydrate de guanazolium ... 125
II.1.1.b. Chlorhydrate de guanazolium deutérié ... 125
II.1.2. Caractérisation des sels étudiés ... 126
II.1.2.a. Dosage potentiométrique ... 126
II.1.2.b. Analyses élémentaires ... 126
II.1.3. Conditions d’enregistrement ... 127
II.2. Présentation des résultats ... 127
II.3. Analyse des spectres ... 127
II.3.1. Vibrations des groupements NH2 ... 131
II.3.2. Vibrations des groupements NH triazoliques ... 134
II.3.3. Vibrations des jonctions C-NH2 ... 137
II.3.4. Vibrations cycliques ... 139
III. EFFET DE LA PROTONATION SUR LES VIBRATIONS DU GUANAZOLE. SITES DE PROTONATION ... 141
IV. ETUDE DE LA LIAISON HYDROGENE DANS LE CHLORHYDRATE DE GUANAZOLIUM .. 144
IV.1. Généralités sur la liaison hydrogène ... 144
IV.2. Liaisons hydrogène dans le chlorhydrate de guanazolium ... 144
IV.2.1. Comparaison du chlorhydrate avec le cation gazeux ... 145
IV.2.2. Comparaison du chlorhydrate avec la molécule de base ... 146
IV.2.3. Comparaison du chlorhydrate avec le bromhydrate ... 147
CONCLUSION ... 150
BIBLIOGRAPHIE ... 152
CONCLUSION GENERALE... 154
ANNEXE ... 157
LISTE DES ABREVIATIONS
TA : 1,2,4-triazole
NH2TA : 5-amino-1,2,4-triazole
(NH2)2TA : 3,5-diamino-1,2,4-triazole ou guanazole
(ND2)2TA : 3,5-diamino-1,2,4-triazole N-deutérié
(NH2)2TA.2HCl : chlorhydrate de guanazolium
(NH2)2TA.2HBr : bromhydrate de guanazolium
(ND2)2TA.2D+ : cation guanazolium N-deutérié
CH3TA : 3-méthyl-1,2,4-triazole (CH3)2TA : 3,5-diméthyl-1,2,4-triazole ClTA : 3-chloro-1,2,4-triazole Cl2TA : 3,5-dichlo-1,2,4-triazole BrTA : 3-bromo-1,2,4-triazole Br2TA : 3,5-dibromo-1,2,4-triazole IR : infrarouge UV : ultaviolet TF : très fort F : fort
M : moyen à fort M : moyen à faible
F : faible Tf : très faible ep: épaulement HF : Hartree-Fock
DFT : Théorie de la Fonctionnelle de la Densité DEP : Distribution d’Energie Potentielle
VEDA 4 : Vibrational Energy Distribution Analysis version 4 u.a : unité atomique. 1 u.a = 2625.5 kJ/mol
LISTE DES TABLEAUX
Tableau I. 1: Distances en Å et angles en ° des contacts intermoléculaires du guanazole dans le cristal. ... 10 Tableau I.2 : Données structurales antérieures publiées pour les sels de guanazolium
monoprotonés. ... 14 Tableau II.1 : Energies totales (ET) et énergies relatives (∆E) du 1H-guanazole et
4H-guanazole calculées aux niveaux HF et DFT/B3LYP avec la base 6-31G* en symétries C1 et CS. ... 26
Tableau II.2 : Populations électroniques de Mulliken et charges nettes des atomes du 1H-guanazole calculées aux niveaux HF et DFT/B3LYP avec la base 6-31G*. . 26 Tableau II.3.a : Longueurs de liaison expérimentales et calculées aux niveaux HF et
DFT/B3LYP avec la base 6-31G* du 1H-guanazole. ... 28 Tableau II.3.b : Angles valentiels expérimentaux et calculés aux niveaux HF et
DFT/B3LYP avec la base 6-31G* du 1H-guanazole. ... 29 Tableau II.3.c : Angles dièdres expérimentaux et calculés aux niveaux HF et
DFT/B3LYP avec la base 6-31G* du 1H-guanazole. ... 30 Tableau II.4 : Nombres d’onde, intensités IR et activités Raman calculés aux niveaux
HF et B3LYP avec la base 6-31G* du 1H-guanazole. ... 35 Tableau II.5 : Coordonnées de symétrie locales du 1H-guanazole. ... 36 Tableau II.6 : Coordonnées internes du 1H-guanazole. ... 37 Tableau II.7 : Nombres d’onde calculés et mis à l’échelle en cm-1 au niveau B3LYP
avec la base 6-31G* du 1H-guanazole. ... 39 Tableau II.8: Nombres d’onde calculés et mis à l’échelle en cm-1 au niveau B3LYP
avec la base 6-31G* du guanazole deutérié. ... 40 Tableau II.9 : Energies des formes 1H-guanazole et 4H-guanazole à l’état gazeux
et en solution dans l’éthanol et dans l’eau calculées au niveau
B3LYP avec la base 6-31G* ... 46 Tableau II.10 : Moments dipolaires du 1H-guanazole à l’état gazeux et en solution dans
l’éthanol et dans l’eau calculés au niveau B3LYP avec la base 6-31G*. ... 47 Tableau II.11 : Charges atomiques du 1H-guanazole à l’état gazeux et en solution dans
l’éthanol et dans l’eau calculées au niveau B3LYP avec la base 6-31G*. ... 47 Tableau II.12 : Paramètres géométriques calculés au niveau B3LYP avec la base 6-31G*
du 1H-guanazole à l’état gazeux et en solution et paramètres géométriques expérimentaux ... 49 Tableau II.13 : Nombres d’onde et intensités IR calculés au niveau B3LYP avec la base
6-31G* du 1H-guanazole en solution dans l’éthanol. ... 52 Tableau II.14 : Nombres d’onde et intensités IR calculés au niveau B3LYP avec la base
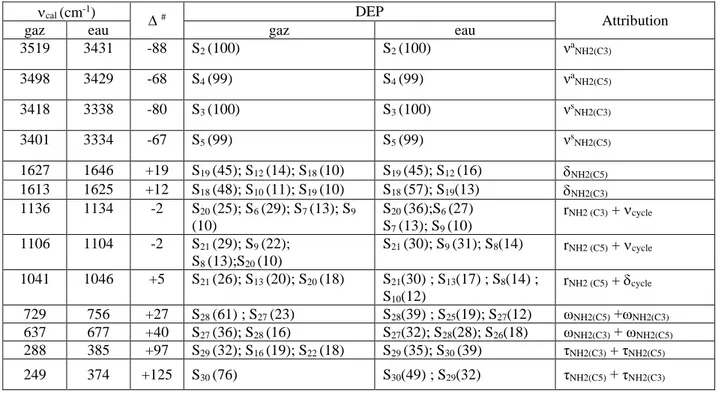
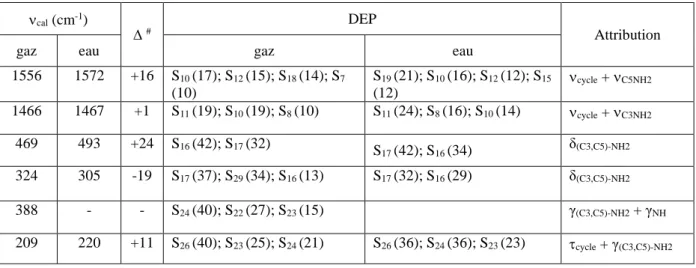
Tableau II.15 : Nombres d’onde, variations des nombres d’onde et DEP des groupements NH2 pour le gaz et la solution aqueuse ... 55
Tableau II.16 : Nombres d’onde, variations des nombres d’onde et DEP du groupement NH pour le gaz et la solution aqueuse ... 56 Tableau II.17 : Nombres d’onde, variations des nombres d’onde et DEP des groupements
C-NH2 pour le gaz et la solution aqueuse... 56
Tableau II.18 : Nombres d’onde, variations des nombres d’onde et DEP des vibrations cycliques pour le gaz et la solution aqueuse ... 57 Tableau III.1 : Nombres d’onde et intensités relatives des bandes infrarouges et des raies
Raman du 1H-guanazole et de son homologue deutérié à l’état solide... 69 Tableau III.2.a : Attribution des vibrations des groupements NH2 du 1H-guanazole.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 70 Tableau III.2.b : Attribution des vibrations des groupements ND2 du guanazole deutérié.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 70 Tableau III.3.a : Attribution des vibrations des groupements C-NH2 du 1H-guanazole.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 74 Tableau III.3.b: Attribution des vibrations des groupements C-ND2 du guanazole deutérié.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 74 Tableau III.4.a: Attribution des vibrations du groupement NH du 1H-guanazole.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 76 Tableau III.4.b: Attribution des vibrations du groupement ND du guanazole deutérié.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 76 Tableau III.5.a: Attribution des vibrations cycliques du 1H-guanazole.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 78 Tableau III.5.b: Attribution des vibrations cycliques du guanazole deutérié.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 78 Tableau III.6 : Comparaison des nombres d’onde NH, du type d’interaction et des distances
intermoléculaires pour le 1H-guanazole et quelques triazoles. ... 82 Tableau III.7 : Nombres d’onde des vibrations cycliques des dérivés C-mono et
C-disubstitués et leurs variations comparativement au 1,2,4-triazole. ... 84 Tableau IV.1 : Energies totales (ET) et énergies relatives (∆E) des différentes formes
diprotonées du 1H-guanazole calculées par la méthode DFT/B3LYP/6-31G*. ... 90 Tableau IV.2 : Charges atomiques du 1H-guanazole et des différentes formes
diprotonées calculées par la méthode DFT/B3LYP/6-31G*. ... 92 Tableau IV.3.a: Longueurs de liaison optimisées du 1H-guanazole et de sa forme
diprotonée (P24) calculées par la méthode DFT/B3LYP/6-31G*. ... 95 Tableau IV.3.b: Angles valentiels optimisées du 1H-guanazole et de sa forme
diprotonée (P24) calculées par la méthode DFT/B3LYP/6-31G*. ... 97 Tableau IV.3.c: Angles dièdres optimisées du 1H-guanazole et de sa forme
Tableau IV.4 : Dénombrement, classement et activité des vibrations du dication
guanazolium. ... 100
Tableau IV.5 : Classes de symétrie des vibrations du dication guanazolium. ... 101
Tableau IV.6 : Coordonnées de symétrie locales du dication guanazolium. ... 102
Tableau IV.7 : Définition des coordonnées internes du dication guanazolium... 104
Tableau IV.8 : Nombres d’onde calculés et mis à l’échelle, intensités IR et activités Raman du dication guanazolium calculés avec B3LYP/6-31G*. ... 106
Tableau IV.9 : Nombres d’onde calculés et mis à l’échelle, intensités IR et activités Raman du dication guanazolium deutérié calculés avec B3LYP/6-31G*. ... 107
Tableau IV. 10 : Attributions des vibrations planes de types A1 et B2 du dication guanazolium. ... 109
Tableau IV. 11 : Attributions des vibrations planes de types A1 et B2 du dication guanazolium deutérié. ... 110
Tableau IV.12 : Attributions des vibrations hors du plan de types B1 et A2 du dication guanazolium. ... 115
Tableau IV.13 : Attributions des vibrations hors du plan de types B1 et A2 du dication ... 115
Tableau IV.14 : Nombres d’onde calculés du 1H-guanazole et du dication-guanazolium et leurs variations par protonation. ... 117
Tableau V.1 : Données expérimentales et calculées des dosages élémentaires du chlorhydrate de guanazolium. ... 126
Tableau V.2 : Nombres d’onde, intensités relatives des bandes infrarouges et des raies Raman du chlorhydrate de guanazolium et de son homologue deutérié. ... 130
Tableau V.3.a : Attribution des vibrations des groupements NH2 du dication guanazolium Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 131
Tableau V.3.b : Attribution des vibrations des groupements ND2 du dication guanazolium deutérié. Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 131
Tableau V.4.a : Attribution des vibrations des groupements NH du dication guanazolium. Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 134
Tableau V.4.b : Attribution des vibrations des groupements ND du dication guanazolium deutérié Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 135
Tableau V.5.a : Attribution des vibrations des groupements C-NH2 du dication guanazolium Nombres d’onde expérimentaux, nombres d’onde calculés et DEP. ... 137
Tableau V.5.b : Attribution des vibrations des groupements C-ND2 du dication guanazolium deutérié. Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 137
Tableau V.6.a : Attribution des vibrations cycliques du dication guanazolium. Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 139
Tableau V.6.b : Attribution des vibrations cycliques du dication guanazolium deutérié.
Nombres d’onde expérimentaux, nombres d’onde calculés et DEP ... 139 Tableau V.7 : Variations des nombres d’onde des vibrations cycliques et de jonction dans le chlorhydrate de guanazolium par rapport à la molécule de base. .... 142 Tableau V.8 : Variations des nombres d’onde des vibrations des groupements NH2 et NH
dans le chlorhydrate de guanazolium par rapport à ceux calculés. ... 145 Tableau V.9 : Variations des nombres d’onde des vibrations des groupements NH2 et NH
dans le chlorhydrate de guanazolium par rapport au guanazole à l'état solide. ... 146 Tableau V.10 : Variations des nombres d’onde des vibrations des groupements NH2 et NH
LISTE DES ILLUSTRATIONS
Figure I.1 : Formes tautomères possibles pour le guanazole. ... 7
Figure I.2 : Données géométriques du guanazole. ... 9
Figure I.3 : Projection de la structure du guanazole selon le plan (001). ... 10
Figure I.4 : Vue de la structure du sel (GuanH)2.Cl2.H2O suivant la direction [100]. ... 16
Figure I.5 : Liaisons hydrogène dans le sel (GuanH)2.Cl2.H2O ... 16
Figure II.1 : Paramètres géométriques expérimentaux de la molécule de guanazole. ... 22
Figure II.2 : Formes tautomères du guanazole avec numérotation des atomes. ... 25
Figure II.3 : Déplacements cartésiens des modes de vibration du groupement amino dans l’aniline. ... 33
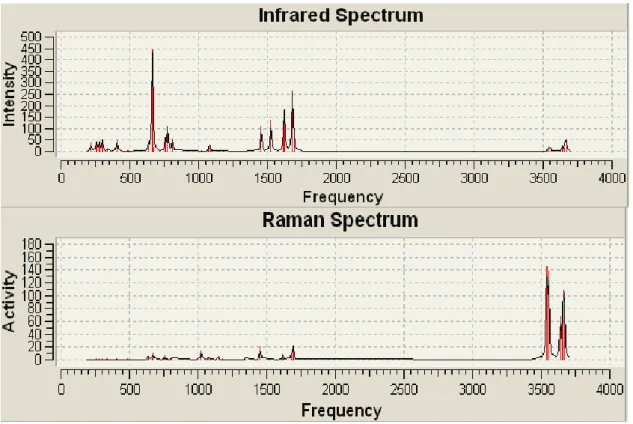
Figure II.4 : Spectres IR et Raman du 1H-guanazole calculés au niveau HF avec la base 6-31G*. ... 34
Figure II.5 : Spectres IR et Raman du 1H-guanazole calculés au niveau DFT/B3LYP/6-31G*. ... 34
Figure II.6 : Variations (eau – gaz) des charges atomiques du 1H-guanazole à l’état gazeux et en solution dans l’eau. ... 47
Figure II.7 : Variations (eau – gaz) des longueurs de liaison en Å du 1H-guanazole à l’état gazeux et en solution dans l’eau. ... 50
Figure II.8 : Variations (eau – gaz) des angles en degré du 1H-guanazole à l’état gazeux et en solution dans l’eau. ... 50
Figure III.1 : Spectre IR du 1H-guanazole à l’état solide dans la région 4000-400 cm-1. ... ….……67
Figure III.2 : Spectre IR du guanazole deutérié à l’état solide dans la région 4000-500 cm-1. ... 67
Figure III.3 : Spectre Raman du 1H-guanazole à l’état solide dans la région 3600-200 cm-1 ... 68
Figure III.4 : Distances intermoléculaires du 1H-guanazole dans le cristal. ... 72
Figure IV.1 : Formes possibles pour le 1H-guanazole diprotoné avec numérotation des atomes. ... 90
Figure IV.2 : Charges atomiques des différentes formes diprotonées et leurs variations (dication - base) par protonation de la molécule 1H-guanazole. ... 93
Figure IV.3.a : Longueurs de liaison en Å de la forme protonée P24 et leurs variations (dication - base) par protonation de la molécule 1H-guanazole. ... 96
Figure IV.3.b : Angles valentiels en ° de la forme protonée P24 et leurs variations (dication - base) par protonation de la molécule 1H-guanazole. ... 96
Figure IV.4 : Structure optimisée de la forme protonée P24 et valeurs de densités de charge ... 99
Figure V.1 : Courbe de titrage du chlorhydrate de guanazolium avec le borax N/10. ... 126 Figure V.2 : Spectre IR du chlorhydrate de guanazolium à l’état solide dans la région
4000-400 cm-1. ... 128 Figure V.3 : Spectre IR du chlorhydrate de guanazolium deutérié à l’état solide dans la
région 4000-500 cm-1. ... 128 Figure V.4 : Spectre Raman du chlorhydrate de guanazolium à l’état solide dans la région
3600-200 cm-1. ... 129
Figure V.5: Spectre Raman du bromhydrate de guanazolium à l’état solide dans la région 3600-200 cm-1. ... 148
1
INTRODUCTION GENERALE
Les 1,2,4-triazoles constituent une classe importante de composés hétérocycliques et font l’objet d’une recherche très active dans le monde en raison de leurs diverses propriétés [1]. Principalement, ils sont omniprésents dans de nombreux agents pharmaceutiques ayant notamment des propriétés analgésiques [2-4], anti-inflammatoires [2,4], anti-dépressantes [5], anti-tumorales [6-9], anti-microbiennes [10,11], bactériennes [12-14] ou anti-tuberculeuses [15]. Concernant d'autres activités biologiques, ces systèmes possèdent en agrochimie un large spectre d'activités fongicidales [16-18] et herbicidales [19,20]. Leur efficacité en tant qu'insecticides [21] ou bactéricides [22,23] a également été mise en évidence. Par ailleurs, les 1,2,4-triazoles sont largement utilisés comme précurseurs dans la synthèse de nombreux hétérocycles à propriétés spécifiques [24-28].
En génie industriel, des dérivés triazoliques ont été testés pour leurs efficacités comme inhibiteurs de corrosion [29-35] et constituent une classe prometteuse utilisable dans les nouvelles technologies comme matériaux potentiels de stockage d’information, en optique non linéaire [36,37] ou encore comme nouveaux matériaux énergétiques pour des applications civiles et militaires [38-48].
Suite à cet intérêt croissant pour le noyau 1,2,4-triazole, et dans le cadre des travaux entrepris au laboratoire depuis plusieurs décennies, nous nous sommes proposée d’étudier le 3,5-diamino-1,2,4-triazole connu aussi sous le nom de "guanazole", particulièrement intéressant pour ses diverses activités.
A titre d’exemple, cette molécule possède des effets avérés, en milieu acide ou basique, pour la prévention contre la corrosion de métaux comme le cuivre et alliages tel que l’alliage d’aluminium 3003 [49,50].
Dans le domaine médical, le guanazole est un agent cytotoxique utilisé dans le traitement de l’anémie[51]. En outre, il a été le premier triazole utilisé en chimiothérapie. En effet, dès 1970, plusieurs auteurs avaient montré que le guanazole présentait une activité anti-tumorale contre les leucémies L-1210 et K-1964 [52,53]. D’autres travaux ont confirmé ultérieurement ces propriétés [54-59].
En astrobiologie, le guanazole est un candidat important en tant que molécule prébiotique, servant de base alternative (réplique ARN) dans l’élaboration de systèmes génétiquesprimordiaux [60,61].
2 En synthèse organique, le guanazole constitue un réactif de base pour l’élaboration de plusieurs molécules hétérocycliques ayant des activités biologiques importantes [62-64].
Par ailleurs, le guanazole est utilisé dans la synthèse de nouveaux matériaux hybrides organiques-inorganiques, servant à l’élaboration d’architectures tubulaires à propriétés spécifiques (absorption de gaz, échange ionique, catalyse hétérogène, électromagnétisme, optique…) [65-68]. En particulier, de nouveaux matériaux énergétiques à base de guanazole ont impliqué ces systèmes dans plusieurs programmes de recherche où une nouvelle approche dans le champ des explosifs et de la balistique est abordée afin de remplacer les explosifs conventionnels [69-73].
Ces nombreuses applications rendent les études visant à obtenir plus d’informations sur la structure et la réactivité de cette molécule particulièrement intéressantes. Ce travail comprend une étude des propriétés spectroscopiques et structurales de la molécule de guanazole et d'un sel protoné de guanazolium par des méthodes de chimie quantique ab-initio (HF, DFT) et par spectroscopie de vibration dans l'infrarouge et le Raman.
Après avoir donné dans un premier chapitre une revue bibliographique concernant le guanazole et ses sels protonés, nous exposerons dans un second chapitre les résultats d'une étude théorique des propriétés énergétiques, géométriques, électroniques et vibrationnelles du guanazole à l’état gazeux et en solution.
Le troisième chapitre sera consacré à une étude expérimentale par spectroscopie de vibration du guanazole et de son homologue deutérié et une comparaison avec les données théoriques afin de confirmer les attributions des vibrations fondamentales et de préciser les éventuels couplages mis en jeu.
Dans le quatrième chapitre, nous présenterons les résultats du calcul théorique réalisé pour le guanazole protoné afin d'approcher ses propriétés énergétiques, géométriques, électroniques et vibrationnelles.
Le cinquième chapitre fera l’objet d’une analyse approfondie des spectres expérimentaux infrarouges et Raman du chlorhydrate de guanazolium, dans le but de déterminer les sites d’interaction du guanazole vis-à-vis du proton, de relever l'effet de la protonation sur les vibrations fondamentales et de préciser la nature et la force de la liaison hydrogène dans le chlorhydrate sur la base d'une comparaison avec le guanazole, les calculs théoriques relatifs au cation et le spectre vibrationnel du bromhydrate de guanazolium.
3
BIBLIOGRAPHIE
1- A.D.M. Curtis, N. Jennings - Reference Module in Chemistry, Molecular Sciences and Chemical Engineering Comprehensive Heterocyclic Chemistry III - Volume 5: Five-membered Rings: Triazoles, Oxadiazoles, Thiadiazoles and their Fused Carbocyclic Derivatives 2008, Pages 159-209.
2- B. Tozkoparan, E. Kupeli, E. Yesilada, M. Ertan, Bioorg. Med. Chem. 15 (2007) 1808. 3- A.M. Vijesh, A.M. Isloor, P. Shetty, S. Sundershan, H. Kun Fun, Eur. J. Med. Chem. 62
(2013) 410.
4- D. Sarigol, A. Uzgoren-Baran, B.C. Tel, E.I. Somuncuoglu, I. Kazkayasi, K. Ozadali-Sari, O. Unsal-Tan, G. Okay, M. Ertan, B. Tozkoparan, Bioorg. Med. Chem. Lett. 23 (2015) 2518.
5- J.K. Sahu, S. Ganguly, A. Kaushik, Chin. J. Nat. Med. 11 (2013) 456.
6- K. Sztanke, T. Tuzimski, J. Rzymowska, K. Pasternak, M. Kandefer-Szerszeń, Eur. J. Med. Chem. 43 (2008) 404.
7- B.N.P. Kumar, K.N. Mohana, L. Mallesha, J. Fluor. Chem. 156 (2013) 15. 8- M.M. Kamel, N.Y. Megally Abdo, Eur. J. Med. Chem. 86 ( 2014) 75.
9- H. Cai, X. Huang, S. Xu, H. Shen, P. Zhang, Y. Huang, J. Jiang, Y. Sun, B. Jiang, X. Wu, H. Yao, J. Xu, Eur. J. Med. Chem. 108 (2016) 89.
10- V. Padmavathi, G.S. Reddy, A. Padmaja, P. Kondaiah, Ali-Shazia, Eur. J. Med. Chem. 44 (2009) 2106.
11- H. Bekta, N. Karaali , D. Şahin, A. Demirba, Ş.A. Karaoglu, N. Demirba, Molecules 15 (2010) 2427.
12- B. Kahveci, O. Bekircan, M. Serdar, A.A. Ikizler, Indian J. Chem., Section B 42 (2003) 1527.
13- P. Zoumpoulakis, C. Camoutsis, G. Pairas, M. Soković, J. Glamočlija, C. Potamitis, A. Pitsas, Bioorg. Med. Chem. Lett. 20 (2012) 1569.
14- Z. Shi, Z. Zhao, M. Huang, X. Fu, C. R. Chimie 18 (2015) 1320.
15- K.M. Krishna, B. Inturi, G.V. Pujar, M.N. Purohit, G.S. Vijaykumar, Eur. J. Med. Chem. 84 (2014) 516.
16- H.A. Torres, R.Y. Hachem, R.F. Chemaly, D. Kontoyiannis, I.I. Raad, Lancet Infect. Dis. 12 (2005) 775.
4 17- X. Chai, J. Zhang, Y. Cao, Y. Zou, Q. Wu, D. Zhang, Y. Jiang, Q. Sun, Eur. J. Med. Chem. 46 (2011) 3167.
18- L.Y. Zhang, B.L. Wang, Y.Z. Zhan, Y. Zhang, X. Zhang, Z.M. Li, Chin. Chem. Lett. 27 (2016) 163.
19- M. Hartmann, H.J. Bauer, K. Wermann, Biocide Polym. (1985) 195.
20- H.G. Hahn, J.S. Choi, H.K. Lim, K.I. Lee, I.T. Hwang, Pestic. Biochem. Physiol.125 (2015) 78.
21- Y.D. Li, W.T. Mao, Z.J. Fan, J.J. Li, Z. Fang, X.T. Ji, X.W. Hua, G.N. Zong, F.Y. Li, C.L. Liu, J.H. Yu, Chin. Chem. Lett. 24 (2013) 1134.
22- S.R. El-Zemity, A.M. El-Shazly, E.A. Kadous, J. Appl. Sci. Res. 2 (2006) 1314.
23- O.Y. Vasylkiv, O.I. Kubrak, K.B. Storey, V.I. Lushchak, Pestic. Biochem. Physiol. 101 (2011) 1.
24- R.M. Zemama, I. Amari, R. Bouhfid , E.M. Essassi , S.W. Ng, Acta Cryst. E65 (2009) 2152.
25- V.M. Chernyshev, A.V. Astakhov, Z.A. Starikova, Tetrahedron 66 (2010) 330. 26- Y. Sugihara, K. Onda, M. Sato, T. Suzuki, Tetrahedron Lett. 51 (2010) 4110. 27- B. Balan, D. Bahulayan, Tetrahedron Lett. 55 (2014) 227
28- D.M. Khomenko, R.O. Doroschuk, V.V. Trachevskii, S. Shova, R.D. Lampeka, Tetrahedron Lett. 57 (2016) 990.
29- F. Bentiss, M. Lagrenee, M. Traisnel , J.C. Hornez, Corros. Sci. 41 (1999) 789.
30- A. Lgamri, H. Abou El Makarim, A. Guenbour, A. Ben Bachir, L. Aries, S. El Hajjaji, Prog. Org. Coat. 48 (2003) 63.
31- E.S.M. Sherif, J. Ind. Eng. Chem. 19 (2013) 1884.
32- K.R. Ansari, M.A. Quraishi, A. Singh, Corros. Sci. 79 (2014) 5.
33- L. Jiang, Y. Lan, Y. He, Y. Li, Y. Li, J. Luo, Thin Solid Films 556 (2014) 395.
34- M. El Belghiti, Y. Karzazi, A. Dafali, B. Hammouti, F. Bentiss, I.B. Obot, I. Bahadur, E.E. Ebenso, J. Mol. Liq. 218 (2016) 281
35- M.E. Belghiti, Y. Karzazi, A. Dafali, I.B. Obot, E.E. Ebenso, K.M. Emran, I. Bahadur, B. Hammouti, F. Bentiss, J. Mol. Liq. 216 (2016) 874.
36- I. Matulková, I. Němec, K. Teubner, P. Němec, Z. Mička, J. Mol. Struct. 873 (2008) 46. 37- M. Abbas, G. Cakmak, N. Tekin, A. Kara, H.Y. Guney, E. Arici, N.S. Sariciftci, Org.
Elect. 12 (2011) 497.
38- A.K Sikder, M Geetha, D.B Sarwade, J.P. Agrawal, J. Hazard. Mater. 82 (2001) 1. 39- S.S. Yun, J.K. Kim, C.H. Kim, J. Alloys Compd.408 (2006) 945.
5 40- C. Darwich, T.M. Klapötke, C.M. Sabate, Chem. Eur. J. 14 (2008) 5756.
41- L. Xue, F.Q. Zhao, X.L. Xing, Z.M. Zhou, K. Wang, H.X. Gao, J.H. Yi, R.Z. Hu, Thermochim. Acta 511 (2010) 174.
42- S. Cudziło, M. Nita, J. Hazard. Mater. 177 (2010) 146.
43- L. Wallace, C.J. Underwood, A.I. Day, D.P. Buck, New J. Chem. 35 (2011) 2894. 44- V. Thottempudi, J.M. Shreeve, J. Am. Chem. Soc. 133 (2011) 19981.
45- V.D. Ghule, J. Comput. Theor. Chem. 992 (2012) 92.
46- H. Zhou, Z.L. Ma, J.L. Wang, D. Wang, Defence Technology 10 (2014) 384. 47- Y. Tang, H. Gao, D.A. Parrish, J.M. Shreeve, Chem. A Euro. J. 27 (2015) 11401. 48- L. Türker, Defence Technology 12 (2016) 1.
49- R. Salghi, L. Bazzi, B. Hammouti, A. Bouchtart, S. Kertit, Z.A. Ait Addi, Z. El Alami, Ann. Chim. Sci. Mat. 25 (2000) 187.
50- S. El Issami, L. Bazzi, M. Hilali, R. Salghi, S. Kertit, Ann. Chim. Sci. Mat. 27 (2002) 63. 51- J.A. Ho, C.V. Pickens, P.M. Gamscik, O.M. Colvin, R.E.Ware, Exp. Hematol. 31 (2003)
586.
52- M. Hahn, R. Adamson, Pharmacologist 12 (1970) 282.
53- J.A. Montgomery, Annual Reports in Medicinal Chemistry 5 (1970) 144. 54- M.F. Stanton, V.J. Heston, CA-Cancer J. Clin. 22 (1972) 127.
55-W.A. Oddy, Stud. Conserv. 19 (1974) 188. 56- J.A. Levi, P.H. Wiernik, Cancer 38 (1976) 36.
57- R.N. Feinstein, R.J.M. Fry, E.E. Staffled, J. Environ. Pathol. Toxicol. 1 (1978) 779. 58- M.K. Islyaikin, E.A. Danilova, E.V. Kudrik, R.P. Smirnov, A.P. Budunova, A.S.
Kinzirskii, Khim. Farm. Zh. 31 (1997) 19.
59- M. Kruger, O. Petrov, K.H. Thierauch, G. Siemeister, United State, Patent Application, 20040186288, 23 septembre, 2004.
60- V.M. Kolb, J.P. Dworkin, S.L. Miller, J. Mol. Evol. 38 (1994) 549.
61- R.S. Perry, V.M. Kolb, Instrument, Methods and Missions for Astrobiology VII (2003). 62- F. Fernández-Lázaro, S. Rodriguez-Morgade, T. Torres, Synth. Met. 62 (1994) 281. 63- S. Leyva, E. Leyva, Tetrahedron 63 (2007) 2093.
64- S.M. Sondhi, S. Jain, M. Dinodia, R. Shukla , R. Raghubir, Bioorg. Med. Chem. Lett. 15 (2007) 3334.
65- A.M. Goforth, C.Y. Su, R. Hipp. R.B. Macquart, M.D. Smith, H.C. zur Loye, J. Solid State Chem. 178 (2005) 2511.
6 67- Q. Zhai, C. Lu, Q. Zhang, X. Wu, X. Xu, S. Chen, L. Chen, Inorg. Chim. Acta 359 (2006)
3875.
68- H. Zhao, H.X. Li, Z.Y. Liu, E.C. Yang, X.J. Zhao, Polyhedron 101 (2015) 29.
69- D.E. Chavez, B.C. Tappan, B.A. Mason, D.A. Parrish, Propellants Explos. Pyrotech. 34 (2009) 475.
70- I. Matulková, I. Císařová, P. Němec, J. Kroupa, P. Vaněk, N. Tesařová, I. Němec, J. Mol. Struct. 1044 (2013) 239.
71- Y.H. Ren, W. Li, F.Q. Zhao, J.H. Yi, B. Yan, H.X. Ma, K.Z. Xu, J.R. Song, R.Z. Hu, J. Anal. Appl. Pyrolysis 102 (2013) 89.
72- J. Ge, Q. Yang, G. Xie, S. Chen, S. Gao, J. Chem. Thermodyn.80 (2015) 1. 73- Q. Yang, X. Song, J. Ge, G. Zhao, W. Zhang, G. Xie, S. Chen, S. Gao, J. Chem.
7
CHAPITRE I
REVUE BIBLIOGRAPHIQUE
INTRODUCTION
La structure moléculaire du guanazole a été le sujet de controverses car différentes formes tautomères sont supposées exister comme on peut le noter sur la figure I.1.
Figure I.1 : Formes tautomères possibles pour le guanazole.
Certains travaux antérieurs, consacrés à cette molécule, ont permis de conclure à la forme tautomère prédominante dans différents états physiques. Selon la forme tautomère envisagée, la molécule présente un caractère amphotère résultant de la présence simultanée d’un groupement NH "pyrrolique", d’azotes tertiaires "pyridiniques" et d’azotes du groupement amino ou imino.
Plusieurs sites d’interaction vis-à-vis de divers agents électrophiles tels que le proton ou les ions métalliques sont possibles d’après les travaux antérieurs relatifs à cette molécule.
Nous présenterons, dans un premier temps, les divers résultats obtenus pour la molécule de guanazole étudiée. Nous traiterons ensuite des données de la littérature concernant l’interaction qui nous intéresse à savoir celle du guanazole avec le proton.
8 I. MOLECULE DE GUANAZOLE
I.1. Synthèse
Le guanazole a été synthétisé pour la première fois par Pellizzari en 1894 [1]. L’auteur a obtenu, avec un faible rendement, le produit par chauffage dans l’alcool du dicyanodiamide avec le monochlorhydrate d’hydrazine selon le schéma réactionnel suivant :
Il a conclu que la molécule de guanazole existe dans ce milieu sous la forme 3,5-di-imino-1,2,4-triazole (Figure I.1, Forme III).
Ultérieurement, en 1953, en faisant réagir le dicyanodiamide avec un dihalogénohydrate d’hydrazine dans l’eau, Kaiser et al. [2] ont pu nettement améliorer le rendement jusqu’à approcher une valeur sensiblement théorique. Jusqu’à ce jour, cette combinaison entre le composé d’hydrazine et l'eau comme solvant est la seule à aboutir à un rendement élevé en guanazole avec le dicyanodiamide.
I.2. Etude par spectroscopie UV-visible
Les premiers travaux par spectroscopie UV-visible portant sur la structure du guanazole ont été réalisés par Davidson en 1969 [3]. Ce composé existe selon cet auteur sous deux formes tautomères : forme 4H-3,5-diamino-1,2,4-triazole (Figure I.1, Forme II) et la forme 3,5-di-imino-1,2,4-triazole (Figure I.1, Forme III).
Par contre, Reiter et al. en 1986 [4], en examinant les spectres UV du guanazole et de certains de ses dérivés de formes bloquées, ont conclu que cette molécule existe en solution sous l’une ou l’autre des formes 1H équivalentes (Figure I.1, Forme I).
I.3. Etude par spectroscopie RMN
Une étude des spectres RMN1H et RMN13C, effectuée par Reiter et al. pour le guanazole et ses dérivés N-substitués [4] , a permis d’aboutir à la même conclusion que celle obtenue par ces mêmes auteurs par spectroscopie UV-visible, à savoir que le guanazole existe sous la forme 1H à l’état dissous.
9 Dans le cadre des travaux effectués par Klapötke et al. en 2010 concernant la protonation du guanazole, les auteurs ont étudié au préalable les spectres de résonance magnétique nucléaire de la molécule non protonée [5]. En solution dans DMSO-d6, pour la
forme 1H, les auteurs ont conclu à l’existence d’un échange lent de l’atome d’hydrogène "pyrrolique" entre les atomes d’azote N1 et N2 (Figure I.1, Forme I). En effet, la largeur du signal à 10.68 ppm en RMN1H attribuable à l’hydrogène "pyrrolique" indique bien un échange prototropique à la température ambiante. Concernant les groupements amino, on note la présence de deux signaux à 5.56 et 4.68 ppm en RMN1H et à 161.8 et 156.4 ppm en RMN13C.
I.4. Etude par diffraction des rayons X
La structure cristalline du guanazole à l’état solide a été résolue par Starova et al. en 1979 [6]. Cette molécule existe sous la forme 1H-3,5-diamino-1,2,4-triazole et cristallise dans le système monoclinique, de groupe d’espace P21/b. Les paramètres de la maille unitaire
sont:
a = 10,658(2) Å b = 10,837(2) Å c = 4,339 (1) Å γ = 118,83 (2) Å
Le nombre de groupements formulaires Z est égal à 4.
Les données géométriques, reportées sur la figure I.2, montrent que dans le cristal, les longueurs de liaison C-N ont une valeur intermédiaire entre une simple et une double liaison et que la liaison N-N a une même valeur qu’une liaison simple.
10 Le noyau triazolique est plan ; les deux groupes amino portés par les carbones C3 et C5 sont inclinés de 21,5° et 29,5° respectivement par rapport au plan de ce dernier. Les valeurs des longueurs de liaison et des angles obtenues pour le guanazole sont voisines de celles trouvées pour le 1,2,4-triazole [7] et son dérivé monosubstitué 1H-5-amino-1,2,4-triazole [8]. De plus, parmi les deux groupements C3-NH2 et C5-NH2, c'est ce dernier, situé à
proximité du NH "pyrrolique", qui possède les données géométriques les plus proches de celles trouvées dans le cas de la molécule 1H-5-amino-1,2,4-triazole [8].
Sur la figure I.3, nous avons reproduit la maille élémentaire du 1H-3,5-diamino-1,2,4-triazole [6] projetée sur le plan (001), montrant le type d’interactions moléculaires que présente ce composé dans le cristal.
Figure I.3 : Projection de la structure du guanazole selon le plan (001) [6].
Les contacts intermoléculaires existants pour cette molécule dans le cristal sont regroupés dans le tableau I.1. La numérotation des atomes est celle adoptée sur la figure I.3.
Tableau I. 1: Distances en Å et angles en ° des contacts intermoléculaires du guanazole dans le cristal [6].
Liaisons Distances (Å) Angles (°)
N……..N N……H N…H...N
N1-H1……N2′ 2.947(2) 2.12(2) 181(2)
N5-H52′…..N4′ 2.992(2) 2.11(2) 168(2)
N3-H31…...N2″ 3.101(2) 2.29(2) 146(2)
11 I.5. Etude théorique
A l’état gazeux, le problème de la tautomérie du guanazole a été analysé par des calculs de chimie théorique. Ainsi, Makarskii et al. [9] ont calculé, par les méthodes semi-empiriques CNDO/2 et MINDO/2, les moments dipolaires et les chaleurs de formation pour les cinq formes tautomères probables pour le guanazole (Figure I.1). Ils ont montré que la forme dans laquelle l’hydrogène est lié à l’azote N1 est la plus stable (Figure I.1, Forme I).
En 2008, Karpinska et al. [10] ont étudié plus de 30 tautomères, conformères et zwitterions de cette molécule en utilisant la méthode DFT au niveau B3LYP/aug-cc-pVDZ à l’état gazeux et la méthode IEF-PCM en solution aqueuse. Les quatre structures les plus stables ont été reoptimisées au niveau B3LYP/aug-cc-pVTZ à l’état gazeux et au niveau IEF-PCM/B3LYP/ aug-cc-pVDZ en solution dans l’eau. Les résultats du calcul ont montré que le tautomère 1H (Figure I.1, Forme I) semble être l'unique forme stable à la fois dans la phase gazeuse et en solution. Le tautomère dont la stabilité suit celle de la forme 1H est le 4H-guanazole (Figure I.1, Forme II).
I.6. Etude vibrationnelle
Les analyses spectroscopiques vibrationnelles (IR, Raman) effectuées antérieurement restent fragmentaires et souvent discutables. Ainsi, Barmin et al. [11] ont, à titre d’exemple, situé le mode νNH à 3000 cm-1, fréquence qui nous semble relativement basse pour la molécule
de guanazole, si on se réfère aux données radiocristallographiques concernant les contacts intermoléculaires [6] (Figure I.3 ; Tableau I.1).
En ce qui concerne les travaux de Lopyrev et al. [12], Desseyn et al. [13], Gabryszewski et al. [14,15], les attributions se sont pour la plupart limitées aux vibrations des groupements NH et NH2.
En 2005, Kumar et al. [16] ont effectué des calculs d’énergie et de fréquences de vibration pour le guanazole à l’état libre par application de la méthode de Théorie de la Fonctionnelle de la Densité (DFT/B3LYP /6-31G*). Les calculs des énergies ont été menés en considérant uniquement la molécule de forme 4H (Figure I.1, forme II) sous les symétries C1,
Cs, C2 et C2v et ont permis de conclure qu’elle existe sous la symétrie C2. Les spectres
vibrationnels calculés sous cette symétrie ont été étudiés par une analyse en coordonnées normales basée sur le champ de force de la fonctionnelle de la densité. D’autre part, ces auteurs ont utilisé les résultats de ces calculs pour interpréter les spectres vibrationnels expérimentaux du guanazole à l'état solide. Ceci nous semble discutable car les résultats
12 théoriques obtenus pour la forme 4H ne peuvent pas être exploités afin d’analyser les spectres expérimentaux du guanazole solide existant sous la forme 1H [6].
I.7. Etude électrochimique
Les 1,2,4-triazoles sont des inhibiteurs de corrosion peu toxiques et pourraient remplacer les inhibiteurs utilisés actuellement dans l’industrie (oxychromate de Zn et Sr) menacés d’interdiction à cause de leur caractère cancérigène. Dans ce sens, des études électrochimiques de l’inhibition de la corrosion par le 3,5-diamino-1,2,4-triazole ont été réalisées. Les matériaux utilisés sont l’alliage d’aluminium 3003 en milieu basique [17] et le cuivre en milieu acide [18]. Les résultats ont été comparés aux dérivés triazoliques tels que le 1,2,4-triazole, le 5-amino-1,2,4-triazole et le 3,4’-bi-1,2,4-triazole et ont permis de montrer que, dans cette série triazolique, le guanazole présente la meilleure efficacité inhibitrice de corrosion à la fois dans le cas de l’alliage et dans celui du cuivre. Pour ce dernier, les auteurs ont expliqué ce comportement par la présence des groupements amino qui semblent jouer un rôle déterminant dans l’inhibition de la corrosion car l’adsorption pourrait être favorisée par l’établissement de liaisons de type hydrogène.
II. SELS DE GUANAZOLIUM
Dans le cas des sels du guanazolium, peu d’études ont été réalisées dans la littérature. II.1. Etude par spectroscopie UV-visible
Une étude des spectres UV-visibles réalisée par Voronkov et al. [19] a montré que l'absorption observée pour le guanazole dans l’éthanol à 208 nm (ε = 6320) reste pratiquement inchangée en solution de HCl 0,1M mais subit un effet hyperchrome. Ce résultat semble indiquer que le système n’est pas touché par la protonation. En outre, un calcul des constantes d’ionisation du guanazole et d’un certain nombre de triazoles 3,5-disubstitués, a montré que la protonation du guanazole a lieu sur l’azote N4 du noyau et non sur un azote du groupement amino [19].
II.2. Etude par spectroscopie RMN
Des études RMN ont été réalisées par Klapötke et al. [5] pour des sels de guanazolium obtenus par protonation du guanazole avec les acides minéraux dilués HCl, HNO3 et HClO4. Le
spectre RMN1H du cation guanazolium montre un déplacement des signaux des groupements
13 à 5.56 et 4.68 ppm et correspondant aux groupements NH2 dans le guanazole apparaissent sous
forme d’un seul signal localisé vers 7.07 ppm pour le chlorhydrate de guanazolium, 6.97 ppm dans le cas du nitrate de guanazolium et 6.94 ppm pour le perchlorate de guanazolium. Concernant les groupements NH, un seul signal large est observé dans le cation comme dans le cas du guanazole mais il est déplacé de 10.68 ppm à 12.03 ppm, 12.08 ppm et 11.93 ppm pour le chlorhydrate, le nitrate et le perchlorate respectivement. Une même constatation a été faite en RMN13C : un seul signal correspondant aux deux carbones a été observé mais déplacé vers les champs forts par rapport à celui du guanazole.
Sur le spectre RMN1H du sel de fluoroaluminate de guanazolium [20], il a été constaté la présence de deux signaux correspondant aux deux groupements NH2 comme dans le cas du
guanazole mais avec un déplacement vers les champs faibles. Pour les groupements NH, deux signaux ont été notés dans le sel de guanazolium vers 10.2 ppm et 11.6 ppm.
II.3. Etude par diffraction des rayons X
L’étude par diffraction des rayons X réalisée par Fabretti [21] en 1999 pour le sel [Pt(3,5-diamino-1,2,4-triazolium)2Br2]Br2 a montré que la protonation s’effectue sur l’azote
N4 "pyridinique" et que le cation guanazolium est de forme 1H, 4H et coordiné au platine via l’azote N2. Les groupements NH et NH2 du cation guanazolium sont engagés dans des
liaisons hydrogène de type NH….Br. Les contacts intermoléculaires varient entre 2.395 et 2.564 Å.
Plus récemment, une série de sels de guanazolium a été synthétisée par action de solutions diluées de différents acides minéraux (HCl, HNO3, HClO4, H2SO4, H3PO4, HBF4,
H2SiF6) et organiques (malonique, glutarique, adipique, L-malique) et analysée par
radiocristallographie [5,20,22-24]. Les résultats des RX ont montré que, pour tous les sels étudiés, seuls les monocations ont été obtenus et que la protonation s’effectue sur l’azote "pyridinique" N4 du noyau triazolique. De plus, des liaisons hydrogène sont formées pour tous ces sels entre les hydrogènes des azotes N1, N4 et ceux des groupements amino avec les différents anions (Cl-, NO3-, ClO-4, SO42-…..). Les données des contacts intermoléculaires
14 Tableau I.2 : Données structurales antérieures publiées pour les sels de guanazolium monoprotonés.
Sel de guanazolium * Synthèse Liaison hydrogène Référence
Type de contact ** Distance en Å
(GuanH)2Cl2.H2O
0.99g de guanazole ajouté sous agitation à10 mL d’une solution d’HCl (1M) et chauffé pendant 10 mn à 50°C. Recristallisation dans l’eau. NH.….N′2 NH2.…N′2 NH2…Cl -NH.…Cl -d(N…. N′2) = 2.834 d( N…. N′2) = 3.038 [5] ; [22] (GuanH)NO3
0.99g de guanazole ajouté sous agitation à 5 mL d’une solution de HNO3 (2M) et chauffé pendant 10
mn à 50°C. Recristallisation dans un mélange d’eau et d’éthanol (1:1).
N4H….O NH2… O N1H….O d(H….O) = 1.945 d(H….O) = [2.057 – 2.643] d(H….O) = 2.048 (GuanH)ClO4 0.99g de guanazole ajouté à 10 mL d’une solution de HClO4 (1M) et
chauffé pendant 10 mn à 50°C. Recristallisation dans l’éthanol chaud.
NH2… N′2
NH2…O
NH.…O
d(N…. N′2) = [2.997 -3.032]
(GuanH)2SO4.2H2O Dissolution du guanazole dans une
solution aqueuse de H2SO4 ou
H3PO4 avec un rapport molaire
1 :1. NH2…. N′2 NH.….O d(N…. N′2) = [2.979 – 3.039] d(N……O) = [2.896 – 3.095] [22] (GuanH)H2PO4 NH 2…. N′2 NH.….O d(N…. N′2) = 2.906 d(N……O) = [2.710 – 3.001] (GuanH)Malonique a
Dissolution du guanazole dans une solution aqueuse d’acides
malonique ou glutarique avec un rapport molaire 1 :1. NH2…. N′2 NH.…..O d(N…… N′2) = 2.970 d(N…….O) = [2.694– 3.048] [23] (GuanH)Glutarique b NH2…. N′2 NH.….O OH…..O d(N…. N′2) = 2.880 d(N….O) = [2.694 – 3.048] d(H….O) = 2.970
15
* (GuanH) : guanazole monoprotoné ** N′ : azote d’une seconde molécule
(GuanH)Adipique c Dissolution du guanazole dans une solution aqueuse d’acide adipique + méthanol avec un rapport molaire 1 :1. NH….. N′2 OH…..O d(N…. N′2) = 3.017 d(H….O) = 2.568 [23] (GuanH)L-malique d Dissolution du guanazole dans une
solution aqueuse d’acide L-malique avec un rapport molaire 1 :1.
NH2…. N′2
OH……O
d(N…. N′2) = [2.974– 3.039]
(GuanH)BF4 Dissolution du guanazole dans une
solution de méthanol avec HBF4
(40%) avec un rapport molaire 1:3
NH2…. N′2 NH2…..F N4H…..F N1H…..F d(H…. N′2) = 2.16 d(H….F) = [2.29 – 2.38] d(H….F) = 1.98 d(H….F) = 2.24 [24]
(GuanH)2SiF6 Dissolution du guanazole dans une
solution de méthanol avec H2SiF6
(48%) avec un rapport molaire1:3
NH2….N2 NH2….F N4H…..F N1H…..F d(H….N) = 2.17 d(H….F) = [2.00 – 2.11] d(H….F) = 1.90 d(H….F) = 2.01 (GuanH)2AlF5 Mélange de Al(OH)3, guanazole,
acide fluorhydrique (40%) et éthanol avec un rapport molaire : 1/3/6/17 N1H…..F N4H…..F NH2…. F NH2…. N′2 d(N1….F) = 2.642 d(N4….F) = 2.620 d(N….F) = [2.756 – 2.877] d(N…. N′2) = 2.964 [20]
(GuanH)2Al2F8 Mélange de Al(OH)3, guanazole,
acide fluorhydrique(40%) et éthanol avec un rapport molaire :
1/0.7/3.7/17 N1H…..F N4H…..F NH2…..F NH2…. N′2 d(N1….F) = 2.716 d(N4….F) = 2.626 d(N….F) = 2.820 d(N…. N′2) = 3.081
[GuanH)2](AlF5(H2O)).2H2O Mélange de Al(OH)3, guanazole,
acide fluorhydrique(40%) et éthanol avec un rapport molaire : 1/5/4/17
N1H….OH2O N4H…..F NH2…..F NH2….OH2O NH2…. N′2 d(N….O) = [2.764 – 2.785] d(N4….F) = [2.638 – 2.664] d(N….F) = [2.764 – 2.979] d(N….O) = 3.044 d(N…. N′2) = 3.077 [20]
16 Parmi les sels de guanazolium étudiés, nous nous sommes particulièrement intéressée à l’attaque du guanazole par la solution diluée d’acide chlorhydrique (milieu dans lequel nous avons travaillé mais dans des conditions différentes). Le composé obtenu est le 3,5-diamino-1,2,4-triazolium semihydraté noté (GuanH)2.Cl2.H2O. Son étude structurale par diffraction des
rayons X a montré que ce composé cristallise dans le système monoclinique avec un groupe d’espace P21/n et 8 molécules par maille. La figure I.4. présente une vue de la structure
suivant la direction [100]. Les traits en pointillés montrent les différents types de liaisons hydrogène NH…N et NH….Cl.
Figure I.4 : Vue de la structure du sel (GuanH)2.Cl2.H2O suivant la direction [100].
Klapötke et al. [5] ont montré que dans ce sel, tous les hydrogènes des vibrateurs NH "pyrroliques" et ceux des groupements amino sont engagés dans de fortes liaisons hydrogène avec les ions chlorures comme c’est représenté sur la figure I.5.
Figure I.5 : Liaisons hydrogène dans le sel (GuanH)2.Cl2.H2O
En outre, des associations intermoléculaires de type NH….N sont également observées. Les distances N…..N sont de l’ordre de 2.834 Å et de 3.038 Å.
17 II.4. Etude vibrationnelle
Une étude vibrationnelle partielle a été faite pour l’ensemble des sels de guanazolium étudiés par Matulková et al. [22,23] et Goreshnik et al. [24]. L’analyse des spectres a montré la présence d’une bande forte et large entre 3500 et 2600 cm-1 décomposée en trois
composantes et attribuée à des vibrations NH des vibrateurs NH engagés dans des liaisons
hydrogène de type NH….N et NH…..O.
Les deux premières absorptions, centrées vers 3100 et 2600 cm-1, sont assignées aux deux vibrations NH dues à des liaisons hydrogène de type NH…N avec des distances
intermoléculaires égales à 3.07 Å et 2.83 Å respectivement. La troisième, localisée dans la région 3350-3100 cm-1, est affectée à la liaison NH…..O avec des distances intermoléculaires
variant de 3.04 à 2.69 Å. Signalons que ces auteurs, lors de leurs attributions, n’ont pas fait de distinction entre les vibrateurs NH "pyrroliques" et ceux des groupements amino. Par ailleurs, des attributions ont été données pour quelques modes cycliques et de déformation des groupements NH2 mais elles restent fragmentaires.
CONCLUSION
D'après cette revue bibliographique, la plupart des études antérieures relatives à la molécule de guanazole ont montré qu'elle existe, dans tous les états physiques, préférentiellement sous la forme 1H. A notre connaissance, aucune étude vibrationnelle approfondie n'a été publiée pour cette forme. Concernant le cation guanazolium, la protonation a permis d'aboutir pour tous les sels étudiés à la formation d'un monocation protoné sur l'azote N4. Aucun sel n'a été étudié de façon systématique et approfondie par la spectroscopie de vibration ou par les méthodes de calcul ab-initio.
18
BIBLIOGRAPHIE
1- G. Pellizzari, J. Chem. Soc. 55 (1894) 517.
2- D.W. Kaiser, G.A. Peters, V.P. Wystrach, J. Org. Chem. 18 (1953) 1610. 3- J.S. Davidson, J. Chem. Soc. C (1969) 194.
4- J. Reiter, L. Pongo, J. Heterocyclic. Chem. 23 (1986) 401.
5- T.M. Klapötke, F.A. Martin, N.T. Mayr, J. Stierstorfer, Z. Anorg. Allg. Chem. 636 (2010) 2555.
6- G.L. Starova, O.V. Frank-Kamenetskaya, E.F. Shibanova, V.A. Lopyrev, M.G. Voronkov, V.V. Makarskii, J. Chem. Heterocyc. Comp. 15 (1979) 1149, Translated from Khim. Geterotsikl. Soedin. 10 (1979) 1422.
7- P. Goldstein, G. Labell, G. Abowitz, Acta Cryst. B25 (1969) 135.
8- G.L. Starova, O.V. Frank-Kamentskaya, V.V. Makarski, A. Lopyrev, Sov . Phys. Cryst. 23 (1978) 478.
9- V.V. Makarskii, V.A. Zubkov, V.A. Lopyev, M.G. Voronkov, Khim. Geterotsikl. Soedin. 4 (1977) 540.
10- G. Karpinska, J.Cz. Dobrowolski, J. Mol. Struct. (Theochem) 853 (2008) 7.
11- M.I. Barmin, E.L. Kassatikova, I.B. Karaaulova, V.V. Mel’Nicov, Russian J. Coord. Chem. 22 (1996) 434.
12- V.A. Lopyrev, N.K. Beresneva, B. Kh. Strelets, Khim. Geterotsikl. Soedin. 5 (1969) 732. 13- H.O. Desseyn, A.C. Fabretti, W. Malavasi, J. Cryst. Spectrosc. Res. 20 (1990) 4.
14- M. Gabryszewski, B. Wieczorek, Pol. J. Chem. 72 (1998) 2352. 15- M. Gabryszewski, B. Wieczorek, Pol. J. Chem. 73 (1999) 2061.
16- V.K. Kumar, G. Keresztury, T. Sundius, R.J. Xavier, Spectrochim. Acta 61A (2005) 261. 17- R. Salghi, L. Bazzi, B. Hammouti, A. Bouchtart, S. Kertit, Z.A. Ait Addi, Z. El Alami,
Ann. Chim. Mat. 25 (2000) 187.
18- S. El Issami, L. Bazzi, M. Hilali, R. Salghi, S. Kertit, Ann. Chim. Mat. 27 (2002) 63. 19- M.G. Voronkov, T.V. Kashik, V.V. Makarskii, V.A. Lopyrev, S.M. Ponomareva, E.F.
Shibanova, Dakl. Acad. Nauk SSSR 227 (1976) 1116.
20- A. Cadiau, A. Le Bail, A. Hémon-Ribaud, M. Leblanc, M. Body, F. Fayon, E. Durand, J. Boulou, V. Maisonneuve, Crystal Growth & Design 10 (2010) 5159.
19 22- I. Matulková, J. Cihelka, M. Pojarová, K. Fejfarová, M. Dušek, P. Vaněk, J. Kroupa, R.
Krupková, J. Fábry, I.Neměc, J. Crystengcomm 14 (2012) 4625.
23- I. Matulková, I. Císarová, P. Neměc, J. Kroupa, P. Vaněk, N. Tesařová, , I. Nemc, J. Mol. Struc. 1044 (2013) 239.
24- E.A. Goreshnik, V.O. Gelmboldt, L.V. Koroeva, E.V. Ganin, J. Fluorine Chem. 132 (2011) 138.
20
CHAPITRE II
ETUDE THEORIQUE PAR LES METHODES AB-INITIO DES
PROPRIETES ENERGETIQUES, GEOMETRIQUES,
ELECTRONIQUES ET VIBRATIONNELLES DU
3,5-DIAMINO-1,2,4-TRIAZOLE
INTRODUCTION
Le présent chapitre porte sur une étude théorique du guanazole dans le but d’analyser les vibrations moléculaires de la forme tautomère la plus stable de cette molécule à l’état gazeux et en solution. Une analyse des propriétés énergétiques, géométriques et électroniques du guanazole a au préalable été réalisée. Pour ce faire, nous avons utilisé les méthodes théoriques de la chimie quantique : ab-initio au niveau Hartree-Fock (HF) et Théorie de la Fonctionnelle de la Densité (DFT) au niveau B3LYP.
Dans ce qui suit, nous exposerons d’abord les deux méthodes de calcul utilisées au cours de ce travail. Avec chacune de ces méthodes, nous avons déterminé la géométrie d’équilibre par une procédure d’optimisation, les caractéristiques énergétiques et les charges atomiques, ainsi que les fréquences vibrationnelles harmoniques. Ensuite, nous présenterons et discuterons les résultats de l’étude des propriétés géométriques, électroniques et vibrationnelles de la molécule 3,5-diamino-1,2,4-triazole en phase gazeuse ainsi que les résultats de l’étude vibrationnelle de son homologue totalement deutérié. La distribution d’énergie potentielle des modes normaux de vibration sera en outre analysée dans le but de servir à l'interprétation, de façon rigoureuse et précise, du spectre de vibration expérimental du 3,5-diamino-1,2,4-triazole donnée dans le chapitre III.
Enfin, nous présenterons les résultats de l'étude théorique à l’état dissous dans
l’éthanol et dans l’eau et nous les comparerons aux données obtenues pour le guanazole gazeux. Une telle étude a été effectuée dans le but d’approcher les conditions de l’état solide.
21 I. MÉTHODES DE CALCUL
Nous avons effectué sur la molécule neutre 3,5-diamino-1,2,4-triazole des études d’optimisation de géométrie et de calcul de fréquences vibrationnelles par les calculs ab-initio et DFT qui connaissent un développement important pour leurs applications en chimie.
Des travaux antérieurs [1,2] ont montré que l’introduction de l’énergie de corrélation électronique dans le calcul du champ de force et des fréquences de vibration donnait des résultats plus fiables et plus précis. L’utilisation de la méthode perturbationnelle Mller-Plesset au second ordre (MP2) [2] a permis de corriger les énergies Hartree-Fock en introduisant l’énergie de corrélation électronique. Cependant, cette méthode reste limitée à l’étude des molécules de petite taille. Les méthodes de la Théorie de la Fonctionnelle de la Densité permettent de considérer une grande partie de la corrélation électronique et constituent donc une alternative intéressante pour incorporer cette corrélation dans les calculs quantiques et cela avec un temps de calcul réduit. L’accord remarquable obtenu entre les calculs DFT et les valeurs expérimentales fait de cette méthode un outil très intéressant pour étudier les molécules de tailles moyenne et grande.
Les calculs théoriques ab-initio au niveau Hartree-Fock (HF) et Théorie de la Fonctionnelle de la Densité (DFT) ont été réalisés avec le programme de calcul quantique Gaussian 03 [3] pour la molécule de guanazole et son homologue totalement deutérié. Parmi les méthodes DFT, nous avons choisi d’utiliser la fonctionnelle hybride B3LYP [4-6] qui associe les trois paramètres de la fonctionnelle d’échange corrigée en gradient de Becke [4] à celle de corrélation corrigée en gradient de Lee, Yang et Parr (LYP) [5], combinée avec la base standard d’atomes 6-31G* [7-11].
Une présentation des différentes méthodes de calcul utilisées au cours de ce travail est donnée dans l’annexe I.
I.1. Structure et symétrie de la molécule
La structure de la molécule de guanazole a été déterminée par Starova et al. [12]. Les paramètres géométriques utilisés sont indiqués sur la figure II.1.
![Tableau I. 1: Distances en Å et angles en ° des contacts intermoléculaires du guanazole dans le cristal [6].](https://thumb-eu.123doks.com/thumbv2/123doknet/2183613.10669/27.892.127.760.983.1135/tableau-distances-a-angles-contacts-intermoleculaires-guanazole-cristal.webp)
![Figure II.1 : Paramètres géométriques expérimentaux de la molécule de guanazole [12].](https://thumb-eu.123doks.com/thumbv2/123doknet/2183613.10669/39.892.124.771.108.315/figure-ii-parametres-geometriques-experimentaux-de-molecule-guanazole.webp)